

Abstract
Primitive neuroectodermal tumor (PNET) is a pathologic diagnosis that encompasses several different tumor types, including central nervous system tumors and Ewing’s sarcomas. Teratoma, a common element of germ cell tumor (GCT), has the ability to transform to malignant PNET in a small number of patients. Making a definitive diagnosis of PNET is difficult given its deviation from elements of GCT and its non-specific pathologic findings. Establishing the diagnosis is crucial as PNETs respond poorly to standard platinum-based chemotherapy used for treatment of GCT. Primary treatment for PNET is surgical, though this is often not feasible in many patients due to extensive disease at diagnosis. As an alternative, chemotherapy regimens traditionally used for Ewing’s sarcoma, such as vincristine, doxorubicin and cyclophosphamide alternating with ifosfamide and etoposide, have shown limited efficacy in the neoadjuvant, adjuvant, and palliative settings. Future research should delineate the genetic underpinnings of PNET and develop therapeutic options accordingly.
Key words: primitive neuroectodermal tumor, PNET, teratoma, chemotherapy
Introduction
Germ cell tumor (GCT) can differentiate into multiple different elements.1 Teratoma, often a benign component found in GCT, is capable of malignant transformation across ectodermal, endodermal, or mesodermal germ cell lines into non-GCT as well.2 Malignant transformation of teratoma occurs in about 3-6% of metastatic GCT, with most of these becoming carcinomas and sarcomas.3 Transformation of teratoma to primitive neuroectodermal tumor (PNET) is a rare event that occurs across ectodermal cell lines.4 PNET is a pathologic term applied to several different tumors. It classically can develop in the central or autonomic nervous system in children, classified as central PNET, or peripherally as in Ewing’s sarcomas.5
PNET is generally not responsive to cisplatin-based chemotherapy, but may be cured with surgical resection.6 Over the last 10 years, however, chemotherapy regimens traditionally used for Ewing’s sarcomas have been shown to be efficacious in treating PNET transformed from teratoma.7 This regimen, which includes vincristine, doxorubicin and cyclophosphamide (VAC) alternating with ifosfamide and etoposide (IE), has been solidified as a treatment option by a single-center study.8 Here, we present the management of three patients with PNET that underwent malignant transformation from GCT (Table 1), all of which included chemotherapy with VAC/IE.
Table 1.
Baseline characteristics of three patients with primitive neuroectodermal tumor.
| Patient characteristics | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Germ cell tumor | |||
| Age at dx, years | 33 | 37 | 24 |
| Stage at dx | III | IA | IA |
| Tumor markers at dx | |||
| BHCG (mIU/mL) | <1 | NL | unk |
| AFP (IU/mL) | 18 | 52.1 | unk |
| LDH (U/L) | 2482 | NL | unk |
| Initial chemotherapy | BEP ×3 | BEP ×4 | BEP ×3, EP ×1 |
| PNET | |||
| Age at dx, years | 33 | 39 | 26 |
| Chemotherapy | VAC/IE ×1, VAC ×4 | VAC/IE ×4 | VAC/IE ×5, IE ×2, Irinotecan ×2 |
| Surgery | Yes | No | No |
| Other therapies | No | CDK inhibitor, Palliative XRT | CDK inhibitor, SCT |
| Clinical outcome | NED | Residual disease | Deceased |
PNET, primitive neuroectodermal tumor; dx, diagnosis; NL, normal; unk, unknown; BEP, bleomycin, etoposide, and cisplatin; EP, etoposide and cisplatin; VAC, vincristine, doxorubicin and cyclophosphamide; IE, ifosfamide and etoposide; CDK, cyclin-dependence kinase; XRT, radiation therapy; SCT, stem cell transplant; NED, no evidence of disease.
Case Report #1
A 33 year-old man underwent right radical orchiectomy after presenting with severe back pain and right testicular swelling. A pre-operative computed tomography (CT) of the chest, abdomen, and pelvis revealed extensive retroperitoneal lymphadenopathy, including a confluence of tissue measuring 9.7×7.4 cm, as well as scattered mediastinal lymphadenopathy. His pathology demonstrated a mixed GCT composed of embryonal carcinoma (40%), teratoma (40%) and yolk sac tumor (20%). Surgery was followed by three cycles of bleomycin, etoposide and cisplatin (BEP). Follow-up imaging revealed a response to chemotherapy, however his disease persisted in the retroperitoneal lymph nodes. He then underwent retroperitoneal lymph node dissection (RPLND), which showed metastatic mature teratoma in multiple lymph nodes without any residual GCT.
Six weeks following RPLND, he presented with acute kidney injury, fatigue, and an LDH >20,000 U/L. Imaging studies showed widespread recurrent disease, including supraclavicular lymphadenopathy, a new right inguinal soft tissue mass and an enlarging conglomerate of retroperitoneal lymphadenopathy in the para-aortic region (Figure 1A). The supraclavicular node was biopsied, which demonstrated a poorly differentiated high grade neoplasm though the definitive diagnosis remained in question. As a result, his original tumor and the RPLND specimens were reviewed at another institution in conjunction with the supraclavicular specimen. Upon further review, his original tumor was deemed primarily PNET (80%) with teratoma (10%), yolk sac tumor (8%) and embryonal carcinoma (2%). The RPLND, as well as the supraclavicular lymph node biopsy, confirmed PNET in both specimens (Figure 2).
Figure 1.
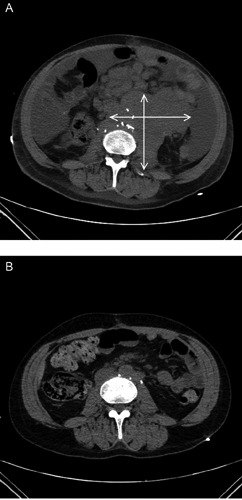
A) Demonstrates conglomerate of para-aortic retroperitoneal lymphadenopathy at diagnosis of primitive neuroectodermal tumor. Patient went on to receive cheomtherapy with VAC/IE. B) Demonstrates parital response to chemotherapy prior to potentially curative surgical resection.
Figure 2.
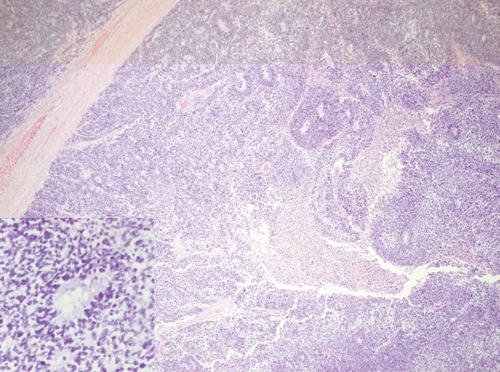
Primitive neuroectodermal tumor (Case 1) consisting of a large confluent aggregate of immature neuroepithelium that forms tubules lined by stratified cells (inset picture) was identified in the orchiectomy sample.
The patient was not initially a candidate for surgical resection. Therefore, neoadjuvant chemotherapy with VAC/IE was initiated. During his first cycle of chemotherapy, he developed ifosfamide-induced encephalopathy and renal tubular acidosis. As a result, the IE component of this regimen was held for his subsequent treatment and he received an additional four cycles of VAC thereafter.
At completion of his neoadjuvant chemotherapy, his imaging studies demonstrated complete response in his neck and mediastinal lymphadenopathy and partial response of his retroperitoneal lymphadenopathy (Figure 1B) and right inguinal mass. Subsequently, the patient underwent repeat RPLND and excision of the right inguinal soft tissue mass. Pathology of both specimens showed extensive necrosis with rare atypical cells consistent with PNET. Adjuvant chemotherapy was not administered after surgery since the patient has already received maximum cumulative dosing of doxorubicin and due to his history of ifosfamide-induced encephalopathy. His last imaging studies two months after his surgery showed no recurrent disease.
Case Report #2
A 37 year-old man underwent a right radical orchiectomy for testicular GCT with pathology demonstrating elements of yolk sac tumor (50%), seminoma (30%), and teratoma (20%). At presentation, he was found to have large retroperitoneal lymphadenopathy as well as a 1.4 cm subcarinal lymph node. After completion of 4 cycles of BEP, he had persistent retroperitoneal adenopathy and his subcarinal lymph node grew to 1.8 cm. He then underwent RPLND followed by resection of his subcarinal lymph node, which all revealed metastatic mature teratoma.
Two years later, the patient presented to the hospital with chest tightness, hemoptysis, and weight loss after he was lost to medical follow-up. A chest CT showed a 11×8.5×4 cm mediastinal mass surrounding the ascending aorta and extending to the posterior mediastinum. A biopsy revealed small round blue cells consistent with PNET. Due to extent of disease burden, the mediastinal mass was not surgically resectable and he was initiated on VAC/IE.
Despite interval partial response, his disease progressed at completion of 4 cycles of VAC/IE. He then enrolled in a phase II clinical study examining PD0332991, a cyclin-dependent kinase (CDK) inhibitor, for tumors that express the retinoblastoma protein (pRB). On this study, his disease was found to have progressed at 4 months, complicated by the development of superior vena cava syndrome. Subsequently, he underwent palliative radiation to the mediastinum with relief of his symptoms. The size of his mediastinal mass decreased by approximately 40% and he is currently being evaluated for salvage surgical resection.
Case Report #3
A 24 year-old man underwent a left radical orchiectomy for testicular GCT composed of mature and immature teratoma (50%), with remaining elements of embryonal and yolk sac tumor. After orchiectomy, his tumor markers returned to normal and he elected to undergo active surveillance but was lost to follow-up for over 1 year. After developing flank pain and weight loss, a repeat CT showed a 11×9×20 cm mass in the left retroperitoneum that displaced the left kidney and the aorta. Additionally, imaging revealed 7×5 cm left external iliac nodal mass. For his recurrent disease, he received three cycles of BEP, followed by one cycle of EP due to bleomycin-induced pulmonary toxicity. CT scan after completion of chemotherapy revealed marked regression of the left retroperitoneal and left external iliac nodal mass.
To surgically resect his residual disease in the retroperitoneum, the patient underwent RPLND, left radical nephrectomy, left partial adrenalectomy and left pelvic mass excision. Pathology of the retroperitoneal mass demonstrated predominantly mature teratoma with a small component of immature teratoma.
A CT to evaluate recurrent chylous ascites 5 months after RPLND showed extensive peritoneal carcinomatosis and new retroperitoneal and mediastinal masses. A biopsy of his retroperitoneal mass was performed and revealed immature teratoma. Upon further review of the specimens from both his RPLND and retroperitoneal biopsy by another institution, the immature teratomatous components were confirmed to be PNET.
The patient initially received one cycle of chemotherapy with VAC/IE but was complicated by grade IV myelosuppression. Since his tumor expressed the pRB, he participated in the PD0332991 clinical study instead of proceeding with further chemotherapy. On his first re-staging scan after initiation of PD0332991, however, his disease progressed. At that time, he completed four additional cycles of VAC/IE chemotherapy without doxorubicin in the last cycle due to hypokinesis of his right ventricle. His disease showed significant response at completion of chemotherapy.
Since myelosuppression limited his ability to tolerate further chemotherapy, he underwent high dose chemotherapy with busulfan and melphalan followed by autologous stem cell transplant. Unfortunately, he rapidly progressed despite this approach. Subsequently, he received two cycles of IE alone, but again his disease progressed while on therapy. After he was admitted to the hospital following two additional cycles of third-line chemotherapy with irinotecan, he was transitioned to comfort care and passed away approximately two years after his PNET diagnosis.
Discussion
Malignant transformation of teratoma to PNET is a rare but described phenomenon. Paramount is establishing a pathologic diagnosis, which can prove to be difficult as demonstrated in our case series. The diagnosis is established when the majority of a 4X field is occupied by a pure proliferation of a neuroepithelium, but this is an arbitrary definition.6,9 On microscopic examination, the tumors in case 1 (Figure 2), 2, and 3 showed similar histology of small, hyperchromatic cells arranged in tubules and rosettes consistent with neuroepithelium. PNETs derived from malignant transformation of teratoma, however, fail to have a consistent immunostaining profile. CD 57 is the most consistent immuno-histochemical marker that is positive for PNET. In addition, PNET derived from teratoma generally lacks distinctive markers for peripheral PNET, such as nuclear expression of WT1, CD99 and Fli-1.10 This variability in tumor profile makes the clinical history extremely helpful in making a pathologic diagnosis.
Once the diagnosis is established, standard curative treatment for PNET is surgical resection.6 However, surgery may not be technically feasible for many patients at presentation due to extensive disease or difficult anatomy. Although metastatic GCT has shown an excellent cure rate of >80% with platinum-based chemotherapy,11 PNET tumors have been described in the past as chemo-resistant tumors.6 Traditional chemotherapy regimens used for Ewing’s sarcoma are emerging as viable therapeutics used for treatment of transformed teratoma to PNET in various settings.7 In a large case series by Al-Hader and Einhorn8 that examined 86 cases of PNET after testicular teratoma, 18 patients received chemotherapy with VAC/IE, which consists of cyclophosphamide (1000-1200 mg/m2), doxorubicin (50-75 mg/m2) and vincristine (2 mg) alternating with ifosfamide (1.8 mg/m2) plus etoposide (100 mg/m2) for 5 consecutive days given 3 weeks after VAC. Twelve of the 18 patients who received this regimen had surgically unresectable disease at presentation. The remaining 6 patients received this regimen in the adjuvant setting after surgical resection. Six cycles of VAC/IE were administered in patients with unresectable disease and four cycles were used in the adjuvant setting. Among the patients with unresectable PNET, partial response by RECIST criteria occurred in 9 patients and 6 had a significant enough of a response that they were able to undergo surgery. The median survival for these 12 patients was 36 months and a 50% disease-specific survival was demonstrated at 2 years. In this case series,8 the 6 patients who received 4 cycles of adjuvant VAC/IE after surgical resection were all still alive without evidence of disease at the time of publication. The findings are proof of concept that VAC/IE can be used to treat PNET in the neoadjuvant, adjuvant, and palliative settings.
In our case series, all three patients with PNET were deemed unresectable at presentation and received VAC/IE chemotherapy with varying toxicities necessitating dose or regimen adjustments. All of them had initial disease response and one patient was able to undergo potentially curative surgery. Two of the patients, patient 1 and 2, are still alive at 14 and 10 months after completion of VAC/IE, respectively.
There remains scant literature regarding second or third-line systemic therapies for PNET not amenable to surgery. Vaughn et al.12 described a case series of three young men with recurrent growing teratoma syndrome whose tumors had strong expression of pRB. Since CDK 4/6 phosphorylates pRB to stimulate cell growth, PD0332991, a CDK inhibitor, was tested in these patients. All of these men experienced durable responses, ranging from 18 to 24 months, which led to the current clinical trial which examined PD0332991 in growing teratoma syndrome that expresses pRB. Two of our patients in the case series participated in this clinical trial with very modest disease control (only 2 and 4 months of stable disease).
Conclusions
Our case series demonstrates the efficacy of VAC/IE for patients with PNET. Based on a recent retrospective study, 4 cycles of adjuvant VAC/IE should be considered for patients after surgical resection. Enrollment on a clinical trial using a CDK inhibitor is an option for PNET that expresses pRB. Future research should delineate the genetic underpinnings of PNET and develop therapeutic options accordingly.
References
- 1.Loehrer PJ, Sr., Sledge GW, Jr., Einhorn LH. Heterogeneity among germ cell tumors of the testis. Semin Oncol 1985;12:304-16 [PubMed] [Google Scholar]
- 2.Ulbright TM, Loehrer PJ, Roth LM, et al. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer 1984; 54:1824-33 [DOI] [PubMed] [Google Scholar]
- 3.Comiter CV, Kibel AS, Richie JP, et al. Prognostic features of teratomas with malignant transformation: a clinicopathological study of 21 cases. J Urol 1998;159:859-63 [PubMed] [Google Scholar]
- 4.Ahmed T, Bosl GJ, Hajdu SI. Teratoma with malignant transformation in germ cell tumors in men. Cancer 1985;56:860-3 [DOI] [PubMed] [Google Scholar]
- 5.Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors - a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294-9 [DOI] [PubMed] [Google Scholar]
- 6.Ganjoo KN, Foster RS, Michael H, et al. Germ cell tumor associated primitive neuroectodermal tumors. J Urol 2001;165:1514-6 [PubMed] [Google Scholar]
- 7.Donadio AC, Motzer RJ, Bajorin DF, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol 2003;21:4285-91 [DOI] [PubMed] [Google Scholar]
- 8.Al-Hader AA, Jain A, Al-Nasrallah N, Einhorn LH. Metastatic malignant transformation of teratoma to primitive neuroectodermal tumor (PNET): results with PNET-based chemotherapy. Am J Clin Oncol 2013Jun 24. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Michael H, Hull MT, Ulbright TM, et al. Primitive neuroectodermal tumors arising in testicular germ cell neoplasms. Am J Surg Pathol 1997;21:896-904 [DOI] [PubMed] [Google Scholar]
- 10.Ulbright TM, Hattab EM, Zhang S, et al. Primitive neuroectodermal tumors in patients with testicular germ cell tumors usually resemble pediatric-type central nervous system embryonal neoplasms and lack chromosome 22 rearrangements. Mod Pathol 2010;23:972-80 [DOI] [PubMed] [Google Scholar]
- 11.Roth BJ, Greist A, Kubilis PS, et al. Cisplatin-based combination chemotherapy for disseminated germ cell tumors: long-term follow-up. J Clin Oncol 1988;6:1239-47 [DOI] [PubMed] [Google Scholar]
- 12.Vaughn DJ, Flaherty K, Lal P, et al. Treatment of growing teratoma syndrome. N Engl J Med 2009;360:423-4 [DOI] [PubMed] [Google Scholar]