

Abstract
The combination of chemical cross-linking and mass spectrometry has recently been shown to constitute a powerful tool for studying protein–protein interactions and elucidating the structure of large protein complexes. However, computational methods for interpreting the complex MS/MS spectra from linked peptides are still in their infancy, making the high-throughput application of this approach largely impractical. Because of the lack of large annotated datasets, most current approaches do not capture the specific fragmentation patterns of linked peptides and therefore are not optimal for the identification of cross-linked peptides. Here we propose a generic approach to address this problem and demonstrate it using disulfide-bridged peptide libraries to (i) efficiently generate large mass spectral reference data for linked peptides at a low cost and (ii) automatically train an algorithm that can efficiently and accurately identify linked peptides from MS/MS spectra. We show that using this approach we were able to identify thousands of MS/MS spectra from disulfide-bridged peptides through comparison with proteome-scale sequence databases and significantly improve the sensitivity of cross-linked peptide identification. This allowed us to identify 60% more direct pairwise interactions between the protein subunits in the 20S proteasome complex than existing tools on cross-linking studies of the proteasome complexes. The basic framework of this approach and the MS/MS reference dataset generated should be valuable resources for the future development of new tools for the identification of linked peptides.
The study of protein–protein interactions is crucial to understanding how cellular systems function because proteins act in concert through a highly organized set of interactions. Most cellular processes are carried out by large macromolecular assemblies and regulated through complex cascades of transient protein–protein interactions (1). In the past several years numerous high-throughput studies have pioneered the systematic characterization of protein–protein interactions in model organisms (2–4). Such studies mainly utilize two techniques: the yeast two-hybrid system, which aims at identifying binary interactions (5), and affinity purification combined with tandem mass spectrometry analysis for the identification of multi-protein assemblies (6–8). Together these led to a rapid expansion of known protein–protein interactions in human and other model organisms. Patche and Aloy recently estimated that there are more than one million interactions catalogued to date (9).
But despite rapid progress, most current techniques allow one to determine only whether proteins interact, which is only the first step toward understanding how proteins interact. A more complete picture comes from characterizing the three-dimensional structures of protein complexes, which provide mechanistic insights that govern how interactions occur and the high specificity observed inside the cell. Traditionally the gold-standard methods used to solve protein structures are x-ray crystallography and NMR, and there have been several efforts similar to structural genomics (10) aiming to comprehensively solve the structures of protein complexes (11, 12). Although there has been accelerated growth of structures for protein monomers in the Protein Data Bank in recent years (11), the growth of structures for protein complexes has remained relatively small (9). Many factors, including their large size, transient nature, and dynamics of interactions, have prevented many complexes from being solved via traditional approaches in structural biology. Thus, the development of complementary analytical techniques with which to probe the structure of large protein complexes continues to evolve (13–18).
Recent developments have advanced the analysis of protein structures and interaction by combining cross-linking and tandem mass spectrometry (17, 19–24). The basic idea behind this technique is to capture and identify pairs of amino acid residues that are spatially close to each other. When these linked pairs of residues are from the same protein (intraprotein cross-links), they provide distance constraints that help one infer the possible conformations of protein structures. Conversely, when pairs of residues come from different proteins (interprotein cross-links), they provide information about how proteins interact with one another. Although cross-linking strategies date back almost a decade (25, 26), difficulty in analyzing the complex MS/MS spectrum generated from linked peptides made this approach challenging, and therefore it was not widely used. With recent advances in mass spectrometry instrumentation, there has been renewed interest in employing this strategy to determine protein structures and identify protein–protein interactions. However, most studies thus far have been focused on purified protein complexes. With today's mass spectrometers being capable of analyzing tens of thousands of spectra in a single experiment, it is now potentially feasible to extend this approach to the analysis of complex biological samples. Researchers have tried to realize this goal using both experimental and computational approaches. Indeed, a plethora of chemical cross-linking reagents are now available for stabilizing these complexes, and some are designed to allow for easier peptide identification when employed in concert with MS analysis (20, 27, 28). There have also been several recent efforts to develop computational methods for the automatic identification of linked peptides from MS/MS spectra (29–36). However, because of the lack of large annotated training data, most approaches to date either borrow fragmentation models learned from unlinked, linear peptides or learn the fragmentation statistics from training data of limited size (30, 37), which might not generalize well across different samples. In some cases it is possible to generate relatively large training data, but it is often very labor intensive and involves hundreds of separate LC-MS/MS runs (36). Here, employing disulfide-bridged peptides as an example, we propose a novel method that uses a combinatorial peptide library to (a) efficiently generate a large mass spectral reference dataset for linked peptides and (b) use these data to automatically train our new algorithm, MXDB, which can efficiently and accurately identify linked peptides from MS/MS spectra.
MATERIALS AND METHODS
Creating MS/MS Training Data for Linked Peptides
We synthesized three combinatorial peptide libraries with the following sequence patterns:
I. K[AW][DE]F[VSHY]A[DY]SCVA[KR]
II. [TW]A[LE]H[FV]SCVT[PSGY]F[KR]
III. [WA]VK[FL]C[DE]T[VSGY]FA[KR]
The letters in square brackets indicate that multiple amino acids were possible at those positions. For example, in library I, both alanine (A) and tryptophan (W) were possible amino acids at the second position. The sequence patterns were designed to contain several desirable properties to facilitate identification. Firstly, each library contains only one cysteine for disulfide-bond formation, so there is no ambiguity in assigning the linking site. The incorporation of residues such as proline (which is known to produce MS/MS spectra with relatively poor fragmentation (38)) was kept low; the theoretical precursor mass of all possible disulfide-bridged peptide pairs that can be formed from the library peptides was computed and the sequence patterns were chosen to optimize for the generation of disulfide peptides with as many unique precursor masses as possible. Finally, the variable positions (i.e. in square brackets) used amino acid residues with different physicochemical properties (i.e. hydrophobicity, polarity, size, etc.) to increase the chance of separation of the library peptides using liquid chromatography.
Each peptide library was designed to generate 26 = 64 unique unlinked peptides and thus (64 × 64)/2 = 2048 unique disulfide-bridged peptide pairs. This ensures a sufficiently large training dataset to learn the fragmentation patterns of disulfide-bridged peptides while at the same time providing a manageable search space so that MS/MS spectra from disulfide-bridged peptides can readily be identified using even a suboptimal search strategy in which all possible linked peptide pairs are considered.
After synthesis, the peptide libraries were put in conditions that allowed the formation of disulfide-bridged dimers and were analyzed using tandem mass spectrometry. Specifically, 20 μg of library peptides were incubated with 30 mm 2,2′-dipyridyldisulfide for 2 h at room temperature. All samples were dried using a SpeedVac, stage-tipped, brought up in 10 μl of 0.1% TFA, and injected on an Orbitrap-Velos. Samples were injected via an autosampler for separation by reverse-phase chromatography on a NanoAcquity UPLC system (Waters, Dublin, CA). Peptides were loaded onto a Symmetry C18 column (1.7 m BEH-130, 0.1 × 100 mm, Waters, Dublin, CA) with a flow rate of 1 μl/min and a gradient of 2% solvent B to 25% solvent B (where solvent A is 0.1% formic acid/2% acetonitrile/water and solvent B is 0.1% formic acid/2% water/acetonitrile) applied over 60 min with a total analysis time of 90 min. Peptides were eluted directly into an Advance CaptiveSpray ionization source (Michrom BioResources/Bruker, Auburn, CA) with a spray voltage of 1.4 kV and were analyzed using an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher, San Jose, CA). Precursor ions were analyzed in a Fourier transform mass spectrometer at 60,000 resolution. MS/MS was performed in the LTQ with the instrument operated in data-dependent mode with the top 15 most abundant ions subjected for fragmentation.
MS/MS spectra from the disulfide-bridged peptide libraries were identified using a two-step search strategy. As illustrated in Fig. 1, an initial search was done using a scoring model learned from SUMOylated peptides (39) against a database containing all possible library peptides. SUMOylated peptides are a specific type of linked peptide in which the peptide QQQTGG is linked to a lysine residue of a substrate peptide. The details of the identification of SUMOylated peptides are discussed elsewhere (40). An initial set of MS/MS spectra from disulfide-bridged peptides were identified at a 5% FDR.1 From these initial training data, a scoring model specific for disulfide-bridged peptides was built and used to search the spectra from all three peptide libraries one more time to obtain a final list of MS/MS spectra from disulfide-bridged peptides. Unless otherwise noted, all searches with MXDB were performed with a 0.05-Da parent mass tolerance and 0.5-Da fragment mass tolerance. Then results were filtered for a precursor mass error of less than 10 ppm, and an FDR of 5% was enforced using a target/decoy approach (41) (see details below).
Fig. 1.
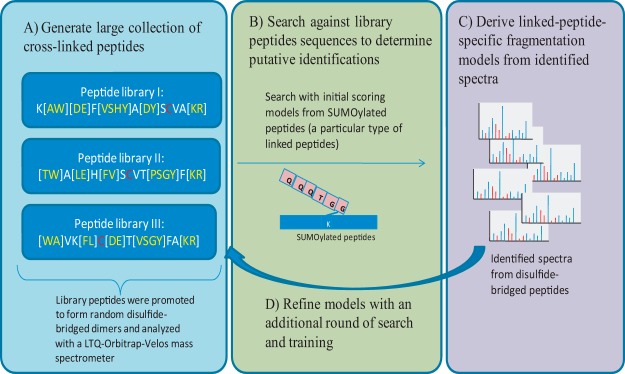
Generating training data using combinatorial synthetic peptide libraries. A, in order to generate a sufficiently large training dataset for linked peptides, we designed and synthesized three combinatorial peptide libraries, each with a cysteine residue at a different position along the peptide sequence. Amino acids in square brackets indicate that multiple residues are possible at that position. The peptide libraries were allowed to form disulfide-bridged dimers and were analyzed using an LTQ-Orbitrap-Velos mass spectrometer. MS/MS spectra from the disulfide-bridged peptide libraries were identified using a two-step strategy. B, an initial set of MS/MS spectra from disulfide-bridged peptides was identified using scoring models learned from SUMOylated peptides (a special type of linked peptide in which the C termini of QQQTGG is linked to the lysine of another peptide). C, from these initial training data, we built a scoring model specific for disulfide-bridged peptides and (D) used the improved scoring models to search the data again to obtain a final set of spectra from disulfide-bridged peptides.
Scoring Models for Linked Peptides
In order to evaluate the match between a cross-linked peptide pair and an observed MS/MS spectrum, we conceptually considered a linked peptide as a mixture of two peptides, each carrying a modification at the cross-linked residue with mass equal to that of the parent mass of the other peptide plus the mass of the linker (see supplemental Fig. S1). In a regular database search, one tries to evaluate how well a single candidate peptide matches to an MS/MS spectrum. For cross-linked peptides one evaluates how well a pair of peptides matches to an MS/MS spectrum. In our previous method, MixDB (42), we introduced a probabilistic model to score how well a pair of peptides matches to a mixture MS/MS spectrum from co-eluting peptides. The statistical framework used here extends that of MixDB by further capturing the specific fragmentation pattern of linked peptides.
Briefly, an MS/MS spectrum is represented as a vector of n bins, each representing a mass interval of width δ Da (δ depends on instrument resolution). An experimental MS/MS spectrum is represented as a vector S = s1,s2, …, sn, where si represents the spectrum-wide peak intensity rank (ranked from most to least intense) in each bin. Similarly, a theoretical spectrum of a peptide P = p1,p2, …, pn is represented as a vector, where pi indicates the ion type of the fragment ion (e.g. b-ion or y-ion) with mass in that bin. The model captures peptide fragmentation statistics by using a set of annotated MS/MS spectra to learn the probability that each type of ion generates an observed peak with a given rank, Prob(s p). Similarly, a noise model Prob(s 0) can be learned using unmatched peaks in the spectrum (where the symbol 0 represents noise). The scoring function for a peptide spectrum match is thus defined as the likelihood ratio of the probability that the observed spectrum S is generated from the candidate peptide P versus the probability that the observed spectrum is generated from noise.
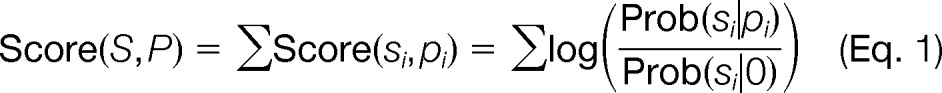 |
This is a model similar to that introduced by Kim et al. in MS-Dictionary (43) and since used in MS-GF (44) and MSGFDB (45). Because linked peptides are represented as pairs of peptides, we can represent a linked peptide as two vectors (P,Q). The vector P = p1,p2, …, pn contains all possible fragment ions from the first peptide, and the vector Q = q1,q2, …, qn contains all possible fragment ions from the second peptide. Without a loss of generality, we define the “first peptide” as the peptide that accounts for the most ion intensity in the MS/MS spectrum, and we define the other peptide as the “second” peptide. This is done to account for possible differences in fragmentation patterns between the first and second peptides. The score of a spectrum S matching to a pair of peptides (P,Q) is thus defined as
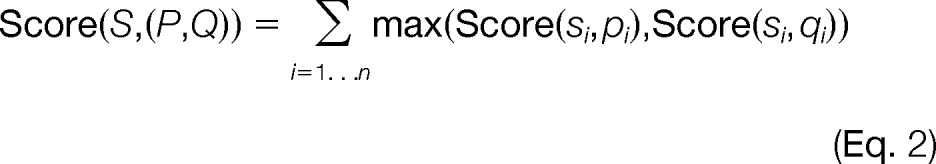 |
where “max” is used to model the dependence between the two peptides. When theoretical fragment ions from both P and Q match the same observed spectrum peak, the model uses only the fragment ion with higher probability, thereby avoiding using the same peak twice to support the identification of linked peptides. If not explicitly prevented, such double counting will incorrectly bias unusually high scores toward pairs of peptide candidates sharing many theoretical fragment ions.
In order to further capture the fragmentation statistics of cross-linked peptides, the set of possible fragment ions was divided into linked and unlinked fragments (supplemental Fig. S1), where linked fragments are fragment ions that are covalently linked to a second peptide. Thus for every ion type that is used to describe linear peptides, we introduce its corresponding linked ion type in MXDB's probabilistic models. For example, in the current implementation, the ion types b, b(iso), b − H2O, b − NH3, y, y(iso)y − H2O, y − NH3 were considered for linear, unlinked peptides, where b(iso) indicates the first 13C isotopic peak of a b-ion. For linked peptides the ion types bx, b(iso)x, b − H2Ox, b − NH3x, yx, y(iso)x, y − NH3x were added to represent the corresponding linked-fragment ions that can be generated from linked peptides. For each ion type, charge states from 1 to the precursor charge of the observed MS/MS spectrum were considered. With these new ion types, the fragmentation statistics specific to linked peptide fragments can be learned from a set of identified linked peptide spectra, and different probabilities/weights thus can be determined for linked and non-linked fragment ions.
Efficient Database Search for Linked Peptides
With a scoring function that properly models the fragmentation characteristics of linked peptides, we can evaluate how well a cross-linked peptide pair matches an observed MS/MS spectrum. However, one still needs to evaluate all possible cross-linked peptide pairs in the sequence database to find the correct match. When the protein sequence database is large, it is not practical to consider all possible peptide pairs in the database. In principle this is similar to the problem of identifying mixture spectra from co-eluting peptides (46–48); however, the parent mass for each of the peptides is known in the case of co-eluting peptides, which can greatly reduce the search space of possible peptides. For linked peptides only the combined parent mass of the two peptides is known; therefore the identification of linked peptides can be thought of as identifying two peptides with an unknown modification at the same time (29, 31). Similar to previous approaches (29–31), MXDB uses a two-step search strategy to find the correct cross-linked peptide match without considering all possible pairs. Because a linked peptide is modeled as a pair of peptides, each of which has a post-translational modification of the mass of the linker plus the other peptide, we capitalize on the fact that for a linked peptide pair that generates the observed spectrum, at least one peptide should have a good score when matched to the spectrum by itself. In brief, in the first stage of the search every candidate peptide is scored against the query spectrum by itself. Then in the second stage only the top-scoring candidate peptides are paired to find the best-scoring peptide pair. Specifically, let S be a query spectrum with parent mass MS and P be a peptide with parent mass MP. Also, let P1,P2,…,Pn be a database containing n peptides. A modified peptide P(Δ,t) is peptide P with a mass offset of Δ Da at the tth residue. For each peptide Pi in the database we consider all of its modified variants Pi(Δ,t), where Δ is the mass difference between the parent mass of the spectrum and the parent mass of the candidate peptide (Δ=Ms−Mpi, s.t.Δ>0) and t spans all possible linking amino acids for the candidate peptide Pi. For example, in the case of disulfide-bridged peptides, all cysteine positions are considered. All modified candidate peptides are scored against the query spectrum S and ranked by decreasing match score. As shown in supplemental Fig. S4, when the training data were searched against a database of all library peptide sequences concatenated with the whole E. coli database (which contains ∼200,000 tryptic peptides), one of the correct peptides ranked in the top 50 highest scoring peptides and the other peptide ranked in the top 200 highest scoring candidates in more than 99% of cases. Thus, rather than consider (200,000 × 200,000)/2 = 2 × 1010 peptide pairs, MXDB can consider 50 × 200 = 10,000 peptide pairs and still find the correct matches in more than 99% of cases. In MXDB the search space is further reduced because peptide pairs are considered such that their combined masses match the precursor mass of the MS/MS spectrum.
Separation of Linked-peptide Matches from False Positives
After the best match to each spectrum has been found, the top-scoring linked-peptide-spectrum matches (LPSMs) are scored using a support vector machine (SVM) to separate true matches from false-positive matches. Features used in the SVM are as follows: (i) normalized score: likelihood score (as in Eq. 1) divided by the number of amino acids in the candidate peptide; (ii) explained intensity: total intensity of matched MS/MS peaks divided by the summed intensity in the MS/MS spectrum; (iii, iv) fraction of b-/y-ions: number of b- or y-ions present in the spectrum divided by the number of b- or y-ions possible from the peptide (two features); (v, vi) length of the longest consecutive series of b- and y-ions (two features); and (vii) average mass error between theoretical and observed fragment ion masses.
Note that we can separately compute the above features for each of the linked peptides. These plus the combined likelihood score that considers matched peaks from both cross-linked peptides (as in Eq. 2) constitute the final set of 15 features used in the SVM. The parameters of the SVM were trained from the identified MS/MS spectra from the combinatorial library of disulfide peptides as described in the previous section. For each training dataset, the correct LPSMs were used as positive training data and top-scoring LPSMs from the decoy database were used as negative training data.
All LPSMs were sorted according to their SVM scores and were accepted as correct matches if their scores passed a certain threshold. The SVM score threshold was chosen to enforce a particular FDR. For a set of LPSMs, the FDR is estimated using a target-decoy approach (41, 49). Briefly, because each LPSM has two peptide matches, it can fall into one of the following categories: TT, in which both peptide matches are from the target database; TD/DT, in which one peptide is from the target database and another is from the decoy database; or DD, in which both peptide matches are from the decoy database. If we define Ntype as the number of LPSMs of a particular type (i.e. NTT is the number of matches of type TT), we can then define the FDR for LPSMs as
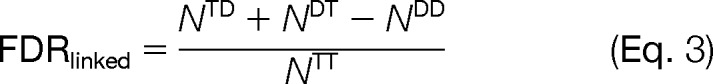 |
Analysis of Spectra from Cross-linked Samples
We analyzed the data from a cross-linked sample of Schizosaccharomyces pombe and rabbit proteasome from a previous study (50). The search results from xQuest were obtained from the original publication (50), and the pLink search results were obtained by running the search with a 50-ppm precursor mass tolerance and the default (0.5-Da) fragment mass tolerance for collision-induced dissociation spectra. The search results were filtered with a 10-ppm precursor mass tolerance and 5% FDR. Protein sequences for the proteasome complex were downloaded from UniProt (51) by extracting all proteins from the corresponding species that contained the keyword “Proteasome” using the “advanced search” function of UniProt. To validate the identified cross-linked peptides, we obtained crystal structures of the proteasome complexes of related species from the Protein Data Bank (52). Because the crystal structures for both rabbit and S. pombe are not available, the S. pombe proteasome sequences were mapped to the crystal structure of the Saccharomyces cerevisiae proteasome (Protein Data Bank I.D.: 1FNT) (53) and the rabbit proteasome sequences were mapped to the crystal structure of mouse proteasome (Protein Data Bank I.D.: 3UNE) (54). The mapping was done by aligning pairs of orthologous sequences from the proteasome complex of the two related species using the sequence alignment function in UCSF Chimera (55). The three-dimensional coordinates of the sequence of known structure were then transferred to aligned residues of the sequence with unknown structure. Then the distance between the Cα atoms of the linking residues in each identified cross-linked peptide was computed. Distances of less than 25 Å were considered to be within the distance constraints of the cross-linker, as determined based on the maximum distance that can be spanned by the cross-linker, the length of the side-chain of a lysine residue and the deviation between the Cα atoms of homologous protein structures. For the two-pass searches we first identified unlinked peptides by searching the data against all rabbit protein sequences using MSGFDB with the following variable modifications: +42.010 (acetylation) on N terminus; +15.994 (oxidation) on methionine; and +138.068, +150.143, +156.080, and +168.155 on lysine (for identification of peptides with a hydrolyzed linker and intrapeptide cross-linked peptides; there are four possible mass offsets because the cross-linker used had light and heavy isotopic variants). Search results were filtered at a 1% FDR, and all proteins containing at least one identified peptide were considered as candidate proteins for the second part of the search. Degenerate peptides shared among multiple proteins resulted in all proteins being considered as candidates.
RESULTS
Building a Linked-peptide Specific Search Method for Disulfide-bridged Peptides
There are three major computational challenges in the identification of linked peptides. First, the covalent linkage of two peptides changes the physicochemical properties of the peptides and generates fragment ions that display substantially different fragmentation patterns than those captured by existing models for linear, unlinked peptides. Second, although spectra from linked peptides contain fragment ions from two peptides, almost all MS/MS database search tools assume that each spectrum comes from a single peptide. The presence of two peptides in the same spectrum creates a quadratic search space for peptide candidates. Efficient techniques are therefore needed to search this vast search space. Finally, there are usually only a small number of reliably identified spectra that are available to learn the fragmentation models for linked peptides, significantly constraining the development of advanced approaches as previously proposed for unlinked peptides (45, 56, 57). In order to address these challenges, we designed and synthesized three combinatorial peptide libraries, each with a cysteine residue at varying positions along the library peptides. The peptide libraries were then allowed to form random disulfide-bridged dimers and were analyzed with an LTQ-Orbitrap-Velos mass spectrometer. MS/MS spectra were identified using a two-step search strategy (see Fig. 1). A total of 5952 MS/MS spectra from disulfide-bridged peptides were identified, corresponding to 2976 unique linked-peptide pairs. To our best knowledge, this is the largest MS/MS dataset of linked peptides available for the development of new tools for the identification of linked peptides.
From these training data, the fragmentation patterns of disulfide-bridged peptides were analyzed. We divided fragment ions from linked peptides into linked and unlinked fragments (see supplemental Fig. S1). Linked fragments are fragment ions that remain covalently linked to a second peptide and were observed to have different fragmentation patterns than unlinked fragments. For example, although triply charged y-ions are quite common in linked fragments from a triply charged precursor, they are rarely observed in unlinked fragments from the same precursor. Furthermore, we observed that both linked and unlinked fragments from disulfide-bridged peptides had different fragmentation patterns than fragment ions from conventional, unlinked peptides. In general, unlinked fragments yielded lower intensity peaks in the MS/MS spectra than the same types of fragment ions from unlinked peptides (see supplemental Fig. S2A). In contrast, for linked fragments, highly charged fragments tend to be more prominent than those in unlinked peptides because linked fragments are covalently attached to another peptide that contains additional N and C termini (tryptic peptides have K/R at the C terminus) that have a higher affinity for protonation (see supplemental Fig. S2B). Finally, it was observed that different types of linked peptides also tended to have different fragmentation patterns as well. Supplemental Fig. S2C compares the fragmentation patterns of disulfide-bridged peptides and those of SUMOylated peptides (39), which are a special type of linked peptide in which the C terminus of the peptide QQQTGG is conjugated to a lysine residue of another peptide (see Fig. 1). Even though triply charged y-ions are prominent in both disulfide-bridged peptides and SUMOylated peptides, they are twice as prominent in the former as in the latter. Thus, ultimately, it is necessary to build a probabilistic model for each type of linked peptide in order to maximize the sensitivity of their identification from MS/MS spectra.
In order to account for the fragmentation characteristics of linked peptides, our database search method, MXDB, uses separate ion models for linked and unlinked fragment ions (e.g. linked b-ion fragments versus unlinked b-ion fragments; see “Materials and Methods”). Therefore, different probability-based weights were assigned to linked and unlinked fragments when scoring a candidate linked peptide against an MS/MS spectrum. To address the fact that an MS/MS spectrum from linked peptides contains fragments from two peptides, MXDB uses a mixture fragmentation model similar to that used in MixDB (42) to explicitly account for the co-fragmentation of two peptides (see supplemental Fig. S1A). In addition, MXDB also allows a two-stage search strategy in which it first identifies only one peptide and then identifies the other peptide in the linked pair (see supplemental Figs. S3 and S4). This strategy enables MXDB to reduce the search space of all possible peptide pairs by 3 to 4 orders of magnitude while still identifying the correct linked peptides (see “Materials and Methods” for details).
Identification of Disulfide-bridged Peptides from the Combinatorial Peptide Libraries
In order to test MXDB's performance in the identification of disulfide-bridged peptides, the MS/MS spectra from three disulfide-bridged peptide libraries were searched against all possible library peptide sequences. Starting with an initial scoring model learned from SUMOylated peptides, we identified an initial set of 4232 MS/MS spectra from disulfide-bridged peptides. From this initial training dataset, improved scoring models specific for disulfide-bridged peptides were built and used to identify an additional 31% to 60% more MS/MS spectra from each peptide library (see Fig. 2A). This result supports our hypothesis that different types of linked peptides have differing fragmentation patterns and that properly capturing these fragmentation patterns improves the sensitivity of the identification of linked peptides from mass spectra.
Fig. 2.
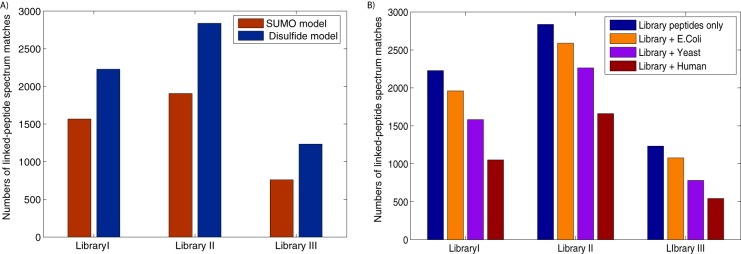
Identification of disulfide-bridged peptides from combinatorial peptide libraries. A, the initial scoring models learned from SUMOylated peptides (shown in red) were compared with the scoring models for disulfide-bridged peptides (shown in blue). The latter models improved the identification of disulfide-bridged peptides by 31% to 60%, confirming the need to model the different fragmentation patterns for different types of linked peptides. B, MXDB's ability to identify disulfide-bridged peptides against whole-proteome sequence databases was tested as follows: the library peptide sequences were concatenated with all E. coli, yeast, and human protein sequences (respectively shown in blue, orange, purple, and red) and the spectra from the peptide libraries were searched against each concatenated database. MXDB was able to identify thousands of spectra from disulfided peptides against these proteome-scale databases, and the general trend shows that as the search space of cross-linked peptides increased by ∼9-fold, the sensitivity of the identification of disulfide-bridged peptides decreased by 20% to 25%, unless a two-pass search strategy was used (see supplementary Fig. S6).
An important goal of devising better approaches for the identification of linked peptides is to enable the identification of linked peptides in more complex biological samples. Thus, we tested whether the MXDB search could scale up to large sequence databases. We tested this by appending E. coli, yeast, and human protein databases in their entirety to the database of library peptide sequences and searching the MS/MS spectra from the peptide libraries against these concatenated databases. The effect of database size on MXDB's sensitivity in identifying disulfide-bridged peptides is shown in Fig. 2B. The trend indicates that as we increased the size of the database by a factor of 3, which roughly corresponded to a 9-fold increase in the search space of linked peptides, there was on average a 20% to 25% drop in sensitivity. In general, it is expected that increases in the size of the search space will result in the decreased sensitivity of database search methods. For comparison, we also searched a trypsin-digested yeast cell lysate dataset (58) using MSGFDB (45), a database search tool for conventional, unlinked peptides. MSGFDB identified 27,337 and 22,168 spectra with 50-ppm and 500-ppm precursor mass tolerance, respectively. Both searches were done using a 0.5-Da fragment mass tolerance. On average, this corresponds to a search space 3.628 times larger due to the fact that the parent masses of peptides do not distribute evenly among all possible mass values. This means there is an ∼18.9% drop in sensitivity when the size of the search space is increased 3.6-fold. MXDB's drop in sensitivity is slightly greater than that observed in traditional database search tools for unlinked peptides, but this is readily explained by the quadratic growth in the search space of linked peptides versus the linear growth for unlinked peptides. Combined with the results discussed below for two-pass searches (illustrated in supplemental Fig. S6), these demonstrate MXDB's ability to identify linked peptides against proteome-scale sequence databases.
Identification of Cross-linked Peptides from Protein Complexes
To illustrate the utility of MXDB in biological applications, we applied it to the identification of chemically cross-linked peptides on two MS/MS datasets: one from the S. pombe 26S proteasome complex, and another from the rabbit 20S proteasome complex (50). Proteasome complexes are responsible for the proteolytic degradation of unneeded proteins inside the cell, and much structural information about the complexes is known because of their functional importance (59). Therefore, they serve as good model complexes for cross-linking studies, as identified linked peptides can be validated using known structural information. As we can see in Fig. 3, in both datasets MXDB was able to identify significantly more cross-linked peptides than the two current state-of-the-art database search tools for cross-linked peptides, xQuest (30) and pLink (36). This in turn allowed MXDB to identify 60% more pairwise interactions between the protein subunits in the proteasome complex (see supplemental Table S1). This shows that the fragmentation models learned from disulfide-bridged peptides are appropriate generic initial models for the identification of linked peptides. To evaluate the quality of the MXDB search results, the identified cross-linked peptides were mapped to homologous proteins with available crystal structures in the Protein Data Bank (11). The distance between the identified cross-linked residue pairs was computed and is shown in supplementary Fig. S5. The expected portion of the identified cross-linked peptides had distances within range of the cross-linker used. Approximately 8% of the identified cross-linked peptides had distances greater than 25 Å, which is considered to exceed the maximum distance range of the cross-linker. This is also consistent with the 5% FDR that was enforced in the search results using the target-decoy approach (41). The slightly higher observed error rate could be due to the distances being computed based on the structure of a homologous protein complex with possible variations in structures between homologous proteins. To evaluate whether MXDB could identify linked peptides against large sequence databases, the rabbit proteasome dataset was searched against the concatenated databases of rabbit proteasome protein sequences appended to the E. coli, yeast, and rabbit protein sequence databases. As shown in supplemental Fig. S6, although MXDB was still able to identify a large number of cross-linked peptides, there was also a noticeable decrease in sensitivity with increasing database size. Thus, in order to improve the sensitivity of linked-peptide identification, one ideally would use the smallest possible database with all the correct proteins. However, in contrast with the analysis of the proteasome complexes (which are relatively well-studied complexes), in many studies involving the identification of protein complexes, the composition of the protein subunits would be unknown. To address this scenario, MXDB capitalizes on the fact that in cross-linking experiments there are usually many unlinked peptides also present in the samples that can be used to determine the protein composition of the complex. Thus we evaluated a two-step search strategy in which a list of candidate proteins was first determined via a search for unlinked peptides and peptides with a hydrolyzed linker using a conventional database search method. MXDB was then used to identify cross-linked peptides against this reduced set of candidate proteins. As shown in supplemental Fig. S6, this two-pass search strategy was able to recover 83% to 95% of the cross-linked peptides in the rabbit proteasome sample when searching proteome-scale databases. Thus, compared with directly searching the whole database, two-pass searches represent a good balance between assumptions about the protein contents of the sample and the sensitivity of identification of linked peptides. This two-pass search strategy is readily applicable to high-throughput protein interaction studies in which protein complexes are extracted from cellular lysates using affinity purification strategies.
Fig. 3.
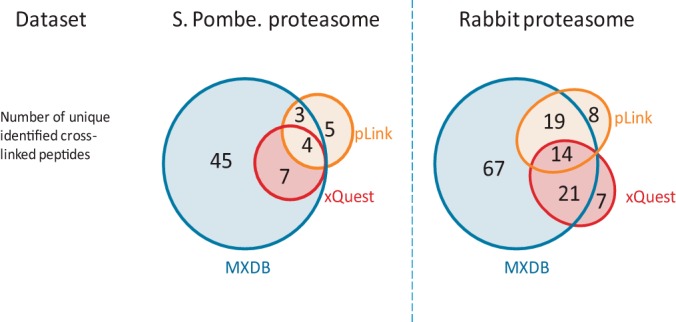
Identification of cross-linked peptides in yeast and rabbit proteasome complexes. We compared the identification of cross-linked peptides by MXDB, pLink, and xQuest on datasets from a cross-linking study of proteasome complexes from S. pombe and rabbit. MXDB was able to identify significantly more cross-linked peptides than pLink or xQuest. The overlap in the identifications also shows that MXDB was able to identify on average 80% of the linked peptides that were identified via the other methods. The additional cross-linked peptides also allowed MXDB to map 60% more pairwise interactions between the protein subunits in the proteasome complex (see supplemental Table S1).
DISCUSSION
Chemical cross-linking followed by tandem mass spectrometry is a versatile strategy for the analysis of protein structures and protein–protein interactions. However, there are several challenges that need to be addressed before it can be routinely applied on a large scale. The development of novel cross-linkers (27, 28) has improved our ability to separate linked peptides from a large background of other analytes in complex samples and facilitates their analysis using mass spectrometry. Nevertheless, the different functional groups on these novel cross-linkers will certainly affect how the linked peptides fragment in mass spectrometers. Here we show that having computational methods that properly capture the fragmentation patterns of linked peptides can substantially improve our ability to identify them from MS/MS spectra. It was recently shown that even linear, unlinked peptides that are products of different enzymatic digestions display rather unique fragmentation patterns in MS/MS spectra, and properly modeling these fragmentation characteristics can greatly improve the identification of peptides from MS/MS spectra (45). Because of the different physicochemical properties of linked peptides, the development of appropriate fragmentation models is even more crucial for their identification. However, this task is challenging because there are no sufficiently large, publicly available reference datasets to learn their fragmentation patterns. Using an integrated experimental and computational strategy based on combinatorial peptide synthesis, we have shown that it is possible to efficiently generate a large mass spectral reference dataset for linked peptides at low cost. These reference data revealed that linked peptides do have substantially different fragmentation patterns than unlinked, linear peptides, meaning most current tools trained from unlinked peptides are suboptimal for the identification of linked peptides. By incorporating linked-peptide specific fragmentation statistics and efficient filtration strategies in MXDB, we have shown that it is possible to identify disulfide-bridged peptides against proteome-scale sequence databases. Beyond addressing the core challenges in the development of sensitive and accurate methods for the identification of linked peptides, our framework, based on combinatorial peptide synthesis, can also be adapted to any type of linked peptide, and thus could simplify and expedite the development of computational tools for the identification of other types of linked peptides. The training data generated in this study have been made publicly available in the MassIVE repository, and we expect them to be a valuable resource for the future development of advanced algorithms for the identification of linked peptides.
Supplementary Material
Footnotes
Author contributions: J.W., V.G.A., J.R.L., and N.B. designed research; J.W., V.G.A., and J.R.L. performed research; J.W., V.G.A., J.K., J.R., and J.R.L. contributed new reagents or analytic tools; J.W., P.E.B., and N.B. analyzed data; J.W., V.G.A., J.R.L., P.E.B., and N.B. wrote the paper.
* This work was partly supported by National Institutes of Health Grant No. GM078596 (J.W. and P.E.B.) and P41 GM103485-05 (J.W. and N.B.) from NIGMS, National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- FDR
- false discovery rate
- MS/MS
- tandem mass spectrometry
- SUMO
- small ubiquitin-like modifier
- LPSM
- linked-peptide-spectrum match
- SVM
- support vector machine.
REFERENCES
- 1. Robinson C. V., Sali A., Baumeister W. (2007) The molecular sociology of the cell. Nature 450(7172), 973–982 [DOI] [PubMed] [Google Scholar]
- 2. Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., Vijayadamodar G., Yang M., Johnston M., Fields S., Rothberg J. M. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403(6770), 623–627 [DOI] [PubMed] [Google Scholar]
- 3. Giot L., Bader J. S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y. L., Ooi C. E., Godwin B., Vitols E., Vijayadamodar G., Pochart P., Machineni H., Welsh M., Kong Y., Zerhusen B., Malcolm R., Varrone Z., Collis A., Minto M., Burgess S., McDaniel L., Stimpson E., Spriggs F., Williams J., Neurath K., Ioime N., Agee M., Voss E., Furtak K., Renzulli R., Aanensen N., Carrolla S., Bickelhaupt E., Lazovatsky Y., DaSilva A., Zhong J., Stanyon C. A., Finley R. L., White K. P., Braverman M., Jarvie T., Gold S., Leach M., Knight J., Shimkets R. A., McKenna M. P., Chant J., Rothberg J. M. (2003) A protein interaction map of Drosophila melanogaster. Science 302(5651), 1727–1736 [DOI] [PubMed] [Google Scholar]
- 4. Rual J.-F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437(7062), 1173–1178 [DOI] [PubMed] [Google Scholar]
- 5. Fields S., Song O. (1989) A novel genetic system to detect protein protein interactions. Nature 340, 245–246 [DOI] [PubMed] [Google Scholar]
- 6. Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17(10), 1030–1032 [DOI] [PubMed] [Google Scholar]
- 7. Collins M. O., Choudhary J. S. (2008) Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr. Opin. Biotechnol. 19(4), 324–330 [DOI] [PubMed] [Google Scholar]
- 8. Gingras A.-C., Gstaiger M., Raught B., Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8(8), 645–654 [DOI] [PubMed] [Google Scholar]
- 9. Pache R. A., Aloy P. (2008) Incorporating high-throughput proteomics experiments into structural biology pipelines: identification of the low-hanging fruits. Proteomics 8(10), 1959–1964 [DOI] [PubMed] [Google Scholar]
- 10. Stevens R. C., Yokoyama S., Wilson I. A. (2001) Global efforts in structural genomics. Sci. Signal. 294(5540), 89. [DOI] [PubMed] [Google Scholar]
- 11. Berman H., Henrick K., Nakamura H., Markley J. L. (2007) The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 35, D301–D303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. http://www.spine2.eu/SPINE2/index.jsp.
- 13. Haydyn D., Mertens T., Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172(1), 128–141 [DOI] [PubMed] [Google Scholar]
- 14. Joo C., Balci H., Ishitsuka Y., Buranachai C., Ha T. (2008) Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 77(1), 51–76 [DOI] [PubMed] [Google Scholar]
- 15. Takamoto K., Chance M. R. (2006) Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 35, 251–276 [DOI] [PubMed] [Google Scholar]
- 16. Stahlberg H., Walz T. (2008) Molecular electron microscopy: state of the art and current challenges. ACS Chem. Biol. 3(5), 268–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sinz A. (2006) Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom. Rev. 25, 663–682 [DOI] [PubMed] [Google Scholar]
- 18. Berggård T., Linse S., James P. (2007) Methods for the detection and analysis of protein-protein interactions. Proteomics 7(16), 2833–2842 [DOI] [PubMed] [Google Scholar]
- 19. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F. U., Ban N., Malmström1 L., Aebersold R. (2012) Structural probing of a protein phosphatase 2a network by chemical cross-linking and mass spectrometry. Science 337(6100), 1348–1352 [DOI] [PubMed] [Google Scholar]
- 20. Zhang H., Tang X., Munske G. R., Tolic N., Anderson G. A., Bruce J. E. (2009) Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol. Cell. Proteomics 8(3), 409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Z. A., Jawhari A., Fischer L., Buchen C., Tahir S., Kamenski T., Rasmussen M., Lariviere L., Bukowski-Wills J. C., Nilges M., Cramer P., Rappsilber J. (2010) Architecture of the rna polymerase ii–tfiif complex revealed by cross-linking and mass spectrometry. EMBO J. 29, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Granneman S., Petfalski E., Swiatkowska A., Tollervey D. (2010) Cracking pre-40s ribosomal subunit structure by systematic analyses of rna-protein cross-linking. EMBO J. 29, 2026–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalisman N., Adams C. M., Levitt M. (2012) Subunit order of eukaryotic tric/cct chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A. 109(8), 2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stengel F., Aebersold R., Robinson C. V. (2012) Joining forces: integrating proteomics and cross-linking with the mass spectrometry of intact complexes. Molecular & Cellular Proteomics 11(3), R111-014027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young M. M., Tang N., Hempel J. C., Oshiro C. M., Taylor E. W., Kuntz I. D., Gibson B. W., Dollinger G. (2000) High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97(11), 5802–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hermanson G. T. (1996) Bioconjugate Techniques, Academic Press London [Google Scholar]
- 27. Trnka M. J., Burlingame A. L. (2010) Topographic studies of the groel-groes chaperonin complex by chemical cross-linking using diformyl ethynylbenzene the power of high resolution electron transfer dissociation for determination of both peptide sequences and their attachment sites. Mol. Cell. Proteomics 9(10), 2306–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lauber M. A., Reilly J. P. (2011) Structural analysis of a prokaryotic ribosome using a novel amidinating cross-linker and mass spectrometry. J. Proteome Res. 10(8), 3604–3616 [DOI] [PubMed] [Google Scholar]
- 29. Singh P., Shaffer S. A., Scherl A., Holman C., Pfuetzner R. A., Larson Freeman T. J., Miller S. I., Hernandez P., Appel R. D., Goodlett D. R. (2008) Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal. Chem. 80(22), 8799–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rinner O., Seebacher J., Walzthoeni T., Mueller L., Beck M., Schmidt A., Mueller M., Aebersold R. (2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods 5(4), 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chu F., Baker P. R., Burlingame A. L., Chalkley R. J. (2010) Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol. Cell. Proteomics 9(1), 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koning L. J., Kasper P. T., Back J. W., Nessen M. A., Vanrobaeys F., Beeumen J., Gherardi E., Koster C. G., Jong L. (2005) Computer-assisted mass spectrometric analysis of naturally occurring and artificially introduced cross-links in proteins and protein complexes. FEBS J. 273(2), 281–291 [DOI] [PubMed] [Google Scholar]
- 33. Xu H., Zhang L., Freitas M. A. (2007) Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J. Proteome Res. 7(1), 138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McIlwain S., Draghicescu P., Singh P., Goodlett D. R., Noble W. S. (2010) Detecting cross-linked peptides by searching against a database of cross-linked peptide pairs. J. Proteome Res. 9(5), 2488–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michael G., Pettelkau J., Schaks S., Bosse K., Ihling C. H., Krauth F., Fritzsche R., Kühn U., Sinz A. (2011) “StavroX–a software for analyzing crosslinked products in protein interaction studies.” Journal of The American Society for Mass Spectrometry 23, 1 (2012): 76–87 [DOI] [PubMed] [Google Scholar]
- 36. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., Dong M. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9(9), 904–906 [DOI] [PubMed] [Google Scholar]
- 37. Choi S., Jeong J., Na S., Lee H. S., Kim H. Y., Lee K. J., Paek E. (2009) New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 9(1), 626–635 [DOI] [PubMed] [Google Scholar]
- 38. Schutz F., Kapp E. A., Simpson R. J., Speed T. P. (2003) Deriving statistical models for predicting peptide tandem MS product ion intensities. Biochem. Soc. Trans. 31, 1479–1483 [DOI] [PubMed] [Google Scholar]
- 39. Meulmeester E., Melchior F. (2008) Cell biology: SUMO. Nature 452(7188), 709–711 [DOI] [PubMed] [Google Scholar]
- 40. Wang J., Anania V. G., Knott J., Rush J., Lill J. R., Bourne P. E., Bandeira N. (2014) A turn-key approach for large-scale identification of complex post-translational modifications. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4(3), 207–214 [DOI] [PubMed] [Google Scholar]
- 42. Wang J., Bourne P. E., Bandeira N. (2011) Peptide identification by database search of mixture tandem mass spectra. Mol. Cell. Proteomics 10, M111-010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim S., Gupta N., Bandeira N., Pevzner P. A. (2009) Spectral dictionaries integrating de novo peptide sequencing with database search of tandem mass spectra. Mol. Cell. Proteomics 8(1), 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim S., Gupta N., Pevzner P. A. (2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7(8), 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J. R., Pevzner P. A. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang N., Li X., Ye M., Pan S., Schwikowski B., Aebersold R. (2005) ProbIDtree: an automated software program capable of identifying multiple peptides from a single collision-induced dissociation spectrum collected by a tandem mass spectrometer. Proteomics 5(16), 4096–4106 [DOI] [PubMed] [Google Scholar]
- 47. Wang J., Perez-Santiago J., Katz J. E., Mallick P., Bandeira N. (2010) Peptide identification from mixture tandem mass spectra. Mol. Cell. Proteomics 9(7), 1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10(4), 1794–1805 [DOI] [PubMed] [Google Scholar]
- 49. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Förster F., Beck M., Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 9(9), 901–903 [DOI] [PubMed] [Google Scholar]
- 50. Leitner A., Reischl R., Walzthoeni T., Herzog F., Bohn S., Förster F., Aebersold R. (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteomics 11(3), M111-014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bairoch A., Apweiler R., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., et al. (2005) The universal protein resource (UniProt). Nucleic Acids Res. 33(Suppl 1), D154–D159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The protein data bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whitby F. G., Masters E. I., Kramer L., Knowlton J. R., Yao Y., Wang C. C., Hill C. P. (2000) Structural basis for the activation of 20s proteasomes by 11s regulators. Nature 408, 115–120 [DOI] [PubMed] [Google Scholar]
- 54. Huber E. M., Basler M., Schwab R., Heinemeyer W., Kirk C. J., Groettrup M., Groll M. (2012) Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–738 [DOI] [PubMed] [Google Scholar]
- 55. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25(13), 1605–1612 [DOI] [PubMed] [Google Scholar]
- 56. Frank A. M. (2009) A ranking-based scoring function for peptide-spectrum matches. J. Proteome Res. 8(5), 2241–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baker P. R., Medzihradszky K. F., Chalkley R. J. (2010) Improving software performance for peptide ETD data analysis by implementation of charge-state and sequence-dependent scoring. Mol. Cell. Proteomics 9, 1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li J., Zimmerman L. J., Park B. H., Tabb D. L., Liebler D. C., Zhang B. (2009) Network-assisted protein identification and data interpretation in shotgun proteomics. Mol. Syst. Biol. 5(1) 5, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.