

Abstract
Analysis of the molecular basis of learning and memory has revealed details of the roles played by many genes and the proteins they encode. Because most individual studies focus on a small number of proteins, many complexities of the relationships among proteins and their dynamic responses to stimulation are not known. We have used the technique of reverse phase protein arrays (RPPA) to assess the levels of more than 80 proteins/protein modifications in subcellular fractions from hippocampus and cortex of mice trained in Context Fear Conditioning (CFC). Proteins include components of signaling pathways, several encoded by immediate early genes or involved in apoptosis and inflammation, and subunits of glutamate receptors. At one hour after training, levels of more than half the proteins had changed in one or more fractions, among them multiple components of the Mitogen-activated protein kinase, MAPK, and Mechanistic Target of Rapamycin, MTOR, pathways, subunits of glutamate receptors, and the NOTCH pathway modulator, NUMB homolog (Drosophila). Levels of 37 proteins changed in the nuclear fraction of hippocampus alone. Abnormalities in levels of thirteen proteins analyzed have been reported in brains of patients with Alzheimer's Disease. We therefore further investigated the protein profiles of mice treated with memantine, a drug approved for treatment of AD. In hippocampus, memantine alone induced many changes similar to those seen after CFC and altered the levels of seven proteins associated with Alzheimer's Disease abnormalities. Lastly, to further explore the relevance of these datasets, we superimposed responses to CFC and memantine onto components of the long term potentiation pathway, a process subserving learning and memory formation. Fourteen components of the long term potentiation pathway and 26 proteins interacting with components responded to CFC and/or memantine. Together, these datasets provide a novel view of the diversity and complexity in protein responses and interactions following normal learning.
The molecular underpinnings of learning and memory (L/M)1 are complex and involve transcription, translation and epigenetic responses (1, 2). At the protein level, they can involve changes in levels and patterns of post translational modifications (PTM), dynamic and reversible processes that regulate protein activity and localization. The best studied of PTMs is phosphorylation and, relevant to L/M, phosphorylations/dephosphorylations of N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor subunits are associated with induction of long term potentiation (LTP) and are critical for initiation of molecular responses to stimulation (3, 4). The cascade of phosphorylation increases of the protein kinases making up the classical MAP kinase pathway has been well documented for roles in L/M (5, 6), as has the MTOR signaling pathway (7). More recently, the roles in L/M of the complex PTMs of histone proteins, involving phosphorylation, acetylation and methylation, and their regulation of chromatin configuration, have been elucidated (8). Protein responses can also involve redistribution among intra-cellular compartments, for example, translocation from cytosol to nucleus or from membrane to cytosol, and these are frequently driven by phosphorylation/dephosphorylation and other PTMs (9). PTM-independent protein levels also change with L/M, and while these can result from initiation of transcription, they also arise from translation of existing compartmentalized mRNAs, e.g. localized within dendrites (10–12), and alteration of rates of protein degradation (13). Rapid increases in levels of proteins in responses to L/M have been documented for immediate early genes (IEGs), such as ARC (14).
Current knowledge of molecular responses to L/M has been assembled from multiple experiments that each measured a small number of proteins. Experiments have differed in L/M task, specifics of the task protocols, the timing of measurements, the brain region analyzed and method of analysis. Observed protein responses will be influenced by each of these variables. Analysis of pathways also is often not comprehensive, for example, measurements of the level of phosphorylation of the MAPK component ERK1/2 is used frequently to represent the activity of the MAPK pathway (15). This may not, however, reflect the full complexity of the pathway responses, or of cross talk of MAPK with other pathways or from other inputs.
For analysis of molecular responses to L/M, Context Fear Conditioning (CFC), a form of associative learning that requires a functioning hippocampus, has advantages (16). A few minutes exposure to a novel environment followed by a brief electric shock is sufficient to provoke a fear response, which is measured as “freezing,” that is, lack of all movement except for respiration, when the animal is returned at a later time to the same context. A single trial has been shown to promote robust learning. Protein responses during a few minutes or a few hours and up to one or a few days after CFC have been reported. For example, in hippocampus of mice exposed to CFC, levels of phospho(p)-CREB were elevated at 30 min post training, normal at 60 min, and elevated again between three and six hours (17). In other experiments, hippocampal levels of pERK1 were increased at 60 min post training, while increased levels of pPKC were observed 24 h later (18). Hippocampal region-specific increases in pERK1/2, pELK and pRSK were present at 30 and 60 min post training, but returned to those levels seen in untrained animals by 120 min (19). Thus, CFC provides a well-defined time frame during which learning and some molecular responses are known to occur.
Hippocampal function is impaired in Alzheimer's Disease (AD) (20–21). Hippocampal-based L/M tasks, including CFC, have been used in studies of mouse models of AD to assess effects of AD mutations, the APP Aβ peptide and drug responses (22). The NMDAR antagonist, memantine, has been shown to rescue L/M deficits in mouse models of AD and has been approved for treatment of moderate AD (23, 24). Little is known, however, about the molecular responses that underlie such successes, and it is of interest to understand the consequences of its use, not only on the NMDAR, but also on downstream signaling and other molecular processes.
To elucidate more of the complexities of molecular responses to learning and relationships among these responses, we have measured levels of 84 proteins/protein modifications that are directly required for L/M and/or that are components of pathways and processes known to subserve L/M. Measurements were made in hippocampus and cortex of mice at 60 min after training in CFC with and without pretreatment with memantine.
MATERIALS AND METHODS
Mice
Mice were on a mixed C57BL/6JEi x C3Sn.BLiA (B6EiC3Sn.BLiA F2) background (statistically 50% B6; 50% C3) obtained as the euploid control progeny of B6EiC3Sn.BLiA a/A-Ts65Dn females and B6EiC3Sn.BLiA F1 males (25). These mice represent the wild type littermates of a cohort originally generated for studies on Down syndrome (to be reported elsewhere). Mice were bred at the University of Colorado School of Medicine (Aurora, Colorado) or The Jackson Laboratory (Bar Harbor, Maine). Colonies were maintained in a room with HEPA-filtered air and a 14:10 light:dark cycle, fed a 6% fat diet and acidified (pH 2.5–3.0) water ad libitum. Littermates (supplemental Table S1) were housed in the same cage. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado or The Jackson Laboratory and were performed in accordance with National Institutes of Health guidelines for the care and use of animals in research. Male mice only (age 3–4 months) were used.
Context Fear Conditioning
Context fear conditioning was performed as described (25–27). Briefly, mice were placed in a novel cage (Med Associates, St. Albans, VT, Modular Mouse Test Chamber), allowed to explore for 3 mins and then given an electric shock (2 s, 0.7 mA, constant electric current). These mice are the context-shock (CS) group and learn to associate the context with the aversive stimulus (25). Learning is displayed by “freezing” upon re-exposure to the context, where freezing is defined as a lack of movement except for respiration. A second group of mice were placed in the novel cage, immediately given the electric shock (2 s), and then allowed to explore for 3 min. These mice are the shock-context (SC) group and do not acquire conditioned fear (27). All mice received an injection of memantine (5 mg/kg intraperitoneal) or the equivalent volume of saline 15 min prior to exposure to the novel context. Four groups of mice (9–10 per group) were used: CS mice injected with saline or with memantine and SC mice injected with saline or with memantine, as described in (25). Mice were sacrificed at 60 min post training without measurement of freezing; prior work has demonstrated that CS mice of this age and strain background are successful in learning, SC mice do not learn and memantine does not affect freezing (25).
Tissue Processing and Preparation of Protein Lysates
Mice were sacrificed by cervical dislocation without anesthetic. Brains were rapidly removed and placed in ice-cold artificial cerebral spinal fluid (aCSF) (in mm: 210 Sucrose, 2.5 KCl, 1 CaCl2, 7 MgSO4, 1. 5 NaH2PO4, 26 NaHCO3, and 10 d-glucose, saturated with 95% O2 and 5% CO2) while the hippocampus and cortex were rapidly dissected. Tissues were snap frozen in liquid nitrogen and stored at −80 °C until use. Without thawing, tissue samples were weighed and placed in 10 volumes of hypotonic buffer (10 mm HEPES, pH7.9, 10 mm KCl, 1.5 mm MgCl2) containing protease, phosphatase and kinase inhibitors. Tissues were thoroughly homogenized using 30 strokes at 300 rpm with a Compact Digital Homogenizer (Model BDC2002, Caframo Ltd. Canada) at 4 °C. Following centrifugation for 10 min at 8000 rpm, the supernatant (S1) was retained and the pellet (P1) was resuspended, in ½ the original volume of Urea buffer (8 m urea, 4% CHAPS, 50 mm Tris) and passaged ten times through a 16 G needle. The suspension was incubated for 1 h at 4 °C with gentle rotation and, following centrifugation at 14000 rpm for 30 min, the supernatant S2 was retained as the nuclear-enriched fraction. The original supernatant S1 was centrifuged for 30 min at 40,000 rpm at 4 °C. The resulting supernatant (S3) was retained as the cytosolic fraction and the pellet (P2) was resuspended in 2.5 volumes of Urea buffer to form the crude membrane fraction. Protein concentrations, determined using the 660 nm Protein Assay kit (Pierce), were within the range of 3–4 mg/ml for all samples. Information for each mouse on age, littermates, and weights of individual brain regions is provided in supplemental Table S1.
Antibodies and Validation for RPPA
Antibodies are listed in supplemental Table S2. Prior to use in RPPA, each lot of each antibody was tested on Western blots of mouse brain lysates, as described previously (28), and verified to produce predominant band(s) of explainable size, that is, clean band(s) of the correct size(s) in the absence of significant background and nonspecific bands. All antibodies listed in supplemental Table S2 are suitable for RPPA.
Array Assembly and Printing
For each sample, a five point dilution series in three replicates was printed as described previously (28). Arrays with nuclear-enriched and cytosolic fractions from hippocampus were produced in three print runs and those from cortex, in two print runs. Membrane fractions from cortex and hippocampus were printed on the same slides in two print runs. Slides were stored at 4 °C until use.
Slide Screening and Data Analysis
Slides were screened with antibodies and processed, signal intensities were normalized to the general protein stain SyproRuby (Invitrogen, CA), and protein expression data analyzed as described in (28). Data from antibody screenings that produced normalized signal intensities <0.1 were considered too low to be reliable and were discarded. Additional details of quality control and validation of inter-slide and inter-print run reproducibility are provided in (28).
Statistical Analysis
After exclusion of technical outliers, each SyproRuby-normalized protein value was included in the statistical analyses if the level was within its mean ± 3 standard deviations. This process eliminated on average <1% of the total observed data for each protein. For each protein, median differences were calculated for four pairwise group comparisons: CS-saline versus SC-saline (normal learning, NL), SC-memantine versus SC-saline (memantine effect, M), CS-memantine versus SC-memantine (successful learning plus memantine, NL+M), and CS-memantine versus CS-saline (successful learning end point, EP). Treatment and genotype differences were assessed using a three-level mixed effects model to account for possible correlations among replicates and dilution levels within each mouse, with different mice being the random effects. Bonferroni corrections were applied. supplemental Table S3 contains results (% difference, p value and S.E.) for all proteins in the nuclear, cytosolic and membrane fractions, for each of the four pairwise comparisons. All data analyses were carried out using SAS® version 9.3 (SAS Institute Inc., Cary, NC).
Network Displays and Databases
Components of the long term potentiation (LTP) pathway and their relationships were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG http://www.genome.jp/kegg/) database. Protein interaction partners of each LTP pathway protein component were obtained from the IntACT (http://www.ebi.ac.uk/intact/), HPRD (Human Protein Reference Database, http://www.hprd.org/) and BioGRID (Biological General Repository for Interaction Datasets, http://thebiogrid.org/) databases; all interacting proteins that were analyzed by RPPA were retained, filtered for Gene Ontology (GO) terms, and those sharing Cellular Component GO terms were added to the LTP pathway network. The expanded network was constructed using Cytoscape 3.0.2 (29).
RESULTS
The goals of the protein measurements were first to sample the complexity of changes that occur with learning in CFC and then to determine how these changes are altered by treatment with the NMDAR antagonist, memantine. A total of 84 proteins/protein modifications were screened in nuclear, cytosolic and/or membrane fractions from hippocampus and cortex. Proteins were chosen for their relevance to CFC specifically, or to learning/memory and synaptic plasticity more generally, and also for associations with Alzheimer's Disease. Because RPPA requires highly specific antibodies, many antibodies that can be used in Western blots cannot be used in RPPA. Therefore, not all proteins of interest could be measured. The number of samples, 10 mice in each of four groups, two brain regions and three subcellular fractions, or a total of 240, precludes use of Western blots for practical reasons. Proteins (listed in supplemental Table S2) include phosphorylation dependent and independent forms: 20 components of MAP kinase signaling, 14 related to MTOR signaling, 14 related to NMDAR subunits or their interactions, seven relevant to apoptosis or inflammation, four IEG proteins, three histone modifications, and 12 proteins relevant to AD. Pairs of phosphorylation dependent and independent levels were measured for 20 proteins.
General Protein Profile Features
Using data from the four groups of mice generated in CFC (the SC-sal and SC-mem groups that do not learn the context, and the CS-sal and CS-mem groups that do learn the context), four pairwise group comparisons were carried out. To identify protein responses in normal, successful learning (NL), protein levels in CS-sal were compared with those in SC-sal. To determine the effects of memantine treatment without the stimulation to learn (M), protein levels in SC-mem were compared with those in SC-sal. Comparison of levels in CS-mem to SC-mem identify protein responses in successful learning in the presence of pretreatment with memantine (NL+M). Lastly, CS-mem versus CS-sal comparisons identify similarities and differences in protein levels at the end point, after successful learning (EP). General features of protein responses are summarized in Table I. As discussed in previous work (28), the number of replicate measurements and the reproducibility of RPPA allows accurate determination of protein differences as small as 10%. Of 84 proteins screened in one or more fractions, levels of expression of 72 and 65 were detectable in nuclear and cytosolic fractions, and 28 in membrane fractions. In all three subcellular fractions of hippocampus, ∼one half of proteins responded to one or more stimuli (normal learning, memantine treatment, or normal learning with memantine) and the large majority of the responses were increases. For example, in the nuclear fraction from normal learning, 37 proteins changed in level, and 34 of these increased. Notably, memantine alone, in the nuclear fraction, resulted in changes in levels of 24 proteins, of which 21 were increases. In cortex, proteins in the nuclear fraction were less affected by normal learning and memantine treatment, with only 25 and nine responding, respectively. Cortex also showed overall a larger proportion of proteins decreasing, in particular in the nuclear and membrane fractions.
Table I. Numbers of proteins changed in each treatment group and brain region/subcellular fraction (total number of proteins measured).
| Nuclear (72) |
Cytosol (65) |
Membrane (28) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| NL | M | NL+M | NL | M | NL+M | NL | M | NL+M | |
| Hippocampus | |||||||||
| Total | 37 | 24 | 23 | 36 | 22 | 26 | 14 | 8 | 13 |
| Increased | 34 | 21 | 19 | 21 | 11 | 23 | 8 | 1 | 12 |
| Decreased | 3 | 3 | 4 | 14 | 11 | 3 | 6 | 7 | 1 |
| Cortex | |||||||||
| Total | 25 | 9 | 31 | 37 | 19 | 22 | 15 | 15 | 18 |
| Increased | 13 | 4 | 20 | 25 | 13 | 6 | 3 | 10 | 6 |
| Decreased | 12 | 5 | 11 | 12 | 6 | 16 | 12 | 5 | 12 |
Although on average, proteins changed in level by 15–30%, a small number of protein changes stand out with magnitudes that exceed 50%. These are shown in Table II. Among these are pERK1/2 and pCAMK2A/B; their involvement in learning is well established (5, 6). A novel finding concerns phosphorylation of NUMB, a protein involved in modulation of NOTCH signaling and postnatal neurogenesis (30, 31). pNUMB showed very strong increases in both hippocampus and cortex in cytosolic fractions with successful learning.
Table II. Proteins that change by >50%.
| Protein | Hippocampus |
Cortex |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nuclear |
Cytosolic |
Nuclear |
Cytosolic |
|||||||||
| NL | M | NL+M | NL | M | NL+M | NL | M | NL+M | NL | M | NL+M | |
| BRAF | +60% | +60% | +60% | |||||||||
| MEK1/2 | +50% | |||||||||||
| pMEK1/2 | +60% | |||||||||||
| pERK1/2 | +60% | +195% | +100% | +110% | +90% | +70% | ||||||
| pCAMK2AB | +120% | +70% | +125% | +135% | +115% | −60% | ||||||
| pPRKCAB | +70% | −65% | ||||||||||
| S6 | −80% | −80% | ||||||||||
| DYRK1A | +80% | +70% | ||||||||||
| NR2A | +80% | |||||||||||
| pNR2A | ||||||||||||
| pNUMB | +155% | +60% | +260% | +190% | ||||||||
| Tau | −65% | −75% | ||||||||||
Figs. 1–6 provide graphical comparisons of responses in hippocampus and cortex to normal successful learning (NL), to memantine treatment alone (M), to successful learning with memantine treatment (NL+M), and of levels at the end point of learning with and without memantine (EP). Results for a subset of proteins measured are shown, grouped by pathway or process (additional results are in supplemental Fig. S1–S8). Data for all proteins in the four comparisons in the three fractions of hippocampus and cortex are provided in supplemental Table S3. All measurements were made at a single, 60 min, time point post training. This time point was chosen because it is within the time frame established for maximum increases in the levels of components of the MAPK pathway following associative learning (19) and therefore results here can be directly compared with those from prior work. With only a single time point, however, protein levels and comparisons between groups represent a snapshot of dynamic processes. The data do not indicate if protein levels in any group were continuing to change, and if so, what their rates of change and directions were. Thus, we describe differences and similarities present at 60 min, aware that such comparisons would likely look different at other time points.
Fig. 1.

Response of MAP kinase and MTOR pathway components in the hippocampus, nuclear-enriched fraction. Protein levels were measured by RPPA and signals normalized to SyproRuby. Median differences were calculated for the indicated pairwise comparisons and statistical significance determined using a three-Level Mixed Effects Model. The y axis indicates the percent increase or decrease in protein level caused by treatment. Black bars, significant change; white bars, nonsignificant change. Error bars indicate the S.E. No error bar indicates weak signals and measurements were not made (protein names are included for ease of comparison with other treatments/fractions/brain regions). X, change is specific to that treatment. A, Normal learning: CS saline versus SC saline; B, Memantine: SC memantine versus SC saline; C, Normal learning + memantine: CS memantine versus SC memantine; D, End point - MAPK: CS memantine versus CS saline. E, Changes in the ratio of phosphorylated to whole protein for a subset of MAP kinase pathway components; white bars, SC saline; black bars, CS saline. *, p < 0.05; **, p < 0.01 by the Student's t test.
Fig. 2.

Response of MAP kinase and MTOR pathway components in the hippocampus, cytosolic fraction. A–D, legend as for Fig. 1.
Fig. 3.

Response of IEG proteins and histone H3 modifications in the hippocampus nuclear-enriched and cytosolic fractions. A–C, legend as for Fig. 1.
Fig. 4.

Response of the NMDA receptor subunits and related proteins in the hippocampus membrane and cytosolic fractions. A–D, legend as in Fig. 1. E, Ratios of phosphorylated to whole protein increase for NR2A and NR2B in NL; white bars, SC saline; black bars, CS saline. **, p < 0.01 by the Student's t test.
Fig. 5.

Response of the AD relevant proteins in the hippocampus nuclear, cytosol and membrane fractions. A–C, legend as for Fig. 1.
Fig. 6.

Protein responses in cortex. A, MAP kinase proteins in the nuclear fraction. B, MTOR proteins in the nuclear fraction. C, NMDA receptor subunits and related proteins in the membrane fraction. A–C, legend as for Fig. 1.
Hippocampus Nuclear-enriched Fraction, MAP Kinase and MTOR
Several components of the MAPK pathway have been shown to directly influence L/M. Knockouts and other mutations created in mouse for BRAF, ERK, RSK, CREB, PCK, and CAMKII, each result in impairments in L/M, synaptic plasticity and/or synaptic transmission (15, 32–38). Other studies using pharmaceuticals to inhibit specific kinases, including MEK, PKC and PKA, also cause L/M impairments (39). In Fig. 1, components of the MAPK pathway in the nuclear-enriched fraction are shown. In NL (Fig. 1A), 13 of 20 components of the MAPK pathway are increased; these include six of ten phosphorylated forms. Notably, sequential components of the classical cascade of MEK1/2-ERK1/2-RSK all show increased phosphorylation, although levels are not proportional. For example, pERK1/2 increases by ∼60%, whereas the upstream pMEK1/2 and downstream pRSK each increase only by ∼20%. Phosphorylation of CREB is not altered; this is consistent with the time frame previously reported that the initial increase in pCREB following CFC returns to baseline by 60 min (17). Additional increases in phosphorylation involve CAMKII (120%), PKCA/B (∼30%) and JNK (∼35%), also documented for association with L/M (37, 38, 40). Changes are not confined to phosphorylation-specific forms. Phosphorylation-independent levels increase for BRAF, ERK1/2, RSK, ELK, CAMKII, PKCA, and JNK, implying that translocation from the cytosol, translation, and/or inhibition of degradation are initiated within the 60 min time frame. While most of these increases are modest, on the order of 10–20%, and therefore might not have been detected when assessed by Western blot methods, levels of BRAF increase by 60% and ELK by 34%.
The ratio of phosphorylated to total protein measures the proportion of a protein that is in a specific functional state; changes in this ratio reflect another regulatory mechanism. Fig. 1E, shows that there are significant changes in proportions of phosphorylated forms of multiple components of the MAP kinase pathway. Although both phosphorylated and total levels of CAMKII increase, the proportion of active pCAMKII to CAMKII increases by ∼75%. In contrast, ratios for RSK are unchanged. For BRAF and ELK, because levels of phosphorylated forms did not change while whole protein levels increased, the proportions of activated BRAF and ELK decrease significantly.
Fig. 1B shows the effects of memantine alone on MAPK. Of the 13 components of the MAPK pathway that increased in NL, eight also increase in M. These include BRAF, pERK1/2, ELK, pCAMKII, PKCA, pPKCA/B, JNK, and pJNK. The changes here, however, are more modest: only ∼15–20% for BRAF and pERK1/2, compared with 60% for both in NL. Similarly, the increase in pCAMKII was only ∼70% here, compared with 120% in NL.
Fig. 1C shows results for NL+M, that is, how memantine treatment alters the normal molecular dynamics associated with learning. Increases in levels of BRAF, pERK1/2, pCAMKII, and pJNK still occur as in NL, but again the increases are more modest. Further consideration shows the relationship among the levels of change in the three treatments. For example, levels of pCAMKII increase by 70% with memantine treatment; these levels then increase a further 40% with CFC training; together this produces an increase in pCAMKII from initial levels equivalent to those seen in NL. This conclusion is supported by data in Fig. 1D: when levels of pCAMKII after NL+M are compared with those after NL (i.e. EP levels, CS-mem compared with CS-sal), the difference is only ∼5% and is statistically insignificant. Similarly for pJNK, the increases are 25 and 15% for M and NL+M, respectively, the sum of which is again equivalent to the 35% increases seen in NL. These data show that memantine treatment initiates a subset of molecular responses that are common to those seen in normal learning, at least at the 60 min time point.
A slightly different scenario is seen with ELK, PCKA, pPCKA/B, and JNK. For these proteins, memantine produces changes equivalent in magnitude to those seen in NL, and no further increases are seen in response to NL+M. One possibility is that, for successful learning, rather than a dynamic response, a specific protein level, which is different from the baseline level, is required for each of these proteins. If this level is induced prior to training, no further increase is required for learning. Again, data in Fig. 1D show that EP levels do not differ significantly.
Lastly, there are treatment-specific changes in protein levels. Specific to NL are increases in pMEK, ERK, RSK, pRSK, and CAMKII; specific to NL+M is an increase pPKCG. These differences could be explained if memantine alters the duration or timing of some protein responses. For example, the elevated pPKCG levels in NL+M could be an extension of activation already terminated in NL or an early initiation of an activation that will occur at a later time point in NL. Similarly, the lack of elevated pMEK in NL+M compared with NL could result if memantine caused MEK activation to be terminated early (or initiated later). Additional experiments assessing protein levels at different time points post training could answer this question. Fig. 1D shows that, with the exception of ERK, differences in end point levels of MAPK components do not reach significance, reflecting the general modest levels of treatment-specific differences.
The MTOR pathway is also relevant to synaptic plasticity and L/M (for review see 7). Inhibition of MTOR kinase activity with rapamycin blocks NMDAR-dependent late phase LTP and knockout of the downstream component of MTOR signaling, p70S6K, results in impaired L/M. As shown in Fig. 1, compared with MAPK, increases in NL in MTOR components are modest, averaging 15–20%. However, examples of the same molecular scenarios seen with MAPK proteins are also seen with MTOR. Increases in pMTOR are ∼20% in NL, equivalent to the sum of ∼10% in M and ∼13% in NL+M. Increases in pS6 and GSK3B are seen only in NL and M, and increases in P70S6, AMPKA and pGSK3B-ser9 are seen only in NL and NL+M. Lastly, treatment-specific responses include increases in pEIF4B and pAKT in NL, and S6 in M. As with MAPK, EP levels do not differ.
Hippocampus Cytosolic Fraction, MAP Kinase and MTOR
Fig. 2 shows results of analysis of MAPK and MTOR pathway components in the cytosolic fraction of hippocampus. Not surprisingly, the patterns of protein responses are quite different from those in the nuclear-enriched fraction, in the number and identities of the proteins that change and the direction and magnitude of the changes. Obvious patterns of cytosol-nuclear translocation, i.e. where increases in the nuclear fraction are accompanied by a decrease in the cytosol, were not generally observed. Levels of ERK, RSK and ELK increased in the cytosolic fraction, even though they also increased in the nuclear fraction. For the MTOR pathway components in NL, cytosolic levels of pP70S6 and S6 decrease by ∼25% and if the initial levels in the nucleus are much higher than those in the cytosol, a consequent nuclear increase may be below the level of detection. Other possibilities to account for the observation include dephosphorylation or degradation. Memantine treatment however provides an example of reciprocal increases and decreases: both S6 and pS6 levels decrease in the cytosol, by 20 and 31%, respectively, and increase in the nuclear fraction by 24 and 19%, respectively. This also effectively increases the proportion of activated S6 in the nucleus by ∼25%.
Treatment-induced relationships among changes in protein levels in the cytosol also differ from those seen in the nuclear fractions. For example, levels of pERK increase very strongly in the cytosol in NL and M, while pCAMKII increases in NL and NL+M. Unlike in the nuclear fraction, BRAF levels do not change in any of the three treatments, but pBRAF increases in NL and M. Conversely, levels of pMEK and of ELK increase only in NL and NL+M. In a scenario different from those seen in the nuclear fraction, the 25% increase in MEK seen in NL is countered by a decrease of ∼10% in M which is “rescued” by a more robust increase ∼32% in NL+M. Similarly, memantine produces an ∼20% decrease in the level of pS6 which is countered by a ∼30% increase in pS6 in NL+M, potentially restoring levels of pS6 to those present in NL. Unlike the nuclear fraction, EP comparisons show several differences, notably in pERK and S6. Because learning is equally successful with or without memantine, such differences must not be critical at this 60 min time point.
Hippocampus, IEGs, and Histone Modifications
Changes in levels of IEG proteins, ARC and EGR1 have been reported in CFC (41) and knockouts in mouse result in impaired L/M or synaptic transmission (42, 43). Fig. 3 shows that, in the cytosolic fraction, levels of EGR1 increase in both NL and NL+M, and levels of ARC increase uniquely in NL+M. In the nuclear-enriched fraction, levels of phosphorylated CFOS increase in both NL and NL+M.
Modifications of histone proteins, including acetylation, phosphorylation and methylation on multiple lysine residues in H3 and H4, have been shown to respond to several forms of L/M, including CFC (44). Adequate antibodies were available for acetylation and methylation of histone H3 (K9, K18 and K4). Shown in Fig. 3 are decreases in the levels of acetylation at lysines K9 and K18 uniquely in the nuclear fraction, while levels of methylation at lysine K4 are not affected by any treatment.
Hippocampus, NMDA Receptor, and Related Proteins
Fig. 4 shows changes in levels of subunits of NMDA and AMPA receptors, compared in both membrane and cytosolic fractions. In NL, membrane levels of NR1, NR2A, and NR2B decrease by 25–35%. Cytosolic levels show a corresponding increase only for NR2B. This suggests that, for NR2A (NR1 signals were too low for reliable quantitation), either the increase was proportionately too small to be detected or that active degradation has occurred. Levels of pNR1 and pNR2B also decrease in the membrane. Phosphorylation of NR1 at Ser889 has been reported to promote dispersion within the membrane (45, 46). Functional consequences of the phosphorylation of NR2B at Tyr1336 are unknown but it is known to be carried out in response to CFC by the FYN kinase and to be transient (47). These changes in NMDAR subunits in the membrane result in increases in the proportions of phosphorylated to whole protein for NR2A and NR2B as shown in Fig. 4E. In the cytosol, levels of phosphorylation of all three NMDAR subunits increase by 15–40%.
Memantine treatment alone produces the same pattern of decreases in the membrane in the levels of NR1 and NR2A as seen in NL. The magnitude of the decreases is smaller (Fig. 4B), however, and there are no further changes in NL+M (Fig. 4C). These patterns of responses result in significantly higher levels of NR1, NR2A and NR2B in NL+M compared with NL, that is, in EP (Fig. 4D). Cytosolic patterns of NMDAR subunits in NL+M are the same as those in NL, with the exception of a strong (80%) increase in levels of NR2A. This may partially compensate for the decrease in NR2A levels induced by memantine alone. Memantine alone did not affect levels of AMPAR subunits, but in NL+M there are significant increases in membrane levels of GLUR3 and GLUR4 that are not seen in NL.
Other changes in NMDAR-related proteins in the membrane include decreases of PSD95 by ∼40% in NL and by ∼20% in M, resulting in ∼20% higher levels in EP. There is also a modest increase in membrane levels of BDNF in NL and NL+M. The most striking response in all three comparisons is the very robust increase in cytosolic levels of phospho-NUMB. This includes one of the strongest responses observed, ∼155% in NL, and ∼40 and ∼60% in M and NL+M, respectively. Levels of pNUMB also increase in the membrane. Although the role of NUMB in learning and memory has not been investigated, it has been shown to interact with NMDAR and to modulate dendritic spine development and morphology (48). Another protein that is involved in regulation of dendritic spines and that also interacts with NMDAR is the guanine nucleotide exchange factor, TIAM1 (49, 50). Memantine induces an increase of TIAM1 in the cytosol and a corresponding decrease in the membrane. Lastly, large increases of ∼50–80% are seen in pCAMKII in the membrane fraction in NL and NL+M.
Hippocampus, Alzheimer's Disease-Related Proteins
Although mutations in only a few genes have been shown to be causative of AD and these only in a small number of familial AD, abnormal levels of additional proteins have been found in brains of AD patients and mouse models and their manipulation in mouse models has had positive effects. Because hippocampal L/M is primarily affected in AD (21–22) and because memantine has shown positive effects in mouse models of AD and is in use for moderate to severe AD (51), we determined the responses of 12 AD-related proteins to CFC. Results are shown in Fig. 5. With the exception of ERBB4, each protein changes in at least one treatment/fraction combination and several have inter-related functions affecting NMDA receptor activity and signaling, as well as phosphorylation of Tau, the microtubule-associated protein that is hyperphosphorylated and a component of neurofibrilary tangles in AD (52). Levels of Tau itself were unchanged here except for increases in the cytosol with memantine treatment.
A major protein player in AD is APP, the amyloid precursor protein whose aberrant processing leads to accumulation of the Aβ peptide in neuritic plaques that are a hallmark AD pathology; mutations in or duplications of the APP gene cause some familial AD (53). APP levels are unchanged in NL and in M, however, they increase by ∼15% in the membrane fraction in NL+M.
Increased levels of calcineurin (CaN), a Ca-calmodulin-dependent protein phosphatase, are seen in aging and are suggested to play a role in age-related cognitive decline (reviewed in 54). A number of lines of evidence suggest that CaN is hyperactivated in mouse models of AD, in particular, inhibition of CaN activity in these systems reverses Aβ-induced abnormalities in synaptic plasticity, dendritic spines and CFC performance (55–57). CaN substrates include NMDA receptor subunits and Tau (58, 59). In NL, levels of the CaN catalytic subunit, CaNA, are decreased in the cytosol by ∼30%; this is accompanied by increases of 15% and ∼20% in the membrane and nuclear fractions, respectively. Memantine did not affect the distribution of CaNA, but in NL+M, CaNA increased in both the membrane and nuclear fractions. RCAN1 is an inhibitor of CaN and it is increased in brains of AD patients (60) and has been proposed to have roles in increased Tau phosphorylation and in neurodegeneration (61, 62). In NL, RCAN1 is increased in the membrane fraction, while memantine treatment produced increases in RCAN1 in all three fractions. In NL+M, a decrease in RCAN1 levels in the cytosol counters the increase produced by memantine. Given the patterns of RCAN1 changes, they are not simply a feedback response to changes in CaN, nor are they driving changes in CaN distribution.
Interleukin 1-beta (IL1B) is a proinflammatory cytokine. Its observed up-regulation in AD is suggested to have both positive and negative contributions to disease progression (63). In NL, cytosolic levels of IL1B decrease by ∼20% while nuclear levels increase in M. IL1B has also been shown to increase the phosphorylation of NR2A and NR2B by SRC family kinases (64). Here, we examined expression of the SRC kinase, FYN and its phosphorylated form, pSRC-Tyr416. FYN is required for CFC (47), its levels are elevated in AD, and its targets include NR2B and Tau (reviewed in 65). In NL, total levels of FYN decrease in the cytosol by 30% and both total FYN and pSRC increase in the nuclear fraction. Memantine alone did not affect FYN or pSRC, but in NL+M nuclear levels of pSRC increase by 30%, comparable to increases in NL.
CDK5, a cyclin dependent kinase, is also required for CFC (66). It is up-regulated in AD brains and includes Tau among its targets. Regulatory proteins, p35 and its calpain-cleaved p25 fragment mediate, respectively, transient and extended activation of CDK5 (reviewed in 67). Under all three stimulation conditions, CDK5 and p35/p25 exhibit complex, and differing, patterns of increases and decreases in the membrane, cytosol and nuclear fractions. In NL, CDK5 appears to redistribute from the cytosol to the membrane, and p35/p25 from the cytosol to the nucleus. The redistributions differ in NL+M and are simpler in M.
PP2A is a protein phosphatase that also targets Tau, binding to a sequence that is targeted by the kinase FYN (68). Changes in levels of PP2A activity and subunit proteins have been documented in AD, potentially contributing to the observations of hyperphosphorylated Tau (69, 70). Levels of the PP2A-A regulatory subunit decreased in the cytosol in NL and in M.
ERBB4 is a membrane receptor tyrosine kinase that is activated by the binding of neuregulin-1. It has critical roles in neural differentiation and synapse formation and, in the adult brain, in both excitatory and inhibitory neurotransmission (71). Activation by neuregulin results in ERBB4 cleavage by γ-secretase and release of the C-terminal fragment into the cytoplasm. This fragment retains kinase activity and translocates to the nucleus where it functions as a transcription factor (72). Increased levels of ERBB4 have been reported in brains of both AD and mouse models of AD (73). Here, levels of ERBB4 are unchanged with NL, M or NL+M.
The Wnt/β-catenin signaling pathway functions in synapse formation and remodeling (reviewed in 74). Reduced levels of β-catenin are found in patients with AD caused by mutations in presenilins and in mouse models of AD (75). In the latter, treatment with lithium up-regulates expression of β-catenin and ameliorates impairments (76). Both NL and M appear to result in redistribution of β-catenin among the three fractions.
Cortex Protein Profiles
In many pathways, protein responses in the cortex are simpler than those in the hippocampus. This is particularly true in NL, as shown for MAP kinase proteins in the nuclear fraction in Fig. 6A (compare with Fig. 1A). Only six proteins were affected versus 13 in hippocampus. MTOR components are also less responsive (Fig. 6B). In contrast, responses of the NMDA receptor subunits and related proteins are actually more complex in cortex than in hippocampus (Fig. 6C; compare with Fig. 4A). In the membrane, 11 proteins change in NL and 12 change in NL+M, compared with nine and seven, respectively, in hippocampus. Specific to cortex are decreases in TRKA, TIAM1, and pCAMKII. The only NL-specific change in cortex is a modest increase in pGLUR2, and NL+M-specific changes are increases in GLUR3 and GLUR4. Interestingly, unlike hippocampus, memantine effects on NMDAR in cortex are largely opposite to those seen in NL. Responses in cortex of additional proteins can be found in supplemental Figs. S3–S8.
DISCUSSION
Analysis of molecular events at the protein and protein modification level provides a more direct measure of functional responses than does analysis at the RNA level. We have generated protein profiles from brain regions of mice 60 min after training in a single trial protocol previously shown to result in robust learning/memory in mice of this mixed B6C3 genetic background (25). Use of the higher throughput technique of RPPA allows measurement of a large number of proteins in a single, uniform sample set. Here, the sample set was composed of hippocampus and cortex from a total of 40 mice, ten in each of four groups, the context-shock and shock-context groups, injected with either saline or memantine. With subcellular fractionation, this resulted in 240 samples. An additional advantage of RPPA is the large number of replicate measurements that are possible per sample, here three replicates of a five point dilution series, which allows an accuracy not practical to obtain with Western blots or immunocytochemistry. In previous work, we have shown that differences as low as 10% can be reproducibly measured with our RPPA protocol (28). Exploiting RPPA therefore can provide a different view of molecular responses to L/M, a broad view, surveying multiple components from multiple pathways and allowing detection of more relationships among levels of functionally diverse proteins. Because of the requirement of RPPA for highly specific antibodies, however, not all proteins of interest can be assayed. For example, antibodies against different sites of NMDAR subunit phosphorylation routinely used in Western blots detected apparently nonspecific bands and therefore could not be used in RPPA. Additional technical issues contribute to details of the protein profiles. To optimize preservation of PTM, protein lysates were prepared as rapidly as possible. Thus, nuclear-enriched and crude membrane fractions were used, instead of, for example, synaptic vesicles and synaptic membranes.
Expression levels of 72 and 65 proteins were above background in the nuclear and cytosolic fractions, respectively. This does not imply that the other proteins screened are not present. Antibodies with higher affinities, spotting higher concentrations of lysates, or use of signal amplification protocols might produce detectable expression. After CFC, in hippocampus, levels of 37, 36, and 14 proteins changed in the nuclear, cytosolic and membrane fractions, respectively, providing a complex picture of molecular events associated with normal learning. Validation of these responses, and support for their relevance to L/M, can be obtained from three sets of data: (1) previous experiments with CFC, (2) mutations that disrupt L/M or synaptic plasticity, and (3) consistency of responses between NL and NL+M.
(1) Several reports have shown that increases in pERK2 levels peak at 60 min post training (19, 77) both in whole hippocampus and specifically in the hippocampal subregion, CA1. Such results are consistent with the increases in pERK1/2 observed here in NL and NL+M in both the nuclear and cytosolic fractions. CFC-associated increases in pERK1/2 also have been shown to depend upon activation of PKC (77), results that are qualitatively consistent with observations here of increased levels of pPKCA/B. Also consistent with results here are reports showing that EGR1 increased in dorsal hippocampus (here in the cytosol) at 60 min post CFC training (41), CFOS protein increased in the nucleus (here pCFOS) after NMDAR stimulation (78), and pSRC-418 increased at 40 min after CFC in dorsal hippocampus (47) (here in the nuclear fraction). These consistent protein responses were observed despite differences in technical aspects of the experiments, for example use of rats versus mice; different background strains of mice; training in CFC with a single shock or with multiple shocks (17–19, 41); or measurement of protein levels by Western blots of tissue lysates or immunohistochemistry on brain slices.
The time frame of measurements with respect to training in CFC is an important variable, for example, no change in pCREB levels were seen here at 60 min post training, but this is consistent with observations that pCREB was elevated at 30 min, but had returned to pretraining levels by 60 min (17). A similar difference in experimental timing may have contributed to the failure here to observe increases in ARC protein that were previously reported (79). That CFC training protocol involved 7–12 shocks with intershock intervals of several minutes, thus extending the training period over 30–60 min. Therefore, although mice were sacrificed 60 min after training, this corresponds roughly to 30–60 min later than our measurements. As reviewed in (14), increases in ARC protein are rapid but transient, returning to baseline by 60 min in some experimental paradigms.
One observation, however, is less explicably inconsistent with prior work: the levels of histone H3 acetylation at K9 decreased robustly by ∼40%. This is in contrast to increases in H3 K9 acetylation reported elsewhere (80). We discuss this further below.
(2) Excluding PTM forms, 60 proteins were analyzed. Of these, 16 are encoded by human intellectual disability (ID) genes, defined as genes that result in ID when mutated based on observations in human populations (81). Nine of these ID proteins plus a further 21 are encoded by genes that, when mutated, knocked out or overexpressed in mouse models, result in one or more L/M or synaptic plasticity phenotypes (82). This means that a total of 37 of the 60 proteins have already been directly implicated as critical for L/M in the mammalian brain (indicated in supplemental Table S2). That 23 of 37 responded to training in CFC is one manifestation of this critical association.
(3) Responses consistent in NL and NL+M include increases in components of the MAP kinase and MTOR pathways, decreases in acetylation of histone H3, and changes in cytosolic and nuclear levels of IEG proteins, and in cytosolic levels of NMDAR subunits. The directions of the changes are the same, although the magnitude of the changes sometimes differs (see below).
These three different types of supporting information argue for the technical validity and biological relevance of the RPPA data overall. They do not, of course, prove that every observed protein response is required for learning in CFC or in any other L/M paradigm, or rule out the possibility that additional responses occur at alternative time points.
Novel Observations of Protein Responses to CFC
For many proteins, this is the first time they have been measured individually or in combination with other pathway/complex components in association with CFC, or indeed with any L/M task. Robust increases, to ∼150% of controls, in phosphorylation of NUMB in the cytosol were observed. NUMB has not previously been implicated in learning, but it has clear roles in cortical development. NUMB functions in asymmetric cell division by antagonizing NOTCH signaling. In cultured hippocampal neurons, it has been shown to accumulate in the tip of growing axons and to regulate endocytosis of cell adhesion proteins; it also localizes to dendritic spines and participates in regulation of spine morphology and maturation (48). NUMB has been shown to complex with NR1 and NR2B in rat brain and to influence processing and trafficking of APP (83–85). NUMB is phosphorylated by PKC and CAMKII (86, 87), both of which are activated by CFC and memantine. These data suggest that NUMB may participate in regulation of NMDAR subunit localization in response to CFC.
The role of the MAPK pathway in L/M is well documented. Here, we examined phosphorylation of components of the classical MAPK cascade of BRAF-MEK-ERK-RSK. In the nuclear fraction, we found no changes in pBRAF, and while increased levels of pMEK1/2 and downstream pRSK are qualitatively consistent with the increases in pERK1/2, they are not directly proportional, with pERK increasing by almost 60% compared with modest 15–20% increases in pMEK and pRSK. In the cytosolic fraction, increases in pBRAF and pMEK of 20–40% are associated with an almost 200% increase in levels of pERK. The MAPK pathway is activated, not only upstream by NMDAR, but also by PKC acting on ERK1/2; individual kinase components of MAPK are also inactivated by multiple protein phosphatases. Thus, these differing changes in phosphorylation levels of components of the MAPK cascade likely reflect the complexities of crosstalk among upstream activation by phosphorylation and the interplay of rates of dephosphorylation.
We observed strong 30–40% decreases in the levels of histone H3 acetylations at K9 and K18 in the nuclear fraction from hippocampus, in both NL and NL+M. This is contrary to a prior report of increases at the same sites after CFC (80). Most experimental parameters used here were the same or similar, e.g. in both studies, mice were ∼3 months old, training in CFC consisted of one trial and a single shock; mice were sacrificed 60 min later for protein analysis. There are differences in strain background: C57BL/6 in (80) versus a mixed B6C3. However, if acetylation changes in H3K9 and K18 are important to L/M, such strain differences would not be expected to result in such large and opposite outcomes. A second difference in experimental protocols is the intensity of the training: use of 1 mA versus 0.7mA in the electric shock. Indeed, training strength has been shown to affect the dynamics at least of acetylation at H3K18: strong training resulted in an increase in acetylation while weak training produced a decreased level (88). Use of 0.7 mA and 1 mA in CFC in mouse are both common and both result in successful L/M. Presumably, both stimulations also result in transcriptional activation of largely similar sets of genes. It is therefore difficult to explain the discrepant results. Certainly, patterns in histone modification are complex and both increases and decreases in acetylation participate in transcriptional activation (8). Further experiments with differing CFC protocols could resolve this issue.
Not all changes involved PTM. Increased whole levels of proteins in one subcellular fraction imply that trafficking from another compartment, inhibition of degradation or increased translation take place within the 60 min time frame of this study. The modest, but statistically significant and reproducible increases of ∼20% that occur for several components of the MAP kinase pathway in both cytosol and nuclear fractions of hippocampus are below the sensitivity of more commonly used methods of Western blots and immunohistochemistry. However, translation of mRNAs localized within dendrites has been well documented and may be responsible for increases in MEK, ERK, ELK, JNK, and CAMKII in the cytosol seen in NL and/or NL+M (89).
Decreases in whole protein levels are seen for NMDAR subunits in the hippocampal membrane fraction. This implies trafficking to the cytosol with or without subsequent degradation. In NL, the decrease in NR2B in the membrane is matched by a corresponding increase in the cytosol. For NR2A, however, the cytosolic level does not change. As reviewed in (90), the number and subunit composition of NMDARs can respond rapidly to neuronal activation or sensory stimulation. The dynamics are specific to cell and synapse type, however, and such observations typically have been made in heterologous cell systems or ex vivo slice cultures (e.g. using measurement of LTP and LTD), where conditions, measurements and resolution are very different than here. Synaptic activity has been shown to accelerate turnover in particular of NR1 (91), and furthermore, following removal from the membrane, NR1 and NR2A are localized to degradative endosomes (consistent with their lack of increase in cytosol here) while NR2B is sequestered to recycling endosomes (consistent with its increased levels in the cytosol here) (92).
Effects of Memantine
Memantine treatment caused decreases in the levels of NR1, pNR1 and NR2A in the hippocampal membrane fraction, and an additional decrease in NR2A in the cytosol. Memantine is an open channel blocker of NMDAR with a high off rate (93). Its clinical use to treat AD is based on the prediction that it can decrease the hyperactivation of NMDAR that contributes to neurotoxicity and neuronal death. It is not obvious then that memantine would also reduce membrane levels of NMDAR. Prior studies of the mechanism of action of memantine have not documented such decreases, but also do not rule them out. Experimental systems have included heterologous expression of NMDAR subunits in xenopus oocytes or other cell systems or electrophysiological measurements in brain slices (94). These systems lack endogenous regulatory mechanisms present in vivo in tests of L/M and do not address changes in protein distribution. Conversely, use here of lysates from whole hippocampus masks region and cell type-specificities, that is, decreases in membrane levels of NMDAR subunits are unlikely to be uniform throughout the hippocampus. However, it remains possible that decreases in membrane levels of NMDAR in response to memantine may be an additional mechanism by which memantine produces the beneficial effect in AD and AD mouse models of decreasing excitatory neurotransmission. These observations are not without precedent. Three hours after treatment with the NMDAR antagonist, MK-801, levels of NR1 and NR2A were significantly decreased in synaptosomes (95). Furthermore, the MK-801 effects were more dramatic than those of another NMDAR antagonist, AP5 (96). Phosphorylation levels were not assessed in these experiments, however, results serve to show that effects on NMDAR subunit level and localization are drug specific and not generalizable.
Memantine treatment also abolishes the decreases in membrane levels of NMDAR subunits seen in response to CFC in NL. As a result, levels of NR1, NR2A, and NR2B are elevated in the membrane after NL+M compared with NL. Memantine did not, however, affect the CFC-induced increases in the level of pCAMKII, one function of which is to bind NR2B, facilitate phosphorylation by the casein kinase CK2, and decrease NR2B membrane association (96).
In contrast to effects on NMDAR subunits, memantine treatment did not alter any AMPAR subunit levels. However, after CFC with memantine treatment levels of GLUR4 (but neither pGLUR2 nor GLUR3) were elevated in the membrane compared with NL. GLUR4 levels normally are highest in early postnatal times, but increases have been documented in several rodent model systems, in responses to stress, ischemia and environmental enrichment (97–99). Lastly, memantine uniquely caused apparent degradation of NR2A with a significant decrease in cytosolic levels. This was, however, followed by a 75% increase following CFC. These data together indicate that there are several additional mechanisms governing expression of glutamate receptor subunits that remain to be elucidated.
Memantine treatment affected nuclear levels of 12 components of MAPK and MTOR and cytosolic levels of 10, indirect consequences of memantine-induced perturbations in NMDAR activity. Notably, 11 of 12 and 6 of 10 of these were also affected, although more strongly, by the stimulation of NL. One interpretation in these cases is that, for learning to occur, two protein parameters are required: a specific level of the protein and a dynamic change in protein level. Memantine initiates the change to the required level and subsequent stimulation in CFC then produces a more modest increase. This is the scenario seen in hippocampus with pERK, BRAF, and pCAMKII. Similarly, some changes occur in NL and M but not in NL+M. It may be that memantine alone produces the required protein level to facilitate learning and no further change is then needed for NL+M. This scenario is seen in hippocampus with ELK, PKC, pPCKA/B, JNK, and pS6.
Interpretation of responses to memantine must consider that, in selecting a single time point, 60 min after training, the profiles obtained are a snap shot of molecular events. They lack information on the rate of change and the direction of any continuing changes. Where the magnitude of change in NL+M is less than in NL, protein levels may have not yet peaked or may have peaked earlier and now are decreasing. Expanding the time frame to include measurements at earlier and later time points would provide information that might be more easily interpreted with respect to data obtained from other experimental protocols.
Alzheimer's Disease-related Proteins
Thirteen proteins with reported abnormal levels in brains of patients with AD were measured (Fig. 5 and NUMB in Fig. 4). No previous report has included measurement of more than one or two of these proteins, so results here provide a unique view of the dynamic relationships. Responses of these proteins are complex and many patterns are specific to one treatment. A survey of the literature reveals that functional interactions among these proteins converge on NMDAR and Tau (54–76). As shown in Fig. 7, nine proteins responded in NL, and six of them directly interact with or activate/inhibit the NMDAR or Tau phosphorylation. It is not obvious how perturbations in the levels of all of these proteins might propagate in the AD brain with the stimulation of L/M. Seven proteins responded in M and eight in NL+M. These experiments were of course carried out in control mice where initial protein levels, and learning, are normal. These responses and the effects of memantine on NMDAR and Tau in AD brains, where levels of these proteins are already perturbed, will be interesting to see. Memantine has also been shown to correct deficits in CFC in a mouse model of Down syndrome (25). This provides yet another model system for elucidating dynamic protein responses.
Fig. 7.
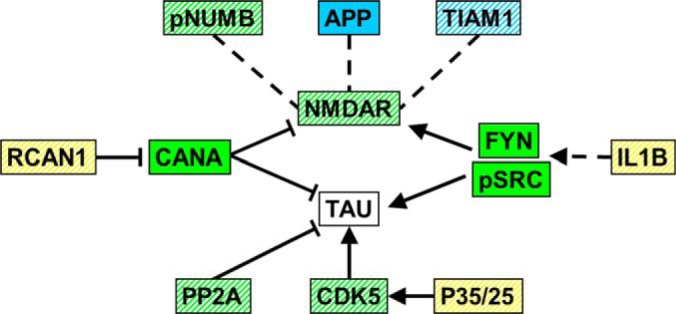
Functional interactions of AD-related proteins. Functions and interactions for ten proteins previously reported to be abnormal in brains from patients with AD converge on NMDAR activity and phosphorylation of Tau. See text for references. Arrows, activation or phosphorylation; blunt lines, inhibition or dephosphorylation; dashed lines, interaction. Yellow, changed in NL only; blue, changed in NL+M only; green, changed in both NL and in NL+M; stripes in any color, changed in M.
Conclusion
To illustrate how the complexities of the observed protein changes might be integrated into a response to L/M, we considered functional relationships between the proteins measured by RPPA and pathways relevant to L/M. We chose long term potentiation (LTP) because it is a major cellular mechanism believed to underlie learning and memory (100). From studies using molecular and genetic manipulation, representations of the LTP pathway at the protein level have been developed and extensively curated (101). The LTP pathway is comprised of 70 proteins, including subunits of protein complexes, and involves signaling through NMDAR to MAPK, plus contributions from and crosstalk with PKC, PKA, and calcineurin complexes, and components of the MTOR pathway. Fifteen RPPA proteins are components of the LTP pathway. We have further expanded the pathway by adding the 30 RPPA proteins that directly interact with one or more components of the LTP pathway. As shown in Fig. 8, a total of 35 RPPA proteins, 11 pathway components and 24 that interact with components, responded in NL. Memantine treatment modulated 22 of these responses and initiated a further six. Thus, normal learning induces considerable modulation of the LTP pathway, and while memantine appears to significantly alter these normal responses, it does so without perturbing the learning process, in this task in these mice. It will be of interest to determine which differences between responses in NL and NL+M are merely differences in timing of the responses. Comparison of these patterns with those associated with failed learning in mouse models of AD, and with patterns associated with rescue of learning by memantine, will be helpful in understanding the critical features of molecular responses to learning and memory. Similar studies are in progress with a mouse model of Down syndrome that also displays deficits in CFC that are rescued by treatment with memantine.
Fig. 8.

LTP pathway components and interacting proteins: response in hippocampus. Components of the LTP pathway imported from the KEGG database are outlined in and their connections are indicated with solid red lines. Complexes are indicated by their components if at least one component was measured by RPPA; otherwise the complex name is used (e.g. CAM). Primary protein interactions involving proteins measured by RPPA are indicated in ovals and connected to pathway components by dashed lines. Pathway components and interacting proteins are color coded according to experimental responses: solid yellow, changed in NL only; solid blue changed in NL+M only; solid green changed in both NL and NL+M. White stripes indicate proteins that were changed in M. Color coding does not discriminate increases and decreases, or nuclear from cytosolic or membrane. See Figs. 1–6 and supplemental Table S3 for magnitude and direction of changes. Gray, no change in any condition. White, components of the LTP pathway that were not measured. *, human ID protein and/or mouse protein that results in L/M or synaptic plasticity deficits when mutated (81).
Supplementary Material
Acknowledgments
We thank Melissa Stasko for technical assistance in context fear conditioning.
Footnotes
Author contributions: K.J.G. designed research; M.A., A.B., and A.C.C. performed research; A.R.D. and S.T. contributed new reagents or analytic tools; M.A., A.R.D., A.B., S.T., and K.J.G. analyzed data; K.J.G. wrote the paper.
* This work was supported by the National Institute of Child Health and Human Development [HD071585], the Linda Crnic Institute for Down Syndrome and the Fondation Jerome Lejeune.
 This article contains supplemental Figs. S1 to S8 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S8 and Tables S1 to S3.
1 The abbreviations used are:
- L/M
- learning and memory
- NMDA
- N-methyl-d-aspartate
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- LTP
- long term potentiation
- PTM
- post-translational modification
- IEG
- immediate early gene
- AD
- Alzheimer's Disease.
REFERENCES
- 1. Weeber E. J., Levenson J. M., Sweatt J. D. (2002) Molecular genetics of human cognition. Mol. Interv. 2, 376–391 [DOI] [PubMed] [Google Scholar]
- 2. Day J. J., Sweatt J. D. (2011) Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiol. Learn Mem. 96, 2–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen B. S., Roche K. W. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee H. K. (2009) Synaptic plasticity and phosphorylation. Pharmacol. Ther. 12, 810–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samuels I. S., Saitta S. C., Landreth G. E. (2009) MAP'ing CNS development and cognition: an ERKsome process. Neuron. 61, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sweatt J. D. (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 76, 1–10 [DOI] [PubMed] [Google Scholar]
- 7. Hoeffer C. A., Klann E. (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gräff J., Tsai L. H. (2013) Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111 [DOI] [PubMed] [Google Scholar]
- 9. Yang S. H., Sharrocks A. D. (2006) Convergence of the SUMO and MAPK pathways on the ETS-domain transcription factor Elk-1. Biochem. Soc. Symp. 73, 121–129 [DOI] [PubMed] [Google Scholar]
- 10. Kindler S., Kreienkamp H. J. (2012) Dendritic mRNA targeting and translation. Adv. Exp. Med. Biol. 970, 285–305 [DOI] [PubMed] [Google Scholar]
- 11. Waung M. W., Huber K. M. (2009) Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol. 19, 319–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blitzer R. D., Iyengar R., Landau E. M. (2005) Postsynaptic signaling networks: cellular cogwheels underlying long-term plasticity. Biol. Psychiatry 57, 113–119 [DOI] [PubMed] [Google Scholar]
- 13. Mabb A. M., Ehlers M. D. (2010) Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell Dev. Biol. 26, 179–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shepherd J. D., Bear M. F. (2011) New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samuels I. S., Karlo J. C., Faruzzi A. N., Pickering K., Herrup K., Sweatt J. D., Saitta S. C., Landreth G. E. (2003) Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 28, 6983–6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanders M. J., Wiltgen B. J., Fanselow M. S. (2003) The place of the hippocampus in fear conditioning. Eur. J. Pharmacol. 463, 217–223 [DOI] [PubMed] [Google Scholar]
- 17. Stanciu M., Radulovic J., Spiess J. (2001) Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res. Mol. Brain Res. 94, 15–24 [DOI] [PubMed] [Google Scholar]
- 18. Atkins C. M., Selcher J. C., Petraitis J. J., Trzaskos J. M., Sweatt J. D. (1998) The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609 [DOI] [PubMed] [Google Scholar]
- 19. Sananbenesi F., Fischer A., Schrick C., Spiess J., Radulovic J. (2002) Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol. Cell Neurosci. 21, 463–476 [DOI] [PubMed] [Google Scholar]
- 20. Mu Y., Gage F. H. (2011) Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol. Neurodegener. 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danysz W., Parsons C. G. (2012) Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br. J. Pharmacol. 167, 324–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuede C. M., Dong H., Csernansky J. G. (2007) Anti-dementia drugs and hippocampal-dependent memory in rodents. Behav. Pharmacol. 18, 347–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parsons C. G., Danysz W., Quack G. (1999) Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist–a review of preclinical data. Neuropharmacology 38, 735–767 [DOI] [PubMed] [Google Scholar]
- 24. Di Santo S. G., Prinelli F., Adorni F., Caltagirone C., Musicco M. (2013) A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer's disease. J. Alzheimers Dis. 35, 349–361 [DOI] [PubMed] [Google Scholar]
- 25. Costa A. C., Scott-McKean J. J., Stasko M. R. (2008) Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 33, 1624–1632 [DOI] [PubMed] [Google Scholar]
- 26. Radulovic J., Rühmann A., Liepold T., Spiess J. (1999) Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J. Neurosci. 19, 5016–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Milanovic S., Radulovic J., Laban O., Stiedl O., Henn F., Spiess J. (1998) Production of the Fos protein after contextual fear conditioning of C57BL/6N mice. Brain Res. 784, 37–47 [DOI] [PubMed] [Google Scholar]
- 28. Ahmed M. M., Sturgeon X., Ellison M., Davisson M. T., Gardiner K. J. (2012) Loss of correlations among proteins in brains of the Ts65Dn mouse model of down syndrome. J. Proteome Res. 11, 1251–1263 [DOI] [PubMed] [Google Scholar]
- 29. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuo C. T., Mirzadeh Z., Soriano-Navarro M., Rasin M., Wang D., Shen J., Sestan N., Garcia-Verdugo J., Alvarez-Buylla A., Jan L. Y., Jan Y. N. (2006) Postnatal deletion of Numb/Numblike reveals repair and remodeling capacity in the subventricular neurogenic niche. Cell 127, 1253–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gulino R., Perciavalle V., Gulisano M. Expression of cell fate determinants and plastic changes after neurotoxic lesion of adult mice spinal cord by cholera toxin-B saporin. Eur. J. Neurosci. 31, 1423–1434 [DOI] [PubMed] [Google Scholar]
- 32. Chen A. P., Ohno M., Giese K. P., Kühn R., Chen R. L., Silva A. J. (2006) Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J. Neurosci. Res. 83, 28–38 [DOI] [PubMed] [Google Scholar]
- 33. Satoh Y., Endo S., Ikeda T., Yamada K., Ito M., Kuroki M., Hiramoto T., Imamura O., Kobayashi Y., Watanabe Y., Itohara S., Takishima K. (2007) Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 27, 10765–10776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dufresne S. D., Bjørbaek C., El-Haschimi K., Zhao Y., Aschenbach W. G., Moller D. E., Goodyear L. J. (2001) Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice. Mol. Cell. Biol. 21, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bourtchuladze R., Frenguelli B., Blendy J., Cioffi D., Schutz G., Silva A. J. (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68 [DOI] [PubMed] [Google Scholar]
- 36. Weeber E. J., Atkins C. M., Selcher J. C., Varga A. W., Mirnikjoo B., Paylor R., Leitges M., Sweatt J. D. (2000) A role for the beta isoform of protein kinase C in fear conditioning. J. Neurosci. 20, 5906–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamagata Y., Kobayashi S., Umeda T., Inoue A., Sakagami H., Fukaya M., Watanabe M., Hatanaka N., Totsuka M., Yagi T., Obata K., Imoto K., Yanagawa Y., Manabe T., Okabe S. (2009) Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J. Neurosci. 29, 7607–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abeliovich A., Paylor R., Chen C., Kim J. J., Wehner J. M., Tonegawa S. (1993) PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell 75, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 39. Ahi J., Radulovic J., Spiess J. (2004) The role of hippocampal signaling cascades in consolidation of fear memory. Behav. Brain Res. 149, 17–31 [DOI] [PubMed] [Google Scholar]
- 40. Sherrin T., Blank T., Hippel C., Rayner M., Davis R. J., Todorovic C. Hippocampal c-Jun-N-terminal kinases serve as negative regulators of associative learning. J. Neurosci. 30, 13348–13361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lonergan M. E., Gafford G. M., Jarome T. J., Helmstetter F. J. Time-dependent expression of Arc and zif268 after acquisition of fear conditioning. Neural. Plast. 2010:139891, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Plath N., Ohana O., Dammermann B., Errington M. L., Schmitz D., Gross C., Mao X., Engelsberg A., Mahlke C., Welzl H., Kobalz U., Stawrakakis A., Fernandez E., Waltereit R., Bick-Sander A., Therstappen E., Cooke S. F., Blanquet V., Wurst W., Salmen B., Bösl M. R., Lipp H. P., Grant S. G., Bliss T. V., Wolfer D. P., Kuhl D. (2006) Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444 [DOI] [PubMed] [Google Scholar]
- 43. Jones M. W., Errington M. L., French P. J., Fine A., Bliss T. V., Garel S., Charnay P., Bozon B., Laroche S., Davis S. (2001) A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296 [DOI] [PubMed] [Google Scholar]
- 44. Levenson J. M., O'Riordan K. J., Brown K. D., Trinh M. A., Molfese D. L., Sweatt J. D. (2004) Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559 [DOI] [PubMed] [Google Scholar]
- 45. Tingley W. G., Ehlers M. D., Kameyama K., Doherty C., Ptak J. B., Riley C. T., Huganir R. L. (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 272, 5157–5166 [DOI] [PubMed] [Google Scholar]
- 46. Suen P. C., Wu K., Xu J. L., Lin S. Y., Levine E. S., Black I. B. (1998) NMDA receptor subunits in the postsynaptic density of rat brain: expression and phosphorylation by endogenous protein kinases. Brain Res. Mol. Brain Res. 59, 215–228 [DOI] [PubMed] [Google Scholar]
- 47. Isosaka T., Hattori K., Kida S., Kohno T., Nakazawa T., Yamamoto T., Yagi T., Yuasa S. (2008) Activation of Fyn tyrosine kinase in the mouse dorsal hippocampus is essential for contextual fear conditioning. Eur. J. Neurosci. 28, 973–981 [DOI] [PubMed] [Google Scholar]
- 48. Nishimura T., Yamaguchi T., Tokunaga A., Hara A., Hamaguchi T., Kato K., Iwamatsu A, Okano H., Kaibuchi K. (2006) Role of numb in dendritic spine development with a Cdc42 GEF intersectin and EphB2. Mol. Biol. Cell 17, 1273–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tolias K. F., Bikoff J. B., Kane C. G., Tolias C. S., Hu L., Greenberg M. E. (2007) The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc. Natl. Acad. Sci. U.S.A. 104, 7265–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tolias K. F., Bikoff J. B., Burette A., Paradis S., Harrar D., Tavazoie S., Weinberg R. J., Greenberg M. E. (2005) The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45, 525–538 [DOI] [PubMed] [Google Scholar]
- 51. Lipton S. A. (2007) Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr. Drug Targets 8, 621–632 [DOI] [PubMed] [Google Scholar]
- 52. Wang J. Z., Xia Y. Y., Grundke-Iqbal I., Iqbal K. (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33, S123–S139 [DOI] [PubMed] [Google Scholar]
- 53. Schellenberg G. D., Montine T. J. (2012) The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol. 124, 305–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reese L. C., Taglialatela G. (2011) A role for calcineurin in Alzheimer's disease. Curr. Neuropharmacol. 9, 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cavallucci V., Berretta N., Nobili A., Nisticò R., Mercuri N. B., D'Amelio M. (2013) Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer's disease. Neuromolecular Med. 15, 541–548 [DOI] [PubMed] [Google Scholar]
- 56. Shankar G. M., Bloodgood B. L., Townsend M., Walsh D. M., Selkoe D. J., Sabatini B. L. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dineley K, T., Hogan D., Zhang W. R., Taglialatela G. (2007) Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol. Learn. Mem 88, 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lieberman D. N., Mody I. (1994) Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature 369, 235–239 [DOI] [PubMed] [Google Scholar]
- 59. Drewes G., Mandelkow E. M., Baumann K., Goris J., Merlevede W., Mandelkow E. (1993) Dephosphorylation of tau protein and Alzheimer paired helical filaments by calcineurin and phosphatase-2A. FEBS Lett. 336, 425–432 [DOI] [PubMed] [Google Scholar]
- 60. Ermak G., Morgan T. E., Davies K. J. (2001) Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J. Biol. Chem. 276, 38787–38794 [DOI] [PubMed] [Google Scholar]
- 61. Lloret A., Badia M. C., Giraldo E., Ermak G., Alonso M. D., Pallardó F. V., Davies K J., Viña J. (2011) Amyloid-β toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer's disease. J. Alzheimers Dis. 27, 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ermak G., Davies K. J. (2013) Chronic high levels of the RCAN1–1 protein may promote neurodegeneration and Alzheimer disease. Free Radic. Biol. Med. 62, 47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shaftel S. S., Griffin W. S., O'Banion M. K. (2008) The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J. Neuroinflammation 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Viviani B., Bartesaghi S., Gardoni F., Vezzani A., Behrens M. M., Bartfai T., Binaglia M., Corsini E., Di Luca M., Galli C. L., Marinovich M. (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boehm J. (2013) A ‘danse macabre’: tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur. J. Neurosci. 37, 1925–1930 [DOI] [PubMed] [Google Scholar]
- 66. Fischer A., Sananbenesi F., Schrick C., Spiess J., Radulovic J. (2002) Cyclin-dependent kinase 5 is required for associative learning. J. Neurosci. 22, 3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shukla V., Skuntz S., Pant H. C. (2012) Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Arch. Med. Res. 43, 655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sontag J. M., Nunbhakdi-Craig V., White C. L., 3rd, Halpain S., Sontag E. (2012) The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: implications for tauopathies. J. Biol. Chem. 287, 14984–14993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martin L., Latypova X., Wilson C. M., Magnaudeix A., Perrin M. L., Terro F. Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Res. Rev. 12, 39–49 [DOI] [PubMed] [Google Scholar]
- 70. Sontag E., Luangpirom A., Hladik C., Mudrak I., Ogris E., Speciale S., White C. L., 3rd (2004) Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 63, 287–301 [DOI] [PubMed] [Google Scholar]
- 71. Rico B., Marín O. (2011) Neuregulin signaling, cortical circuitry development and schizophrenia. Curr. Opin. Genet. Dev. 21, 262–270 [DOI] [PubMed] [Google Scholar]
- 72. Sardi S. P., Murtie J., Koirala S., Patten B. A., Corfas G. (2006) Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 127, 185–197 [DOI] [PubMed] [Google Scholar]
- 73. Woo R. S., Lee J. H., Yu H. N., Song D. Y., Baik T. K. (2011) Expression of ErbB4 in the neurons of Alzheimer's disease brain and APP/PS1 mice, a model of Alzheimer's disease. Anat. Cell Biol. 44, 116–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maguschak K. A., Ressler K. J. (2012) A role for WNT/β-catenin signaling in the neural mechanisms of behavior. J. Neuroimmune. Pharmacol. 7, 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Z., Hartmann H., Do V. M., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Sommer B., van de Wetering M., Clevers H., Saftig P., De Strooper B., He X., Yankner B. A. (1998) Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702 [DOI] [PubMed] [Google Scholar]
- 76. Toledo E. M., Inestrosa N. C. (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol. Psychiatry 15, 272–285 [DOI] [PubMed] [Google Scholar]
- 77. Chwang W. B., O'Riordan K. J., Levenson J. M., Sweatt J. D. (2006) ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn. Mem. 13, 322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gao C., Gill M. B., Tronson N. C., Guedea A. L., Guzmán Y. F., Huh K. H., Corcoran K. A., Swanson G. T., Radulovic J. (2010) Hippocampal NMDA receptor subunits differentially regulate fear memory formation and neuronal signal propagation. Hippocampus 20, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Czerniawski J., Ree F., Chia C., Ramamoorthi K., Kumata Y., Otto T. A. (2011) The importance of having Arc: expression of the immediate-early gene Arc is required for hippocampus-dependent fear conditioning and blocked by NMDA receptor antagonism. J. Neurosci. 31, 11200–11207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Peleg S., Sananbenesi F., Zovoilis A., Burkhardt S., Bahari-Javan S., Agis-Balboa R. C., Cota P., Wittnam J. L., Gogol-Doering A., Opitz L., Salinas-Riester G., Dettenhofer M., Kang H., Farinelli L., Chen W., Fischer A. (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756 [DOI] [PubMed] [Google Scholar]
- 81. Sturgeon X., Le T., Ahmed M. M., Gardiner K. J. (2012) Pathways to cognitive deficits in Down syndrome. Prog. Brain Res. 197, 73–100 [DOI] [PubMed] [Google Scholar]
- 82. Smith C. L., Eppig J. T. (2012) The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome 23, 653–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim S., Walsh C. A. (2007) Numb, neurogenesis and epithelial polarity. Nat. Neurosci. 10, 812–813 [DOI] [PubMed] [Google Scholar]
- 84. Kyriazis G. A., Wei Z., Vandermey M., Jo D. G., Xin O., Mattson M. P., Chan S. L. (2008) Numb endocytic adapter proteins regulate the transport and processing of the amyloid precursor protein in an isoform-dependent manner: implications for Alzheimer disease pathogenesis. J. Biol. Chem. 283, 25492–25502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chigurupati S., Madan M., Okun E., Wei Z., Pattisapu J. V., Mughal M. R., Mattson M. P., Chan S. L. (2011) Evidence for altered Numb isoform levels in Alzheimer's disease patients and a triple transgenic mouse model. J. Alzheimers Dis. 24, 349–361 [DOI] [PubMed] [Google Scholar]
- 86. Tokumitsu H., Hatano N., Inuzuka H., Sueyoshi Y., Yokokura S., Ichimura T., Nozaki N., Kobayashi R. (2005) Phosphorylation of Numb family proteins. Possible involvement of Ca2+/calmodulin-dependent protein kinases. J. Biol. Chem. 280, 35108–35118 [DOI] [PubMed] [Google Scholar]
- 87. Dho S. E., Trejo J., Siderovski D. P., McGlade C. J. (2006) Dynamic regulation of mammalian numb by G protein-coupled receptors and protein kinase C activation: Structural determinants of numb association with the cortical membrane. Mol. Biol. Cell. 17, 4142–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Merschbaecher K., Haettig J., Mueller U. (2012) Acetylation-mediated suppression of transcription-independent memory: bidirectional modulation of memory by acetylation. PLoS One 7, e45131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martin K. C., Zukin R, S. (2006) RNA trafficking and local protein synthesis in dendrites: an overview. J. Neurosci. 26, 7131–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lau C. G., Zukin R. S. (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 [DOI] [PubMed] [Google Scholar]
- 91. Ehlers M. D. (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–342 [DOI] [PubMed] [Google Scholar]
- 92. Scott D. B., Michailidis I., Mu Y., Logothetis D., Ehlers M. D. (2004) Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J. Neurosci. 24, 7096–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lipton S. A. (2007) Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr. Drug Targets 8, 621–632 [DOI] [PubMed] [Google Scholar]
- 94. Chen H. S., Lipton S. A. (2005) Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-D-aspartate-gated channels. J. Pharmacol. Exp. Ther. 314, 961–971 [DOI] [PubMed] [Google Scholar]
- 95. Zhong W. X., Dong Z. F., Tian M., Cao J., Xu L., Luo J. H. (2006) N-methyl-D-aspartate receptor-dependent long-term potentiation in CA1 region affects synaptic expression of glutamate receptor subunits and associated proteins in the whole hippocampus. Neuroscience 141, 1399–1413 [DOI] [PubMed] [Google Scholar]
- 96. Sanz-Clemente A., Gray J. A., Ogilvie K. A., Nicoll R. A., Roche K. W. (2013) Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 3, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Martisova E., Solas M., Horrillo I., Ortega J. E., Meana J. J., Tordera R. M., Ramírez M. J. (2012) Long lasting effects of early-life stress on glutamatergic/GABAergic circuitry in the rat hippocampus. Neuropharmacology 62, 1944–1953 [DOI] [PubMed] [Google Scholar]
- 98. Tang Q., Yang Q., Hu Z., Liu B., Shuai J., Wang G., Liu Z., Xia J., Shen X. (2007) The effects of willed movement therapy on AMPA receptor properties for adult rat following focal cerebral ischemia. Behav. Brain Res. 181, 254–261 [DOI] [PubMed] [Google Scholar]
- 99. Naka F., Narita N., Okado N., Narita M. (2005) Modification of AMPA receptor properties following environmental enrichment. Brain Dev. 27, 275–278 [DOI] [PubMed] [Google Scholar]
- 100. Jensen O., Lisman J. E. (2005) Hippocampal sequence-encoding driven by a cortical multi-item working memory buffer. Trends Neurosci. 28, 67–72 [DOI] [PubMed] [Google Scholar]
- 101. Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.