

Abstract
Exogenous glucocorticoids inhibit neointimal proliferation in animals. We aime to test the hypothesis that endogenous glucocorticoids influence neointimal proliferation; this may be mediated by effects on systemic risk factors or locally in vessels, and modulated either by adrenal secretion or by enzymes expressed in vessels which mediate local inactivation (11β-HSD2 in endothelium) or regeneration (11β-HSD1 in smooth muscle) of glucocorticoids. Femoral artery wire-angioplasty was conducted in C57Bl/6J, Apo-E−/−, 11β-HSD1−/−, Apo-E, 11β-HSD1−/− (double knockout) and 11β-HSD2−/− mice following glucocorticoid administration, adrenalectomy, glucocorticoid or mineralocorticoid receptor antagonism, or selective 11β-HSD1 inhibition. In C57Bl/6J mice, neointimal proliferation was reduced by systemic or local glucocorticoid administration, unaffected by adrenalectomy, reduced by the mineralocorticoid receptor antagonist eplerenone, and increased by the glucocorticoid receptor antagonist RU38486. 11β-HSD2 deletion had no effect on neointimal proliferation, with or without eplerenone. 11β-HSD1 inhibition or deletion had no effect in chow-fed C57Bl/6J mice, but reduced neointimal proliferation in Apo-E−/− mice on Western diet. Reductions in neointimal size were accompanied by reduced macrophage and increased collagen content. We conclude that pharmacological administration of glucocorticoid receptor agonists or of mineralocorticoid receptor antagonists may be useful in reducing neointimal proliferation. Endogenous corticosteroids induce beneficial glucocorticoid receptor activation and adverse mineralocorticoid receptor activation. However, manipulation of glucocorticoid metabolism has beneficial effects only in mice with exaggerated systemic risk factors, suggesting effects mediated primarily in liver and adipose rather than intra-vascular glucocorticoid signalling. Reducing glucocorticoid action with 11β-HSD1 inhibitors that are being developed for type 2 diabetes appears not to risk enhanced neointimal proliferation.
Keywords: Glucocorticoids, mineralocorticoids, Angioplasty, Neointimal proliferation, 11β-hydroxysteroid dehydrogenases (11β-HSDs)
INTRODUCTION
Neointimal proliferation is a key factor determining the success or failure (re-stenosis) of angioplasty in occlusive vascular disease. It also provides a paradigm through which to gain insights into vascular responses to injury that are relevant to other vascular lesions, notably atheroma. Glucocorticoids have anti-proliferative and anti-inflammatory properties that have been exploited pharmacologically to reduce post-angioplasty neointimal proliferation in rats, rabbits and dogs, albeit that human trials of glucocorticoid administration, either systemically or by stent elution, have had mixed results (reviewed in (1)). However, the influence, if any, of endogenous glucocorticoid signalling on neointimal proliferation remains uncertain. This is important since variations in circulating and/or intracellular glucocorticoid concentrations have been associated with cardiovascular disease risk (2), and new therapies targeted at reducing intracellular glucocorticoid action are emerging (3).
Endogenous glucocorticoids (corticosterone in rodents and cortisol in humans) activate intracellular glucocorticoid (GR) and mineralocorticoid (MR) receptors. In many cell types, GR activation is amplified by local regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase type-1 (11β-HSD1) (4), and MR activation is prevented by local inactivation of glucocorticoids by another isozyme, 11β-HSD2 (5). GR, MR and both isozymes of 11β-HSD are expressed in the blood vessel wall, with 11β-HSD1 predominantly localised to vascular smooth muscle cells and 11β-HSD2 present in endothelial cells (1, 6, 7). It has been shown that MR antagonism with spironolactone or eplerenone attenuates post-angioplasty neointimal proliferation in rabbits (8) and pigs (9, 10). Moreover, there is evidence from mice that 11β-HSD2 modifies vascular function by maintaining endothelium-dependent vasodilatation (11), while 11β-HSD1 modifies vascular remodelling by providing a tonic restraint on angiogenesis (12). However, there is a paucity of data concerning interactions between the 11β-HSDs and corticosteroid receptors in modulating neointimal proliferation.
11β-HSD1 is of particular relevance to vascular lesion development. Selective 11β-HSD1 inhibitors are in development for treating type 2 diabetes (3), by reducing intracellular glucocorticoid concentrations in metabolic tissues such as adipose and liver. However, 11β-HSD1 is also expressed in macrophages, where loss of regeneration of glucocorticoids during 11β-HSD1 inhibition may be pro-inflammatory (13, 14). Macrophages have an established pro-inflammatory role in vascular lesion development both in atherogenesis (15) and neointimal proliferation, where they are implicated as targets for the therapeutic effects of glucocorticoids (16). There is, therefore, a concern that 11β-HSD1 inhibitors could adversely affect cardiovascular outcomes. Reassuringly, in atherogenesis models in mice, 11β-HSD1 inhibition is either protective or neutral for progression of atheroma (17, 18), but any influence on neointimal proliferation has not been explored.
We hypothesised that variations in endogenous glucocorticoids influence neointimal proliferation. We investigated neointimal proliferation following intra-vascular femoral artery injury in mice. Using mice with targeted deletion of 11β-HSD1 or 11β-HSD2 in combination with pharmacological receptor antagonists or an 11-HSD1 inhibitor, we tested whether: (1) activation of GR by endogenous glucocorticoids attenuates neointimal proliferation; (2) activation of MR by endogenous glucocorticoids promotes neointimal proliferation; (3) 11β-HSD1 activity amplifies GR activation and hence reduces neointimal proliferation; and (4) 11β-HSD2 activity attenuates MR activation and hence reduces neointimal proliferation.
MATERIALS AND METHODS
Animals
All animal work was carried out under UK Home Office licence in accordance with the Animals (Scientific Procedures) Act, 1986 and conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Male C57Bl/6J mice (Harlan Laboratories, UK), Apo-E−/− mice (Charles River Laboratories, Kent, UK), 11β-HSD2−/− mice homozygous for a disrupted hsd11b2 allele (bred in house (19)) and 11β-HSD1−/− mice homozygous for a disrupted hsd11b1 allele (bred in house (20)) and mice with double 11β-HSD1−/−/ ApoE−/− (generated in-house by crossing the two lines) were studied aged 12 weeks. All lines were congenic on a C57Bl/6J background. Animals were maintained under standard conditions of light (lights on 8am-8pm) and temperature (21-22°C). Systolic blood pressure (BP), where indicated, was measured weekly in conscious, warmed, restrained mice using tail-cuff photo-plethysmography.
Materials
Unless stated otherwise, chemicals were purchased from Sigma-Aldrich, UK. Compound 544 (3-(1-adamantyl)-6,7,8,9-tetrahydro-5H-[1,2,4]triazolo[4,3-α]azepine), a potent and selective inhibitor of 11β-HSD1 (17), was obtained from Enamine Ltd., Ukraine. Standard chow (RM1) was obtained from Special Diet Services (Witham, UK) and Western diet (OpenSource™ D12079B) was obtained from Research Diets Inc. (Brunswick, USA).
Femoral artery wire injury
Neointimal proliferation was induced by insertion of an angioplasty guide-wire into the femoral artery, as described previously (21). Briefly, mice were anaesthetised with isoflurane inhalation (5% for induction and 2% for maintenance, adequacy assessed by loss of pedal withdrawal reflex), and a small skin incision made in the proximal thigh. A fixed core straight 0.015″ diameter guide-wire (COOK Inc., Bloomington, USA) was introduced into the popliteal artery via an arteriotomy, advanced 5-10 mm into the common femoral artery towards the iliac artery, left in place for 1 minute and then removed. The popliteal artery was then ligated proximal to the arteriotomy site and blood flow restored in the common femoral artery. This induced endothelial denudation and stretching of the artery wall, followed by reperfusion of blood over the injured area. For local drug administration, a pocket was created next to the injured artery and a silastic pellet inserted. The wound was closed using silk sutures and the animals were allowed to recover.
Adrenalectomy
Adrenalectomy or sham surgery was conducted under isoflurane anaesthesia 1 week before femoral artery injury. Adrenalectomised mice were maintained with 0.9% NaCl in their drinking water. Plasma corticosterone or cortisol levels were measured on trunk blood samples obtained at sacrifice. Cortisol was measured using a radioimmunoassay kit (ICN Biomedicals Inc, U.S.A.), as per manufacturer’s instructions. Corticosterone was measured using an in-house radioimmunoassay (22), using a rabbit anti-corticosterone primary antibody (diluted 1:100 in borate buffer; kindly provided by C. Kenyon),) and an anti-rabbit secondary antibody linked to scintillation proximity assay (SPA) beads (diluted according to manufacturer’s instructions; GE Healthcare, U.K.). Intra-assay and inter-assay coefficients of variation for this assay were 4.6–6.2% and 6.4–8.2%, respectively.
Drug administration
Dexamethasone was administered daily (in the morning) as a subcutaneous injection of a 0.1 mg/ml solution in 4% ethanol:96% normal saline. Silastic pellets (10 mg) were prepared, as previously described (12, 23), containing silastic medical grade elastomer base with 10% curing agent (Dow Corning, USA) ± 2 mg cortisol or 3.3 mg RU38486. Eplerenone (Pfizer Inc., New York, USA) was mixed with chow (1.7 mg/g) to achieve a dose of ~200 mg/kg/day (24). The 11β-HSD1 inhibitor compound 544 was mixed with chow or western diet, as appropriate, to achieve a daily dose of ~30 mg/kg/day (17). Both eplerenone and compound 544 were administered for 1 week prior to intravascular injury and for 3 weeks thereafter until the animals were sacrificed.
Evaluation of lesions
Animals were killed by decapitation or perfusion fixation, as appropriate, 21 days after intra-vascular injury. Trunk blood was collected and organs dissected and weighed. Femoral arteries were cleaned of connective tissue, and excised from the bifurcation of the iliac artery to the branch with the popliteal artery. Femoral arteries were immediately placed in 10% neutral buffered formalin, fixed for up to 24 hours and then stored in 70% ethanol if necessary.
All histological analyses were conducted by an observer who was blind to the group from which the sections were obtained. The arteries were dehydrated in a graded alcohol series, embedded in paraffin and cut into transverse sections. Sections were stained with United States Trichrome (UST). Images of stained sections were digitised using a Photometrics CoolSnap camera (Roper Scientific, Tucson, USA) coupled to a light microscope via a microcolour liquid crystal turnable RGB filter (Cambridge Research and Instrumentation Inc., Woburn, USA). Microcomputer Imaging Device software (MCID; Imaging Research Inc., St. Catherines, Canada) was used for image analysis of sections every 60 μm along the length of the artery. The area (μm2) inside the lumen and the areas inside the internal and external elastic laminae (IEL and EEL, respectively) were measured. These measurements were then used to calculate the neointimal (NI) area (area inside IEL – lumen area), the area of the media (area inside EEL – area inside IEL) and NI area corrected for vessel size [(NI / IEL) × 100].
Lesion composition was determined by staining sections at the point of maximum neointimal lesion area. Collagen content was identified by staining with Picrosirius red. Macrophage and smooth muscle content were assessed by immunohistochemistry, for which sections were deparaffinised, blocked with normal goat serum, incubated with primary antibodies followed by appropriate secondary antibody and Extravidin-Peroxidase LSAB reagent, developed with DAB substrate and counterstained with haematoxylin. Primary antibodies used were purified monoclonal anti-mouse Mac2 antibodies for macrophages (1:6000; Cedarlane, UK) and monoclonal anti-mouse α-smooth muscle actin antibodies for smooth muscle cells (1:400; Sigma-Aldrich, UK). Photomicrographs of stained sections were acquired as described and quantification performed using a semi-automated colour deconvolution process with Photoshop CS3 Extended software (Adobe Systems Inc., USA).
Statistics
Data are mean ± SEM, where n indicates the number of mice in each group. Statistical analyses were performed using GraphPad prism (GraphPad Software Inc., California, USA). The statistical tests used are indicated in the figure legends.
RESULTS
Pharmacological glucocorticoids act locally to reduce neointimal lesions in C57Bl/6J mice, mimicking the effect of systemically administered synthetic glucocorticoids
To confirm the utility of the model for testing corticosteroid effects, mice were treated systemically with the potent GR agonist dexamethasone (1 mg/kg/day by daily subcutaneous injection). Dexamethasone substantially reduced fibro-proliferative lesion area (Figure 1A), but also delayed wound healing and prevented weight gain following surgery (Table 1) and reduced the weights of glucocorticoid-responsive organs including the adrenal glands (0.011±0.001% vs. 0.018±0.001% of body weight, p=0.001) and thymus (0.05±0.01% vs. 0.17±0.01% of body weight, p<0.0001).
Figure 1. Systemic or local glucocorticoid administration attenuates neointimal lesion formation in C57Bl/6J mice.
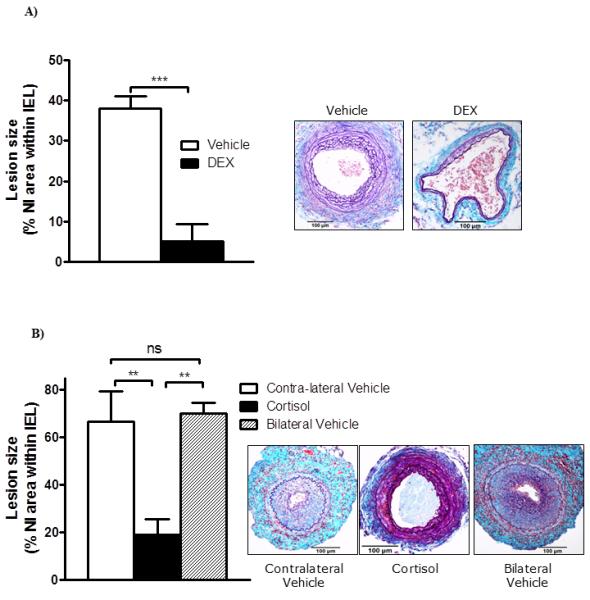
A) Systemic administration of a potent GR agonist, dexamethasone (DEX, 1 mg/kg/day by daily subcutaneous injection), substantially reduced fibroproliferative lesion area. Data are mean±SEM for n= 8/group and were analysed by Student’s un-paired t-test: *** indicates p<0.001 vs. vehicle. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm, arrows indicate internal and external elastic laminae).
B) Local administration of cortisol inhibited neointimal lesion formation. Extensive neointimal lesions developed in vehicle-treated control arteries (either in the contralateral control or in animals with bilateral vehicle pellets), but lesion formation was inhibited when a 2mg cortisol pellet was placed next to the artery. Data are mean±SEM for n= 8-9/group and were analysed by one-way ANOVA with Tukey’s post-hoc test: ** indicates p<0.01 vs. vehicle; ns indicates non-significant. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm, arrows indicate internal and external elastic laminae).
Table 1. Effects of corticosteroid manipulation on body weights.
| Intervention | Time point* | Body Weight (g) | p | |
|---|---|---|---|---|
| Controls | Intervention | |||
| Systemic dexamethasone | Baseline | 26.6±0.6 | 25.8±0.4 | 0.3 |
| 4-weeks | 28.5±0.5 | 25.3±0.3 | <0.0001 | |
| Local cortisol | Baseline | 28.8±1.1 | 27.8±1.9 | 0.7 |
| 4-weeks | 29.3±1.0 | 28.1±1.6 | 0.5 | |
| Adrenalectomy | Baseline | 30.0±0.8 | 29.4±0.6 | 0.6 |
| 4-weeks | 28.4±0.5 | 28.2±0.5 | 0.7 | |
| GR antagonism (RU38486) | Baseline | 28.8±1.1 | 28.7±0.7 | 0.9 |
| 4-weeks | 29.3±1.0 | 29.2±0.6 | 0.9 | |
| MR antagonism (eplerenone) | Baseline | 29.3±0.7 | 28.5±0.5 | 0.4 |
| 4-weeks | 30.9±1.0 | 29.0±0.7 | 0.2 | |
| 11β-HSD2 deletion in C57Bl/6J | Baseline | 29.3±0.7 | 28.7±0.4 | 0.5 |
| 4-weeks | 29.3±1.0 | 28.1±0.6 | 0.3 | |
| 11β-HSD1 deletion in C57Bl/6J | Baseline | 29.5±0.7 | 30.0±0.4 | 0.5 |
| 4-weeks | 30.0±0.5 | 30.7±0.8 | 0.5 | |
| 11β-HSD1 inhibition in C56Bl/6J | Baseline | 29.0±0.7 | 29.1±0.5 | 0.9 |
| 4-weeks | 29.1±0.9 | 28.5±0.4 | 0.5 | |
| 11β-HSD1 deletion in Apo-E−/− | Baseline | 29.9±0.9 | 26.2±0.4 | <0.01 |
| 4-week | 32.8±0.4 | 28.6±0.5 | <0.001 | |
| 11β-HSD1 inhibition in Apo-E−/− | Baseline | 30.2±0.5 | 30.1±0.5 | 0.9 |
| 4-weeks | 30.4±0.7 | 28.6±0.5 | 0.05 | |
Baseline weights were measured one week before angioplasty and terminal weights 3 weeks after angioplasty
GR, Glucocorticoid receptor; MR, Mineralocorticoid Receptor; 11β-HSD, 11beta-hydroxy steroid dehydrogenase; ApoE−/−, apolipoprotein E knockout mice. Date represent Mean ± SEM, for n= 6-8 mice / group.
To avoid these systemic effects, and to test the effects of a non-synthetic glucocorticoid, cortisol was administered by implantation of a silastic pellet next to the injured artery. Cortisol was selected over the endogenous murine glucocorticoid corticosterone to allow assessment of systemic release; no cortisol was detected by radioimmunoassay in the systemic circulation of these animals (data not shown), and there was no effect on body weight (Table 1) or weights of the adrenal glands and thymus (not shown). However, local cortisol release substantially reduced the area of the neointimal lesion following intra-vascular injury, in comparison with vehicle-treated controls either in the contralateral leg or in animals not treated with cortisol (Figure 1B).
Adrenalectomy does not affect neointimal proliferation in C57Bl/6J mice
To test whether endogenous corticosteroids provide a net tonic suppression of vascular proliferation, mice were adrenalectomised or sham-operated. Successful surgery was confirmed by measuring terminal plasma corticosterone levels (Sham 560±41nM vs. Adrenlaectomy 56±5nM; p<0.001). Adrenalectomy had no effect on body weight (Table 1), neointimal lesion area (Figure 2A), smooth muscle content (45.6±11.1% vs. 52.7±9.3%, p=0.5) or macrophage content (11.5±3.4% vs. 14.4±3.0%, p=0.5) of neointimal lesions.
Figure 2. MR antagonism decreases, GR antagonism increases and adrenalectomy does not alter neointimal lesion formation in C57Bl/6J mice.
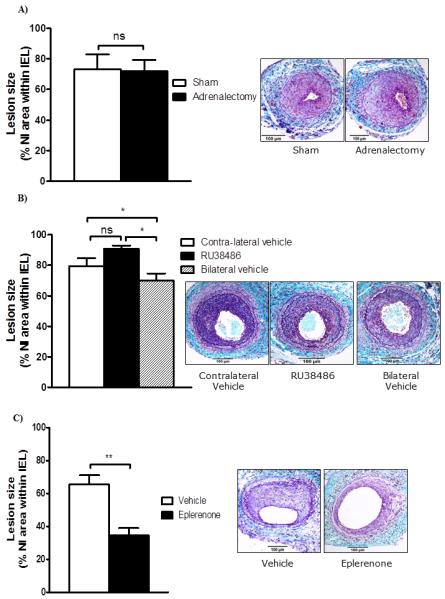
A) Adrenalectomy had no effect on neointimal lesion area in wire-injured femoral arteries in C57Bl/6J mice. Data are mean±SEM for n= 7-8/group and were analysed by Student’s un-paired t-test: ns indicates non-significant. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
B) Locally administered GR antagonist, RU38486 (3.3mg), increased neointimal proliferation in wire-injured arteries when compared with lesions in untreated mice; contralateral control lesions were intermediate, suggesting some systemic effect of RU38486. Data are mean±SEM for n= 8/group and were analysed by one-way ANOVA: ns indicates non-significant, * indicates p<0.05. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
C) Oral administration of an MR antagonist, eplerenone (200mg/Kg/day), reduced neointimal proliferation after wire angioplasty in C57Bl/6J mice. Data are mean±SEM for n= 6/group and were analysed by Student’s un-paired t-test: ** indicates p<0.01. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
MR antagonism attenuates but GR antagonism increases neointimal proliferation in C57Bl/6J mice
To test for independent and distinct effects of MR and GR on neointimal proliferation, we used corticosteroid receptor antagonists. To avoid compensatory activation of the hypothalamic-pituitary-adrenal axis that results from systemic GR antagonism, the GR antagonist RU38486 was administered locally by silastic implant (12) adjacent to the injured artery. Plasma corticosterone levels (Control 89.6±14.1 nmol/L vs. RU38486 54.3±18.8 nmol/L, p=0.16) and body weights (Table 1) were not altered. Neointimal lesion size was increased by local RU38486 administration in comparison with control mice with bilateral vehicle implants (Figure 2B). Contralateral lesions in mice with RU38486 pellets inserted were intermediate in size (Figure 2B), suggesting a degree of systemic leak of RU38486 insufficient to raise plasma corticosterone.
Systemic administration of eplerenone (200 mg/kg/day) did not alter body weight (Table 1) or tail cuff BP (115±3 mmHg vs. 116±2 mmHg, p=0.8), but substantially reduced neointimal lesion size (Figure 2C).
Neither 11β-HSD1 nor 11β-HSD2 influence neointimal proliferation in C57Bl/6J mice
To test the influence of 11β-HSD1 and 11β-HSD2 on GR- and MR-mediated modulation of neointimal proliferation, respectively, we studied mice deficient in either isozyme.
11β-HSD2−/− mice were similar in weight to age matched C57Bl/6J mice (Table 1) but had substantially higher systolic BP (Figure 3A) and, consistent with their chronic hypertension, exhibited medial hypertrophy and outward vascular remodelling in uninjured vessels (Figure 3B). However, after femoral artery injury, there was no difference in size of neointimal lesions between 11β-HSD2−/− and wild type mice (Figure 3C). To test whether the inhibition of lesion size by MR antagonists was enhanced in 11β-HSD2−/− mice, which cannot inactivate the potential MR ligand corticosterone, another group of 11β-HSD2−/− mice was studied with and without eplerenone administration. Notably, eplerenone did not alter tail cuff BP even in hypertensive 11β-HSD2−/− mice (Figure 4A). Body weights were similar in all groups indicating no effect of eplerenone or 11β-HSD2 deletion. Eplerenone substantially reduced neointimal proliferation in wild type as well as 11β-HSD2−/− mice (Figure 4B). This effect of eplerenone on neointimal lesions was of no statistical difference between the two groups, and there was no effect of 11β-HSD2 deficiency in the presence of eplerenone (Figure 4B). Eplerenone reduced the macrophage content in both C57Bl/6J and 11β-HSD2−/− mice but had no effect on collagen and smooth muscle cell content of neointimal lesions (Figure 4C).
Figure 3. Effects of 11β-HSD2 deficiency on blood pressure, vascular remodelling and neointimal lesion formation.
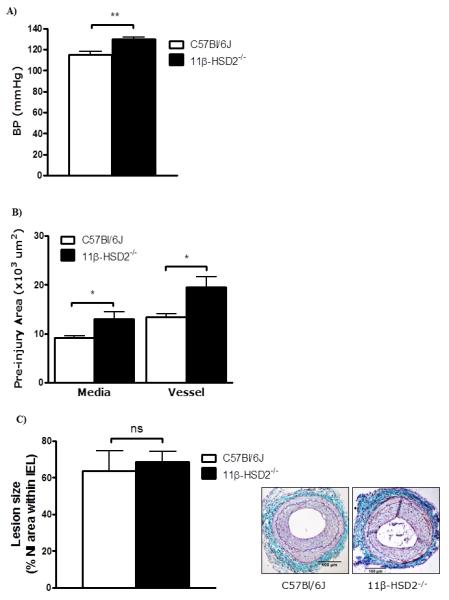
A) 11β-HSD2−/− mice had substantially higher systolic blood pressure, measured by tail cuff plethysmography, than age-matched C57Bl/6J mice.
B) Uninjured femoral arteries showed medial hypertrophy and outward remodelling resulting in a larger total vessel area.
C) Following intra-arterial injury neointimal lesion area was not different between groups. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
Data are mean±SEM for n= 6/group and were analysed by unpaired Student’s t-tests. ns indicates non-significant, * indicates p<0.05, ** indicates p<0.01.
Figure 4. Effects of the MR antagonist eplerenone on neointimal lesions in C57Bl/6J mice and 11β-HSD2−/− mice.
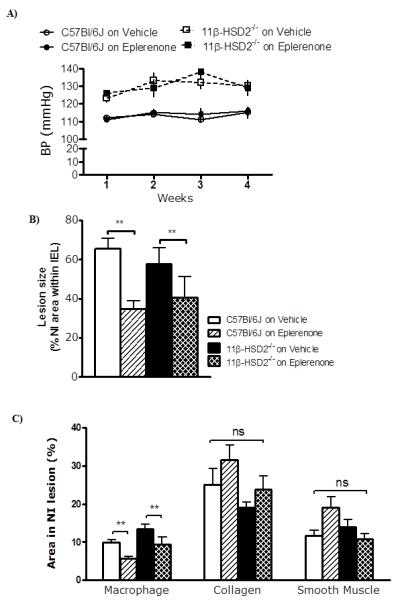
A) Eplerenone had no effect on systolic blood pressure in either normotensive C57Bl/6J or hypertensive 11β-HSD2−/− mice (p=0.6).
B) Eplerenone reduced neointimal area in both C57Bl/6J and 11β-HSD2−/− mice.
C) Eplerenone reduced macrophage content of lesions (staining for Mac-2), but did not affect smooth muscle (α-smooth muscle actin staining) or collagen (Picrosirius red) content.
Data are mean±SEM for n= 6/group and were analysed by two-way ANOVA and post hoc t tests when appropriate. ns indicates non-significant, ** indicates p<0.01.
The effect of 11β-HSD1 deletion or pharmacological inhibition (with compound 544 17) on neointimal proliferation was studied in C57Bl/6J mice. 11β-HSD1 inhibition had no effect on body weight (Table 1) or systolic BP (110±2 mmHg vs. 112±2 mmHg, p=0.2). Deletion of 11β-HSD1 had no effect on neointimal lesion size, an apparent reduction in lesion size following administration of compound 544 was not statistically significant (Figure 5A). However, there was no effect of 11β-HSD1 inhibition on composition of neointimal lesions (Figure 5B).
Figure 5. Effects of 11β-HSD1 inhibition/deletion on neointimal lesion formation in C57Bl/6J mice.
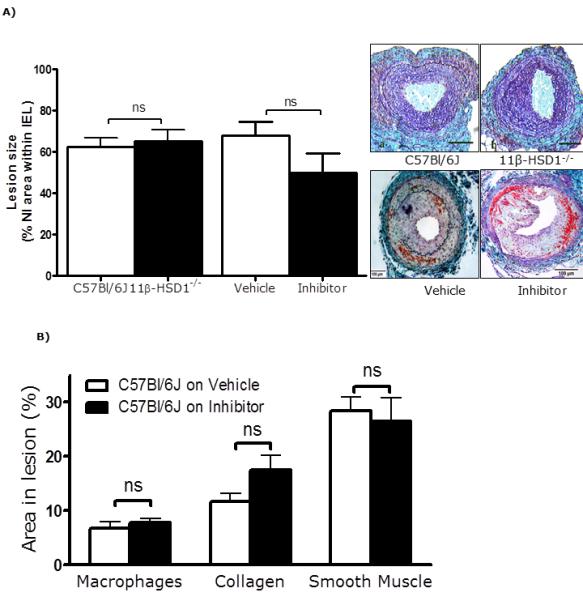
A) In mice on a C57Bl/6J background, neointimal lesions were similar in 11β-HSD1−/− mice and wild type controls, and were not significantly affected by compound 544. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
B) 11β-HSD1 inhibition did not affect macrophage (staining for Mac-2), collagen (Picrosirius red) or smooth muscle (α-smooth muscle actin staining) content of lesions.
Data are mean±SEM for n= 6/group and were analysed by un-paired Student’s t-tests. ns indicates non-significant.
11β-HSD1 inhibition reduces neointimal proliferation in atherosclerosis-prone Apo-E−/− mice
The lack of a significant effect of 11β-HSD1 inhibition or deletion on neointimal lesion development in C57Bl/6J mice contrasts with the atheroprotective effect of 11β-HSD1 inhibition observed in Apo-E−/− mice fed a high cholesterol Western diet (17). It is possible that the pro-inflammatory effects of hyperlipidaemia and Western diet feeding increase susceptibility to manipulations of 11β-HSD1 activity. Therefore, we administered compound 544 to Apo-E−/− mice on Western diet. 11β-HSD1 inhibition induced some weight loss (Table 1), and tended to lower fasting plasma glucose and BP, but had no effect on plasma lipid profile (Table 2). Following 11β-HSD1 inhibition, neointimal lesions were significantly reduced in size (Figure 6A), with reduced macrophage content, elevated collagen content and unchanged smooth muscle content (Figure 6B).
Table 2. Effects of 11β-HSD1 inhibition/deletion on systemic cardiovascular risk factors in Western diet-fed Apo-E−/− mice.
| Parameter* | 11β-HSD1 inhibition | 11β-HSD1 deletion | ||||
|---|---|---|---|---|---|---|
| Vehicle | Inhibitor | p | Apo-E−/− | DKO | p | |
| Systolic BP (mmHg) | 116±6 | 108±3 | 0.09 | 117±3 | 110±4 | <0.01 |
| Fasting plasma glucose (mg/dl) | 338±27 | 258±30 | 0.07 | 272±33 | 181±20 | 0.03 |
| Plasma cholesterol (mmol/L) | 8.1±0.6 | 8.2±0.6 | 0.9 | 8.9±1.4 | 8.8±1.3 | 0.9 |
| Plasma triglycerides (mmol/L) | 1.8±0.1 | 1.7±0.2 | 0.9 | 1.7±0.1 | 1.6±0.1 | 0.3 |
All measurements are at 4 week time point. BP, blood pressure; 11β-HSD, 11beta-hydroxy steroid dehydrogenase; Apo-E−/−, apolipoprotein E knockout mice; DKO, Double (Apo-E & 11β-HSD-1) Knockout mice. Date represent Mean ± SEM, for n= 6-8 mice / group.
Figure 6. Effects of 11β-HSD1 inhibition/deletion on neointimal lesion formation in Apo-E knockout mice.
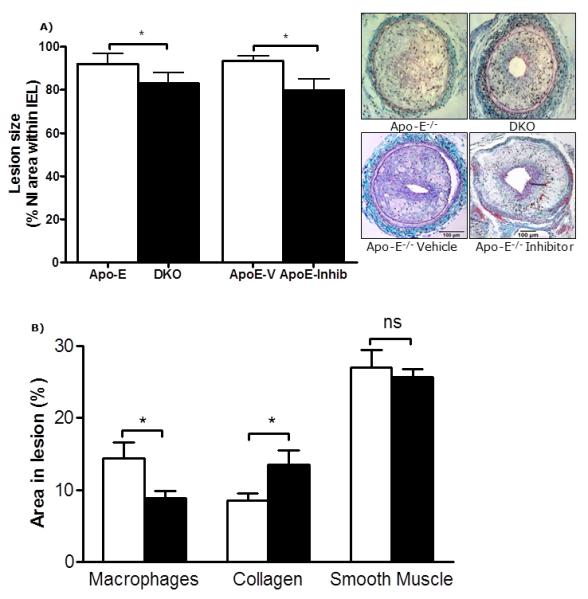
A) In Apo-E−/− mice fed on Western diet, administration of the selective 11β-HSD1 inhibitor compound 544 reduced neointimal lesion size. Mice with transgenic deletion of Apo-E and11β-HSD1 (double knockout, DKO) also had significantly lower neointimal proliferation than Apo-E−/− mice. Representative sections stained with United States Trichrome are shown as insets (scale bar = 100 μm); arrows indicate internal and external elastic laminae.
B) Selective 11β-HSD1 inhibition also reduced macrophage content, elevated collagen content and had no effect on smooth muscle content of lesions.
Data are mean±SEM for n= 6-8/group and were analysed by un-paired Student’s t-tests. ns indicates non-significant, * indicates p<0.05.
To determine whether the effects of compound 544 were due to selective 11β-HSD1 inhibition, post-angioplasty neointimal proliferation was also studied in Apo-E−/− and Apo-E−/−, 11β-HSD1−/− (DKO) mice (n=8/group) fed on western diet for four weeks, starting one week before wire-angioplasty. As with 11β-HSD1 inhibition, deletion of the enzyme on an Apo-E−/− background lowered blood pressure and fasting glucose but had no effect on plasma lipids (Table 2). Transgenic deletion of 11β-HSD1 in Apo-E−/− mice significantly reduced lesion size compared with controls (Figure 6A). As with 11β-HSD1 inhibition, 11β-HSD1 deletion had no effect on smooth muscle content (25±1% vs. 25±3%, p=1) but reduced the macrophage (11±1% vs. 16±2%, p=0.02) and increased the collagen (40±3% vs. 30±3%, p=0.05) content of plaques compared with control mice.
DISCUSSION
Our data confirm that pharmacological administration of GR agonists reduces neointimal proliferation following intra-vascular injury in mice, as previously demonstrated in dog, pig, rat, rabbit and human (1). This pharmacological effect appears to be mediated locally in the vessel wall, since it is recapitulated by implantation of a slow-release device supplying cortisol at the site of injury without increasing circulating glucocorticoid concentrations. GR and MR (but not PR) are expressed in the media and neointima of mouse femoral arteries and it is conceivable that glucocorticoid administration alters this micro-environment by down-regulating GR. Moreover, our data show that MR antagonists reduce neointimal lesion size in mice, as previously demonstrated in pigs (9, 10) and rabbits (8). In addition, we show for the first time that GR antagonism exacerbates neointimal proliferation (although it should be noted that RU38486 can also block progesterone receptors but we have found no evidence that these receptors were present in the mouse femoral artery or its lesions). Removing endogenous corticosteroid receptor ligands by adrenalectomy, however, had no discernible effect on neointimal proliferation. These findings suggest that endogenous corticosteroids sustain a balance between GR-dependent protective effects and MR-dependent adverse effects.
Despite this evidence of tonic GR-mediated suppression of neointimal proliferation by endogenous glucocorticoids, we found that preventing glucocorticoid regeneration by 11β-HSD1 (which has been shown previously to influence GR-dependent vascular phenotypes (11, 12), including atherogenesis (17)), had no significant effect on neointimal proliferation or lesion content in C57Bl/6J mice. Indeed, inhibition or deletion of 11β-HSD1 was paradoxically protective against neointimal proliferation in Apo-E−/− mice.
Endogenous glucocorticoids have well-established systemic effects on cardiovascular and metabolic variables, many of which may influence neointimal proliferation. Manipulating 11β-HSD1 in liver and adipose tissue in mice has been shown to affect plasma glucose, lipid profile and BP (20, 25-28), and pharmacological inhibition of 11β-HSD1 has similar consequences (3). Our data in Apo-E−/− mice suggest that any influence of 11β-HSD1 inhibition on neointimal proliferation is likely to be dependent on systemic effects: although 11β-HSD1 inhibition or deficiency had no statistically significant influence either on neointimal proliferation or on metabolic risk factors in C57Bl/6J mice on normal chow diet, in Apo-E−/− mice challenged with a Western diet inhibition or deletion of 11β-HSD1 tended to improve metabolic risk factors and reduced neointimal proliferation and intravascular inflammation, as measured by macrophage content.
Regarding MR, the reduction in lesion size induced by eplerenone suggests a tonic contribution of MR activation to neointimal proliferation. However, removing ligand by adrenalectomy had no net effect. This could possibly be explained by the loss of opposing GR and MR mediated actions, resulting in no overall effect. We also show that MR are not ‘protected’ from ligand activation by 11β-HSD2 in vessels in relation to vascular remodelling as they are, for example, in the kidney in relation to BP control (5). Whether the beneficial effect of MR antagonists on neointimal lesion formation is mediated by alterations in systemic risk factors or by direct effects in the vessel wall remains a moot point. The effects of eplerenone appear to be dissociated from any antihypertensive effect. Thus, 11β-HSD2−/− mice had substantial systolic hypertension, as previously reported (19), but this did not aggravate neointimal proliferation and was not measurably reduced by eplerenone. Similar observations have been made in Apo-E−/−/11β-HSD2−/− double knockout mice, in which eplerenone is atheroprotective without measurably lowering BP (29). Eplerenone has also been shown to significantly reduce cerebral ischemia/neurological deficit without any effect on blood pressure (30). Thus the results presented here suggest that reduced neointimal proliferation in response to eplerenone treatment is not due to an effect on systolic blood pressure. In other settings, MR activation is pro-inflammatory and MR blockade reduces vascular inflammation (24, 31), observations which, together with the reduced macrophage content observed in neointimal lesions following eplerenone treatment in the current studies, suggests an effect which might be mediated in non-resident inflammatory cells.
11β-HSD1 inhibitors are being developed for use in patients with type 2 diabetes and have beneficial effects on multiple metabolic cardiovascular risk factors (32-34). However, there are increasing demands to document cardiovascular risk reduction for new oral hypoglycaemic agents. There has been concern about pro-inflammatory effects of 11β-HSD1 inhibition, resulting from reduced glucocorticoid concentrations in macrophages (13, 35). Certainly, loss of 11β-HSD1 is associated with prolonged inflammatory responses following chemical peritonitis or serum arthritis (13, 14). However, effects of glucocorticoids on inflammation are complex and context dependent, and loss of 11β-HSD1 has been associated with reduced indices of inflammation in adipose tissue (36). Arguably, the most important conclusion from the current studies is that inhibition of 11β-HSD1 is either neutral or protective, not only for atherogenesis (17) but also for vascular lesion development following intravascular injury.
ACKNOWLEDGEMENTS
We are grateful to Professors John Mullins for access to 11β-HSD2 deficient mice and to Eileen Miller for technical support. The authors are grateful for support from the British Heart Foundation Centre of Excellence Award.
Grant support: This work was supported by research funding from the British Heart Foundation; Carnegie Trust and Wellcome Trust.
Footnotes
Disclosure statement: JI, LJM, LL, CWY and PWFH have no potential conflicts of interest/disclosures relevant to this article. BRW and JRS are inventors on relevant patents owned by the University of Edinburgh and have consulted for several companies developing selective 11βHSD1 inhibitors.
REFERENCES
- 1.Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712. doi: 10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker BR. Glucocorticoids and cardiovascular disease. European Journal of Endocrinology. 2007;157:545–559. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- 3.Hughes KA, Webster SP, Walker BR. 11-Beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity 3390. Expert Opin Investig Drugs. 2008;17:481–496. doi: 10.1517/13543784.17.4.481. [DOI] [PubMed] [Google Scholar]
- 4.Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- 5.Stewart PM, Krozowski ZS. 11Beta hydroxysteroid dehydrogenase. Vitamins and Hormones. 1999;57:249–324. [PubMed] [Google Scholar]
- 6.Walker BR, Yau JL, Brett LP, Seckl JR, Monder C, Williams BC, Edwards CR. 11 beta-hydroxysteroid dehydrogenase in vascular smooth muscle and heart: implications for cardiovascular responses to glucocorticoids. Endocrinology. 1991;129:3305–3312. doi: 10.1210/endo-129-6-3305. [DOI] [PubMed] [Google Scholar]
- 7.Christy C, Hadoke PWF, Paterson JM, Mullins JJ, Seckl JR, Walker BR. Glucocorticoid action in mouse aorta; localisation of 11beta Hydroxysteroid dehydrogenase type 2 and effects on response to glucocorticoid in vitro. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- 8.Van Belle E, Bauters C, Wernert N, Hamon M, McFadden EP, Racadot A, Dupuis B, Lablanche JM, Bertrand ME. Neointimal thickening after balloon denudation is enhanced by aldosterone and inhibited by spironolactone, and aldosterone antagonist. Cardiovasc Res. 1995;29:27–32. [PubMed] [Google Scholar]
- 9.Ward MR, Kanellakis P, Ramsey D, Funder J, Bobik A. Eplerenone suppresses constrictive remodeling and collagen accumulation after angioplasty in porcine coronary arteries. Circulation. 2001;104:467–472. doi: 10.1161/hc3001.091458. [DOI] [PubMed] [Google Scholar]
- 10.Wakabayashi K, Suzuki H, Sato T, Iso Y, Katagiri T, Takeyama Y. Eplerenone suppresses neointimal formation after coronary stent implantation in swine. Int J Cardiol. 2006;107:260–266. doi: 10.1016/j.ijcard.2005.03.078. [DOI] [PubMed] [Google Scholar]
- 11.Hadoke PW, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, Mullins JJ, Walker BR. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11beta-hydroxysteroid dehydrogenase. Circulation. 2001;104:2832–2837. doi: 10.1161/hc4801.100077. [DOI] [PubMed] [Google Scholar]
- 12.Small GR, Hadoke PW, Sharif I, Dover AR, Armour D, Kenyon CJ, Gray GA, Walker BR. Preventing local regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci U S A. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS, Chapman KE. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol. 2006;176:7605–7611. doi: 10.4049/jimmunol.176.12.7605. [DOI] [PubMed] [Google Scholar]
- 14.Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, Gilmour JS, Seckl JR, Savill JS, Chapman KE. 11beta-Hydroxysteroid Dehydrogenase Type 1, But Not Type 2, Deficiency Worsens Acute Inflammation and Experimental Arthritis in Mice. Endocrinology. 2011 doi: 10.1210/en.2011-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 16.Poon M, Gertz SD, Fallon JT, Wiegman P, Berman JW, Sarembock IJ, Taubman MB. Dexamethasoneinhibits macrophage accumulation after balloon arterial injury in cholesterol-fed rabbits. Atherosclerosis. 2001;155:371–380. doi: 10.1016/s0021-9150(00)00605-5. [DOI] [PubMed] [Google Scholar]
- 17.Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C, Fordstrom P, Zhou M, Wang M, Kaufman SA, Veniant MM. Antidiabetic effects of 11beta-HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes Metab. 2009;11:688–699. doi: 10.1111/j.1463-1326.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- 19.Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, Seckl JR, Mullins JJ. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest. 1999;103:683–689. doi: 10.1172/JCI4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dover AR, Hadoke PWF, Macdonald LJ, Miller E, Newby DE, Walker BR. Intravascular glucocorticoid metabolism during inflammation and injury in mice. Endocrinology. 2007;148:166–172. doi: 10.1210/en.2006-0996. [DOI] [PubMed] [Google Scholar]
- 22.Al-Dujaili EA, Forrest GC, Edwards CR. Development and application of an automated direct radioimmunoassay for plasma aldosterone [proceedings] J Endocrinol. 1979;81:111P. [PubMed] [Google Scholar]
- 23.Soro A, Panarelli M, Holloway CD, Fraser R, Kenyon CJ. In vivo and in vitro effects of carbenoxolone on glucocorticoid receptor binding and glucocorticoid activity. Steroids. 1997;62:388–394. doi: 10.1016/s0039-128x(96)00252-8. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki J, Iwai M, Mogi M, Oshita A, Yoshii T, Higaki J, Horiuchi M. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arterioscler Thromb Vasc Biol. 2006;26:917–921. doi: 10.1161/01.ATV.0000204635.75748.0f. [DOI] [PubMed] [Google Scholar]
- 25.Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11á-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- 26.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 27.Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: Hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA. 2004;101:7088–7093. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes. 2004;53:931–938. doi: 10.2337/diabetes.53.4.931. [DOI] [PubMed] [Google Scholar]
- 29.Deuchar GA, McLean D, Hadoke PW, Brownstein DG, Webb DJ, Mullins JJ, Chapman K, Seckl JR, Kotelevtsev YV. 11beta-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe−/− mice. Endocrinology. 2011;152:236–246. doi: 10.1210/en.2010-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwanami J, Mogi M, Okamoto S, Gao XY, Li JM, Min LJ, Ide A, Tsukuda K, Iwai M, Horiuchi M. Pretreatment with eplerenone reduces stroke volume in mouse middle cerebral artery occlusion model. Eur J Pharmacol. 2007;566:153–159. doi: 10.1016/j.ejphar.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 31.Keidar S, Hayek T, Kaplan M, Pavlotzky E, Hamoud S, Coleman R, Aviram M. Effect of eplerenone, a selective aldosterone blocker, on blood pressure, serum and macrophage oxidative stress, and atherosclerosis in apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 2003;41:955–963. doi: 10.1097/00005344-200306000-00019. [DOI] [PubMed] [Google Scholar]
- 32.Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516–1522. doi: 10.2337/dc09-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feig PU, Shah S, Hermanowski-Vosatka A, Plotkin D, Springer MS, Donahue S, Thach C, Klein EJ, Lai E, Kaufman KD. Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab. 2011;13:498–504. doi: 10.1111/j.1463-1326.2011.01375.x. [DOI] [PubMed] [Google Scholar]
- 34.Shah S, Hermanowski-Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J, Hwang PM, Ryan NW, Langdon RB, Feig PU. Efficacy and safety of the selective 11beta-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. J Am Soc Hypertens. 2011;5:166–176. doi: 10.1016/j.jash.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, Hermanowski-Vosatka A. 11 Beta-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol. 2001;167:30–35. doi: 10.4049/jimmunol.167.1.30. [DOI] [PubMed] [Google Scholar]
- 36.Wamil M, Battle JH, Turban S, Kipari T, Seguret D, de Sousa Peixoto R, Nelson YB, Nowakowska D, Ferenbach D, Ramage L, Chapman KE, Hughes J, Dunbar DR, Seckl JR, Morton NM. Novel fat depot-specific mechanisms underlie resistance to visceral obesity and inflammation in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes. 2011;60:1158–1167. doi: 10.2337/db10-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]