

Significance
Guanidinium chloride (GdmCl) has been used to modulate the stability of proteins for more than 50 years; surprisingly, however, the molecular mechanism of its action is still poorly understood. Here, we provide direct kinetic evidence for the hypothesis that GdmCl unfolds proteins by a two-step mechanism. In the first step, it binds to the protein surface, resulting in the formation of a “dry molten globule,” an expanded form of the native protein with a dry core. Core solvation and global structural disruption occur in the second step. The observation of a dry molten globule during unfolding indicates that dispersion forces, and not only the hydrophobic effect, also play an important role in stabilizing proteins.
Keywords: protein unfolding, dry molten globule, steady-state FRET
Abstract
Dry molten globular (DMG) intermediates, an expanded form of the native protein with a dry core, have been observed during denaturant-induced unfolding of many proteins. These observations are counterintuitive because traditional models of chemical denaturation rely on changes in solvent-accessible surface area, and there is no notable change in solvent-accessible surface area during the formation of the DMG. Here we show, using multisite fluorescence resonance energy transfer, far-UV CD, and kinetic thiol-labeling experiments, that the guanidinium chloride (GdmCl)-induced unfolding of RNase H also begins with the formation of the DMG. Population of the DMG occurs within the 5-ms dead time of our measurements. We observe that the size and/or population of the DMG is linearly dependent on [GdmCl], although not as strongly as the second and major step of unfolding, which is accompanied by core solvation and global unfolding. This rapid GdmCl-dependent population of the DMG indicates that GdmCl can interact with the protein before disrupting the hydrophobic core. These results imply that the effect of chemical denaturants cannot be interpreted solely as a disruption of the hydrophobic effect and strongly support recent computational studies, which hypothesize that chemical denaturants first interact directly with the protein surface before completely unfolding the protein in the second step (direct interaction mechanism).
Denaturants such as guanidinium chloride (GdmCl) and urea are classic perturbants used to probe the thermodynamics and kinetics of protein conformational changes, although their mechanism of action is poorly understood (1–15). In some of these studies, a dry molten globular (DMG) state has been observed on the native side of the free-energy barrier (16–18). The DMG is an expanded form of the native protein in which at least some of the side-chain packing interactions are disrupted without solvation of the hydrophobic core; it was originally postulated to explain heat-induced unfolding of proteins (19, 20). Recently, unfolding intermediates resembling the DMG have also been observed in the absence of denaturants, and it has been suggested that the DMG exists in rapid equilibrium with the native state (21–23). The fact that the DMG can be observed by the addition of denaturants is, however, counterintuitive, because traditional models of chemical denaturation rely on changes in solvent-accessible surface area (SASA) (3, 24, 25) and there is no notable change in SASA during the formation of the DMG.
Recent computational studies have suggested an alternative model of chemical denaturation in which denaturants first interact directly with the protein surface, causing the protein to swell, and then penetrate the core (the so-called “direct interaction” mechanism) (9, 11, 12, 26). One of the consequences of this model is that denaturants unfold proteins in two steps (9, 12). In the first step, denaturant molecules displace water molecules within the first solvation shell of the protein or domain. The enhanced presence of denaturant molecules compared with water allows them tighter binding with the protein surface through stronger dispersion interactions (12, 27). This leads to the formation of the dry globule-like intermediate. In the second step, denaturant and water molecules penetrate and solvate the hydrophobic core, resulting in global structural disruption. Experimental support for this model has, however, been scarce and indirect (4, 6, 13, 26).
Here, we demonstrate that the well-studied protein Escherichia coli ribonuclease HI (RNase H) (28–30) also rapidly populates a DMG state when exposed to denaturant and use this observation to explore the denaturant dependence of the DMG. We use multisite fluorescence resonance energy transfer (FRET) and kinetic thiol-labeling measurements to dissect the temporal order of protein expansion, side-chain disruption, and water solvation during GdmCl-induced unfolding. The FRET experiments indicate that unfolding begins with the expansion of the protein into an intermediate state that has an average expansion of 3–4 Å more than the native protein along two measured axes. Thiol-labeling experiments suggest that the core of the intermediate is dry and highly protected from solvent. The kinetics of unfolding measured by both thiol labeling and far-UV CD indicate that solvation and global unfolding take place in the second, slower step. Finally, the size and/or population of the DMG is linearly dependent on [GdmCl], although not as strongly as the second (major) step of unfolding. This rapid GdmCl-dependent population of the DMG indicates that GdmCl can interact with the protein before disrupting the hydrophobic core and strongly supports the two-step direct interaction mechanism of protein denaturation by GdmCl. These results also imply that the effect of chemical denaturants cannot be interpreted solely as a disruption of the hydrophobic effect.
Results
Thionitrobenzoate Quenches the Fluorescence of Tryptophan in a Distance-Dependent Manner.
The absorbance spectrum of thionitrobenzoate (TNB) overlaps with the emission spectrum of tryptophan (Trp) (forming a FRET pair), and this pair has been used to measure distance changes during conformational transitions of proteins (18, 31). Ideally, a single fluorescent donor (Trp) and a single acceptor (TNB) should be used to measure expansion of a protein using FRET. However, in many cases, it is difficult to obtain protein samples containing a single Trp residue. In those cases, multiple donors (Trp residues) and a single acceptor are used (32). RNase H has six tryptophan residues located in one region of the protein (near the so-called basic protrusion area of the protein) (30) (Fig. 1A). In this study, this cluster of Trp residues served as the fluorescence donor (D), and a TNB adduct attached via the thiol of an engineered single cysteine variant (Cys36 or Cys136) (Fig. 1A) served as the FRET acceptor (A). Cys36 is located in the third β-strand, and Cys136 is located in the middle of the C-terminal helix (Fig. 1A), creating two distinct measured axes. The fluorescence emission spectra are similar for the two unlabeled variants, S36C and L136C (Fig. 1 B and C), in both the folded and unfolded states. The fluorescence of the Trp cluster is quenched dramatically in the native state of S36C-TNB and L136C-TNB, but to different extents (Fig. 1 B and C), and is also quenched to a lesser extent in the corresponding unfolded states. The observation that the extent of quenching of fluorescence of the Trp cluster depends on the position of the C-TNB in the protein, and that there is more quenching in the compact native state than in the expanded unfolded form, indicates that the quenching is distance-dependent and hence due to FRET between the donor (Trp cluster) and the acceptor (TNB).
Fig. 1.

TNB quenches the fluorescence of Trp in a distance-dependent manner. (A) Structure of RNase H. The location of I7, A24, S36, I53, and L136 that were replaced by cysteine is shown along with the cluster of Trp residues. The structure was drawn from PDB file 2RN2 by using the program PyMOL (www.pymol.org). Residues 7, 24, and 53 are buried in the native protein and were used for thiol-labeling experiments. Residues 36 and 136 are on the surface of the protein and were used for FRET experiments. S36 and L136 were mutated independently to Cys, to yield two different single-cysteine-containing mutant proteins, S36C and L136C, respectively. The sole thiol moiety in each of the two proteins was labeled with TNB that quenches the fluorescence of Trp in a distance-dependent manner. The TNB-labeled proteins are named as S36C-TNB and L136C-TNB. (B and C) Fluorescence emission spectra of unlabeled and TNB-labeled proteins. In both B and C, the solid cyan line and the solid dark red line denote the fluorescence spectra of unlabeled and TNB-labeled native proteins, respectively. The dashed cyan line and the dashed dark red line denote the fluorescence spectra of unlabeled and TNB-labeled unfolded proteins, respectively. All of the fluorescence spectra in B and C were collected in an identical manner, with the excitation wavelength set to 295 nm.
All four variants, S36C, L136C, S36C-TNB, and L136C-TNB, have similar secondary structure and thermodynamic stability (Fig. S1), as expected because both residues, 36 and 136, are on the surface of the protein (Fig. 1A). Hence, the data on unlabeled and corresponding TNB-labeled proteins can be compared directly for FRET experiments.
GdmCl-Induced Unfolding of RNase H Begins with a Fast Expansion of the Protein.
To probe any fast changes in the dimensions of the protein during GdmCl-induced unfolding, we studied the unfolding kinetics of unlabeled and TNB-labeled proteins. Both the unlabeled proteins, S36C and L136C, have maximum change in fluorescence intensity at 320 nm upon unfolding (Fig. 1 B and C) and show monophasic unfolding kinetics when monitored at this wavelength (Fig. 2 A and B). For both, the kinetic traces extrapolate at t = 0 to the signal of fully native protein, i.e., there is no burst-phase change in fluorescence.
Fig. 2.

Unfolding kinetics of unlabeled and TNB-labeled proteins monitored by change in Trp fluorescence in 3.2 M GdmCl. (A) S36C; (B) L136C; (C) S36C-TNB; and (D) L136C-TNB. In A and B, the solid cyan line represents the change in fluorescence intensity of unlabeled proteins at 320 nm during unfolding. The data are normalized to the value of 1 for the signal of unlabeled native protein at 320 nm (dashed cyan line). A and B (Insets) show the change in fluorescence intensity of unlabeled proteins at 360 nm (solid cyan line) during unfolding, which is normalized to the value of 1 for the signal of unlabeled native protein at 360 nm (dashed cyan line). C and D show the change in fluorescence intensity of TNB-labeled proteins at 360 nm (solid dark red line) during unfolding. The data in C and D are normalized to the value of 1 for the signal of TNB-labeled native protein at 360 nm (dashed dark red line). In A−D, the black solid line is a single exponential fit to the observed data.
To monitor unfolding by FRET, the changes in the fluorescence of the donor alone (FD) and donor−acceptor (FDA) proteins need to be compared (Eq. 1). To avoid complications arising from donor-only fluorescence, it is ideal to perform FRET experiments under conditions where there is minimum change in donor-only fluorescence. Therefore, because for both S36C and L136C the Trp fluorescence in the native and unfolded states is similar near 360 nm (Fig. 1), we chose this wavelength for the FRET experiments. As expected, there is only marginal change in Trp fluorescence intensity during unfolding at 360 nm (Fig. 2 A and B, Insets).
Unlike the unlabeled proteins, both TNB-labeled proteins, S36C-TNB and L136C-TNB, show biphasic unfolding kinetics (Fig. 2 C and D) when monitored using the change in fluorescence at 360 nm. For both, the unfolding traces do not extrapolate at t = 0 to the signal for native protein and a fraction of the total change in fluorescence intensity occurs in an apparent burst phase. This is followed by a single exponential increase in the fluorescence signal (Fig. 2 C and D). The fluorescence intensity increases during the unfolding reaction because the D−A distance is expected to increase as the protein unfolds. The rate constants for the observed phases of unfolding in the unlabeled and corresponding TNB-labeled protein are very similar at all concentrations of GdmCl (Fig. S1 E and F). In total, these results indicate that the GdmCl-induced unfolding of RNase begins with a fast initial expansion of the protein, followed by a slower expansion. The β-strand containing residue 36 and the α-helix containing residue 136 move away from the region containing the Trp cluster in the initially expanded state.
The Average Change in D−A Distances in the Initial Expanded State Is Dependent upon [GdmCl].
If the initial expansion of the protein surface is induced by interaction with GdmCl, then the population, or extent of expansion, in the intermediate is expected to show some dependence on denaturant concentration. The unfolding burst-phase, final amplitudes, and equilibrium denaturation curves, as measured by the change in fluorescence at 360 nm, are shown for S36C and S36C-TNB in Fig. 3A and for L136C and L136C-TNB in Fig. 3D. For the TNB-labeled proteins, a significant fraction of the initial increase in fluorescence during unfolding occurs in the burst phase, and the t = 0 point reflects the amplitude of this phase. This burst-phase amplitude increases linearly with [GdmCl] and extrapolates to the linear GdmCl dependence for the equilibrium GdmCl-dependent fluorescence of the native protein.
Fig. 3.

Dependence of the amplitude of the initial fast phase of unfolding on the concentration of GdmCl. Kinetic versus equilibrium amplitudes of the unfolding of unlabeled and TNB-labeled proteins as monitored by the change in fluorescence at 360 nm. (A) S36C and S36C-TNB; (D) L136C and L136C-TNB. In A and D, the cyan circles represent the equilibrium unfolding transition of the unlabeled protein. The solid cyan line through the data represents a fit to a two-state native (N) ⇌ unfolded (U) model. The cyan inverted triangles and the cyan squares represent the t = 0 and t = ∞ signals, respectively, obtained from fitting the kinetic traces of unfolding of the unlabeled protein to a single exponential equation. The dark red circles represent the equilibrium unfolding transition of the TNB-labeled protein, the continuous dark red line represents a fit to a two-state N ⇌ U model, and the dotted dark red line is a linear extrapolation of the native protein baseline. The dark red squares represent the t = ∞ signal, and the dark red inverted triangles represent the t = 0 signal, obtained from fitting the kinetic traces of unfolding of the TNB-labeled protein to a single exponential equation. The error bars, where shown, represent SDs of the measurements from at least three separate experiments. In B and E, the FRET efficiencies in the forms of each protein, which are populated before commencement of the observable unfolding phase, are plotted (inverted triangles) against the concentration of GdmCl for S36C-TNB and L136C-TNB, respectively. Also shown (circles) are the FRET efficiencies in the native forms of each protein, which are populated in the GdmCl concentrations that define the native protein baseline of the equilibrium unfolding transition. The data in B and E were converted into apparent change in distances by using Eq. 1, and the distances are shown in C and F for S36C-TNB and L136C-TNB, respectively.
RNase H has six Trp residues, making it difficult to assign the Trp moiety that serves as the fluorescence donor in the current study (the Trp nearest to TNB in the native structure is the most likely candidate), and to extract exact changes in distances between the donor and the acceptor. It is, however, possible to determine average changes in distances between the region containing the Trp clusters and the region containing the residues Cys36 and Cys136 [Cys36 and Cys136 are located diagonally opposite to Trp clusters in the structure of RNase H (Fig. 1A)]. We used the data on unlabeled and TNB-labeled proteins (Fig. 3 A and D) to estimate the change in FRET efficiency (E) and apparent change in distances (R) between the two ends of the protein, in the initially expanded state during unfolding, as well as of completely native protein, at each concentration of GdmCl using Eq. 1:
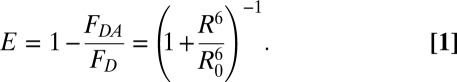 |
For each [GdmCl], the fluorescence at t = 0 for unfolding of the unlabeled protein was taken as FD, and the corresponding fluorescence at t = 0 for unfolding of the labeled protein was taken as FDA. The Forster’s distance, R0, of the Trp-TNB FRET pair has been shown to lie between 22 and 23 Å in [GdmCl] ranging between 0 and 6 M for several different proteins (18, 31). In this study, we used a mean value of 22.5 Å for R0. The donors as well as the acceptors appear to rotate freely at rates faster than fluorescence decay rate of donor tryptophans as required for FRET measurements (SI Text).
The FRET efficiencies of both the burst-phase intermediate and the equilibrium native state decrease in a continuous monotonic manner with an increase in [GdmCl] (Fig. 3 B and E). Therefore, both calculated intramolecular distances increase continuously with [GdmCl] (Fig. 3 C and F).
Initial Expansion Is Not Accompanied by Disruption of Secondary Structure.
For both TNB-labeled proteins, the kinetic traces of far-UV CD-monitored unfolding extrapolate at t = 0 to the signal of the native protein (Fig. 4 A and B). The kinetic amplitudes of unfolding match the equilibrium amplitudes (Fig. 4 A and B, Insets), and the entire expected change in the far-UV CD signal (which represents the dissolution of hydrogen-bonded secondary structure) during unfolding occurs in a single exponential phase the rate of which is similar to the rate of observed unfolding phase by fluorescence. These results indicate that the initially expanded state has native-like secondary structure and amide−amide H-bonds are not replaced by amide−water H-bond. The dissolution of the secondary structure and global structure disruption occurs during the slow, observed phase of unfolding.
Fig. 4.

Kinetics of change in secondary structure during unfolding as monitored by change in far-UV CD signal at 222 nm. (A) S136C-TNB; (B) L136C-TNB. In A and B, the solid dark red line shows the kinetic trace of unfolding in 2.5 M GdmCl, and the solid black line through the data is a fit to a single exponential equation. The data are normalized to the value of 1 for the signal of native protein (solid and dashed blue lines). Insets compare the kinetic versus equilibrium amplitudes of unfolding. The circles represent the equilibrium unfolding transition, and the continuous line through the data represents a fit to a two-state N ⇌ U model. The triangles represent the t = 0 signal, and the squares represent the t = ∞ signal, obtained from fitting the kinetic traces of unfolding to a single exponential equation. The black dotted line is a linear extrapolation of the native protein baseline. The error bars, wherever shown, represent the SDs of measurements from three separate experiments.
Initial Expansion Is Not Accompanied by Solvation of Protein Core.
To probe whether the initial burst-phase expansion is accompanied by solvation of the protein core, we monitored the solvent accessibility of buried thiol groups during unfolding using three single-cysteine-containing variants of RNase H (Fig. 1A). Residues 7 and 24 (Cys7 and Cys24) are buried positions near the surface of the RNase H, and residue 53 is buried inside the hydrophobic core of the protein (Fig. 1A). We used two different thiol-specific labeling reagents, 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) and methyl metahnethiosulfonate (MMTS), both of which react rapidly with exposed thiolate groups (33–35), to measure the changes in solvent accessibility during unfolding (Fig. 5 A−C). DTNB is relatively large, contains aromatic rings, and is negatively charged, whereas MMTS is small, neutral, and highly soluble in water. The use of reagents with different sizes and chemical properties allows us to evaluate the size of the structural opening and the possible penetration of solvent in the initially expanded globule.
Fig. 5.

Kinetics of core solvation during unfolding by monitoring the reactivity of buried thiols against labeling by DTNB and MMTS. Three single-cysteine-containing variants of RNase H, (A) I7C (Cys7), (B) A24C (Cys24), and (C) I53C (Cys53), were generated, and each of them was diluted in solutions containing 1.3 mM DTNB and 0 M (red), 3 M (magenta), and 3.4 M (blue) GdmCl (A−C). The unfolding reaction was monitored by the change in absorbance at 412 nm due to the release of TNB molecules and is shown according to the left y axis. The solid black line through the data is fit to a single exponential equation. The kinetics of change in cysteine accessibility during unfolding as monitored by MMTS labeling is shown in green according to the right y axis. Fractional labeling (green circle) is plotted against the time of application of the labeling pulse (6-ms pulse of 20 mM MMTS) after the commencement of unfolding in 3.4 M GdmCl. The green solid line is a fit of the MMTS labeling data to a single exponential equation. D shows the accessibility (protection factor) of Cys53 to MMTS labeling in the unfolded protein (3.4 M GdmCl) (green triangle and green dashed line), the native protein (0 M GdmCl) (blue circle and blue solid line), and the initially expanded globule (red squares and red dashed line). The lines through the data are drawn only to help visual inspection. The extent of labeling is plotted against the duration of the labeling pulse containing 20 mM MMTS. For the initially expanded globule, labeling pulse was applied 500 ms after the initiation of the unfolding reaction in 3.4 M GdmCl.
For DTNB labeling, under the conditions used for our experiments (pH 8, 1.3 mM DTNB, 25 °C), the unique thiol in all three proteins gets labeled rapidly in the unfolded state [within the dead time (∼10 s) of the manual mixing experiments] but reacts very slowly in the native state (Fig. 5 A−C). When the unfolding process was monitored by DTNB, no unfolded-like reactivity was seen in the dead time of the experiment (Fig. 5 A−C).
For MMTS labeling, we applied a 6-ms pulse of 20 mM MMTS at different time points of unfolding (Fig. 5 A−C). The pulse is ∼15,000-fold shorter than the time constant of global unfolding and fully labels solvent-exposed thiols but does not label buried thiols, such as in the native protein (Fig. 5D). This pulsed approach is ideal for measuring any transient exposure of buried thiols during unfolding. When the unfolding process was monitored by MMTS, none of the three thiols showed any label in the initially expanded globule (Fig. 5 A−C), suggesting that the solvent is unable to penetrate the hydrophobic core in this initially expanded state.
For both the labels, we observed single exponential slow formation of thiol reactivity (Fig. 5 A−C) with a rate that matched the global unfolding as monitored by Trp fluorescence (Fig. S2). Together, these data indicate that the protein core is largely undisrupted and dry (not accessible to solvent) in the initially expanded state during unfolding and the solvation of the protein core occurs upon global unfolding in the slow phase of unfolding.
Protection in the Core of the Protein Is Similar in the Native State and the Initially Expanded State.
To determine the degree to which the initially expanded state is protected against solvation, we measured the level of protection to labeling by MMTS by monitoring the extent of modification as a function of pulse intensity. The duration of the 20 mM labeling pulse was varied from 6 to 600 ms using the Cys53 variant. In unfolded RNase H, Cys53 RNase H is fully labeled by the shortest pulse of 6 ms (Fig. 5D), whereas in native RNase H, Cys53 is highly protected and does not show any measureable label when the duration of pulse is increased to 200 ms, and is labeled only to ∼5% during a 600-ms labeling pulse (Fig. 5D). Fig. 5D also shows the extent of labeling after unfolding for 500 ms in 3.4 M GdmCl, which corresponds to a time of unfolding when the initially expanded globule is fully formed but only ∼0.5% of the global unfolding reaction has occurred. Cys53 does not get labeled at all in the initially expanded state when the duration of pulse is increased 10-fold to 60 ms and gets labeled to only ∼10% when the duration of pulse is increased 100-fold to 600 ms. These results strongly indicate that the core of the initially expanded globule is strongly protected against solvation and its protection is very similar to the native protein.
Discussion
A large body of work in protein folding has focused on understanding how proteins collapse and fold when diluted from a solution containing a high concentration of denaturant (good solvent). The reverse process of unfolding, swelling of the native protein into a random coil, however, has received much less attention and is poorly understood. In particular, we do not fully understand how common denaturing agents, like GdmCl, initiate the unfolding process. It has been proposed theoretically that denaturants unfold structure either in a single step by an indirect mechanism, which involves alteration of water structure (2, 3, 36, 37), or in two stages, by first directly interacting with the protein surface (5, 12, 15) and then unfolding the protein.
The direct interaction mechanism hypothesizes that, initially, denaturant molecules replace water molecules from the first solvation shell of the protein. This helps the denaturant molecules to interact directly with the protein surface, causing the protein to swell into an ensemble of native-like intermediates (12). This initially swollen globule is called a “dry globule” due to the lack of water in the core. In the second step, denaturant molecules enter the hydrophobic core followed by water molecules, causing global structural disruption. Although well studied theoretically, to date, it has not been possible to experimentally distinguish between these two mechanisms because the interaction between denaturants and proteins is very weak.
A DMG has been observed as an early intermediate during the unfolding of several proteins (16–18). It has been suggested that the DMG exists in rapid equilibrium with the native state of proteins (21–23) and that such states may be a general intermediate on the native side of the free energy barrier (38, 39). Here we find that the protein RNase H forms such a DMG and provide direct kinetic evidence that GdmCl-induced unfolding of RNase H occurs via expansion of the protein in two distinct stages. In the first stage, RNase H expands into a DMG state whose dimension or population depends upon the concentration of GdmCl, indicating that protein swelling is dependent on interaction with the denaturant, presumably binding of the denaturant to the protein surface (Figs. 2 and 3). At least two different secondary structural elements (β-strand containing residue 36 and the C-terminal α-helix) appear to move away from the Trp cluster in the initially expanded state (Fig. 3). Lack of accessibility to thiol labeling indicates that the expanded globule does not have a solvated core, and CD suggests that its secondary structure is very similar to the native protein (Figs. 4 and 5). Disruption of secondary structure and solvation of the hydrophobic core occurs during the second, slower and more denaturant-dependent step of unfolding (Figs. 4 and 5).
Although small compared with global unfolding, formation of the initial DMG shows a denaturant dependence. The average dimensions of the transient unfolding intermediate show a linear denaturant dependence, which appears to be continuous with that of the denaturant dependence of the average dimensions of the protein under native conditions (Fig. 3 C and F). Although not fully explored, some denaturant dependence was also observed in the NMR studies on the unfolding of RNase A, which first observed the DMG as an unfolding intermediate (16).
The steady-state FRET experiments used in this study cannot distinguish between denaturant-dependent changes in the size of the DMG versus a change in the population of the two states (N and DMG). Moreover, because formation of the DMG occurs within the 5-ms dead time of our stopped-flow measurements, the DMG conformations of RNase H may be members of the native state ensemble with increasing [GdmCl] biasing the ensemble toward the DMG. Consistent with this hypothesis, we do not observe a sigmoidal transition between the fully native and the DMG state, suggesting that the two might not be separated by a single rate-limiting barrier but by many small, distributed barriers. However, resolving the difference between such a cooperative and noncooperative transition may be outside of the resolution of our experiments. Pressure-induced unfolding of ubiquitin has shown that under native-like conditions, the native protein exists in equilibrium with energetically equivalent dry globules with fluctuating side-chain structures (23); 17O NMR relaxation dispersion experiments on three structurally unrelated proteins have revealed that their molten globule states closely resemble the native proteins with regard to internal hydration and surface topography (40). Triplet−triplet energy transfer experiments on HP35 also suggest that the DMG is in rapid equilibrium with the native state (21). Future time-resolved FRET experiments on RNase H may shed light on the nature of the barrier between these two states.
Nevertheless, our kinetic FRET experiments unequivocally show that the addition of GdmCl populates the DMG and that the average dimension of this state depends on [GdmCl]. These observations indicate that GdmCl interacts with protein surface before the entry of water molecules inside the protein core and strongly support the direct interaction mechanism of protein denaturation by GdmCl.
Direct denaturant binding to amino acid-like model compounds without hydrophobic side chains was first postulated by Robinson and Jencks (26) to explain their data on the effects of guanidinium and urea on the solubility of acetyltetraglycine ethyl ester. Calorimetric data on binding of GdmCl and urea with proteins also point toward a direct interaction between denaturants and proteins (6). The change in heat capacity of unfolding measured with heat unfolding and denaturant-induced unfolding are different, indicating that chemical denaturant may participate directly in the process (41). Discrete denaturant binding sites and low B-factors of surface residues have also been observed in the X-ray crystal structures of proteins in the presence of GdmCl (7, 8, 42).
Neutron diffraction studies have revealed that guanidinium ions interact very weakly with water and have no recognizable hydration shell (10). This weak hydration promotes the partial dehydration required for these ions to interact with the protein surface. It also enhances the solubility of hydrophobic side chains by reducing the local water density and preempting the space in the solvation shell around the hydrophobic side chains that could otherwise be occupied by water molecules (10). Not surprisingly, theoretical studies predict that guanidinium ions are capable of intercalating between nonpolar groups (11, 27). Although we do not observe that the thiol-labeling reagents present in the solvent penetrate the hydrophobic core in the initially expanded state (Fig. 5), it is possible that some of the protein-bound GdmCl in the first solvation shell might enter and interact with hydrophobic groups in the core and facilitate subsequent unfolding, as predicted by a recent theoretical study (12).
The role of packing interactions in the stability of proteins has been widely debated (43–45). Although hydrophobic interactions are considered the major determinant of protein stability, the observation of a state in which the core is loosely packed but dry highlights the importance of packing interactions for the stability of the protein fold. Because initial events during unfolding are similar to the last step during folding, the results of this study support the model that tight side-chain packing might be the last event during the folding of proteins.
Materials and Methods
Protein Expression, Purification, and TNB Labeling.
All of the single-cysteine-containing variants of E. coli RNase H used in this study have been characterized in a previous study (29) and were purified as described earlier (29). The TNB-labeled proteins were obtained as described in SI Text.
Fluorescence and Far-UV CD Spectra.
Fluorescence spectra were collected on a Spex Fluoromax 3 spectrofluorimeter, with the excitation wavelength set to 295 nm. Far-UV CD spectra were collected on an Aviv 410 spectropolarimeter as described earlier (29).
Fluorescence-Monitored Equilibrium and Kinetic Unfolding Experiments.
The equilibrium unfolding reactions of the unlabeled and TNB-labeled proteins were carried out in an identical manner. For S36C and S36C-TNB, a Spex Fluoromax 3 spectrofluorimeter was used, and for L136C and L136C-TNB, the stopped-flow module (SFM-400) from Biologic was used. The native protein was incubated in different concentrations of GdmCl ranging from zero to 5 M for 12 h, and the equilibrium fluorescence signals were measured. Sample excitation was carried out at 295 nm, and emission was monitored at 360 nm. The unfolding kinetics for S36C and S36C-TNB was measured on the Spex Fluoromax 3 spectrofluorimeter, and for L136C and L136C-TNB on SFM-400. The unfolding reactions of the unlabeled and TNB-labeled proteins were carried out in an identical manner. Sample excitation was carried out at 295 nm, and emission was monitored at 360 nm. The unfolding reactions of the unlabeled proteins were also monitored at 320 nm upon excitation at 295 nm. All of the experiments were performed at pH 8 with buffer containing 50 mM Tris and 50 mM KCl.
Thiol-Labeling Experiments.
For DTNB-labeling experiments, the single cysteine containing variants of RNase H were diluted in solutions containing 1.3 mM DTNB and 0 M, 3 M, and 3.4 M GdmCl at pH 8 with buffer containing 50 mM Tris and 50 mM KCl. The unfolding reaction was monitored by the change in absorbance at 412 nm due to the release of TNB molecules.
MMTS-labeling experiments were performed as described previously (33, 35). In brief, RNase H was unfolded in 3.4 M GdmCl inside a quenched-flow machine (SFM-400 Q/S from Biologic), and a 6-ms pulse of 20 mM MMTS was applied at different times of unfolding. The labeling reaction was quenched by excess cysteine hydrochloride. Following this, the reaction mixture was desalted and the total protein concentration was determined by measuring absorbance at 280 nm. The amount of unreacted (free) thiol was determined by absorbance at 412 nm after reaction with excess DTNB.
CD-Monitored Equilibrium and Kinetic Unfolding Experiments.
Equilibrium and kinetic unfolding of all of the proteins using far-UV CD was monitored on an Aviv 410 spectropolarimeter as described earlier (29). The dead time of the measurement for the manual kinetic experiments was ∼8–10 s.
Supplementary Material
Acknowledgments
We thank the members of the S.M. laboratory for helpful discussions and the reviewers for suggested experiments. This work was supported by a grant from the National Institutes of Health (to S.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315453111/-/DCSupplemental.
References
- 1.Hopkins FG. Denaturation of proteins by urea and related substances. Nature. 1930;126(3174):328–330. 383−384. [Google Scholar]
- 2.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 3.Tanford C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 4.Gordon JA. Denaturation of globular proteins. Interaction of guanidinium salts with three proteins. Biochemistry. 1972;11(10):1862–1870. doi: 10.1021/bi00760a021. [DOI] [PubMed] [Google Scholar]
- 5.Schellman JA. Solvent denaturation. Biopolymers. 1978;17(5):1305–1322. doi: 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- 6.Makhatadze GI, Privalov PL. Protein interactions with urea and guanidinium chloride. A calorimetric study. J Mol Biol. 1992;226(2):491–505. doi: 10.1016/0022-2836(92)90963-k. [DOI] [PubMed] [Google Scholar]
- 7.Dunbar J, Yennawar HP, Banerjee S, Luo J, Farber GK. The effect of denaturants on protein structure. Protein Sci. 1997;6(8):1727–1733. doi: 10.1002/pro.5560060813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratnaparkhi GS, Varadarajan R. X-ray crystallographic studies of the denaturation of ribonuclease S. Proteins. 1999;36(3):282–294. [PubMed] [Google Scholar]
- 9.Mountain RD, Thirumalai D. Molecular dynamics simulations of end-to-end contact formation in hydrocarbon chains in water and aqueous urea solution. J Am Chem Soc. 2003;125(7):1950–1957. doi: 10.1021/ja020496f. [DOI] [PubMed] [Google Scholar]
- 10.Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM. The hydration structure of guanidinium and thiocyanate ions: Implications for protein stability in aqueous solution. Proc Natl Acad Sci USA. 2003;100(8):4557–4561. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien EP, Dima RI, Brooks B, Thirumalai D. Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: Lessons for protein denaturation mechanism. J Am Chem Soc. 2007;129(23):7346–7353. doi: 10.1021/ja069232+. [DOI] [PubMed] [Google Scholar]
- 12.Hua L, Zhou R, Thirumalai D, Berne BJ. Urea denaturation by stronger dispersion interactions with proteins than water implies a 2-stage unfolding. Proc Natl Acad Sci USA. 2008;105(44):16928–16933. doi: 10.1073/pnas.0808427105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almarza J, Rincon L, Bahsas A, Brito F. Molecular mechanism for the denaturation of proteins by urea. Biochemistry. 2009;48(32):7608–7613. doi: 10.1021/bi9007116. [DOI] [PubMed] [Google Scholar]
- 14.Lim WK, Rösgen J, Englander SW. Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group. Proc Natl Acad Sci USA. 2009;106(8):2595–2600. doi: 10.1073/pnas.0812588106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.England JL, Haran G. Role of solvation effects in protein denaturation: From thermodynamics to single molecules and back. Annu Rev Phys Chem. 2011;62:257–277. doi: 10.1146/annurev-physchem-032210-103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiefhaber T, Labhardt AM, Baldwin RL. Direct NMR evidence for an intermediate preceding the rate-limiting step in the unfolding of ribonuclease A. Nature. 1995;375(6531):513–515. doi: 10.1038/375513a0. [DOI] [PubMed] [Google Scholar]
- 17.Hoeltzli SD, Frieden C. Stopped-flow NMR spectroscopy: Real-time unfolding studies of 6-19F-tryptophan-labeled Escherichia coli dihydrofolate reductase. Proc Natl Acad Sci USA. 1995;92(20):9318–9322. doi: 10.1073/pnas.92.20.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jha SK, Udgaonkar JB. Direct evidence for a dry molten globule intermediate during the unfolding of a small protein. Proc Natl Acad Sci USA. 2009;106(30):12289–12294. doi: 10.1073/pnas.0905744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shakhnovich EI, Finkelstein AV. Theory of cooperative transitions in protein molecules. I. Why denaturation of globular protein is a first-order phase transition. Biopolymers. 1989;28(10):1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 20.Finkelstein AV, Shakhnovich EI. Theory of cooperative transitions in protein molecules. II. Phase diagram for a protein molecule in solution. Biopolymers. 1989;28(10):1681–1694. doi: 10.1002/bip.360281004. [DOI] [PubMed] [Google Scholar]
- 21.Reiner A, Henklein P, Kiefhaber T. An unlocking/relocking barrier in conformational fluctuations of villin headpiece subdomain. Proc Natl Acad Sci USA. 2010;107(11):4955–4960. doi: 10.1073/pnas.0910001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche J, et al. Cavities determine the pressure unfolding of proteins. Proc Natl Acad Sci USA. 2012;109(18):6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Y, et al. Coupled motion in proteins revealed by pressure perturbation. J Am Chem Soc. 2012;134(20):8543–8550. doi: 10.1021/ja3004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nozaki Y, Tanford C. The solubility of amino acids and related compounds in aqueous urea solutions. J Biol Chem. 1963;238:4074–4081. [PubMed] [Google Scholar]
- 25.Auton M, Holthauzen LM, Bolen DW. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc Natl Acad Sci USA. 2007;104(39):15317–15322. doi: 10.1073/pnas.0706251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson DR, Jencks WP. Effect of compounds of urea-guanidium class on activity coefficient of acetyltetraglycine ethyl ester and related compounds. J Am Chem Soc. 1965;87(11):2462–2470. doi: 10.1021/ja01089a028. [DOI] [PubMed] [Google Scholar]
- 27.Godawat R, Jamadagni SN, Garde S. Unfolding of hydrophobic polymers in guanidinium chloride solutions. J Phys Chem B. 2010;114(6):2246–2254. doi: 10.1021/jp906976q. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat Struct Biol. 1996;3(9):782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein R, Schmidt KL, Harbury PB, Marqusee S. Structural and kinetic mapping of side-chain exposure onto the protein energy landscape. Proc Natl Acad Sci USA. 2011;108(26):10532–10537. doi: 10.1073/pnas.1103629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dabora JM, Marqusee S. Equilibrium unfolding of Escherichia coli ribonuclease H: Characterization of a partially folded state. Protein Sci. 1994;3(9):1401–1408. doi: 10.1002/pro.5560030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jha SK, Dhar D, Krishnamoorthy G, Udgaonkar JB. Continuous dissolution of structure during the unfolding of a small protein. Proc Natl Acad Sci USA. 2009;106(27):11113–11118. doi: 10.1073/pnas.0812564106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammarstrom P, Persson M, Carlsson U. Protein compactness measured by fluorescence resonance energy transfer. Human carbonic anhydrase II is considerably expanded by the interaction of GroEL. J Biol Chem. 2001;276(24):21765–21775. doi: 10.1074/jbc.M010858200. [DOI] [PubMed] [Google Scholar]
- 33.Ha JH, Loh SN. Changes in side chain packing during apomyoglobin folding characterized by pulsed thiol-disulfide exchange. Nat Struct Biol. 1998;5(8):730–737. doi: 10.1038/1436. [DOI] [PubMed] [Google Scholar]
- 34.Jha SK, Udgaonkar JB. Exploring the cooperativity of the fast folding reaction of a small protein using pulsed thiol labeling and mass spectrometry. J Biol Chem. 2007;282(52):37479–37491. doi: 10.1074/jbc.M706714200. [DOI] [PubMed] [Google Scholar]
- 35.Jha SK, Dasgupta A, Malhotra P, Udgaonkar JB. Identification of multiple folding pathways of monellin using pulsed thiol labeling and mass spectrometry. Biochemistry. 2011;50(15):3062–3074. doi: 10.1021/bi1006332. [DOI] [PubMed] [Google Scholar]
- 36.Frank HS, Evans MW. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J Chem Phys. 1945;13:507–532. [Google Scholar]
- 37.Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11(4):281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 38.Jha SK, Udgaonkar JB. Free energy barriers in protein folding and unfolding reactions. Curr Sci. 2010;99(4):457–475. [Google Scholar]
- 39.Baldwin RL, Frieden C, Rose GD. Dry molten globule intermediates and the mechanism of protein unfolding. Proteins. 2010;78(13):2725–2737. doi: 10.1002/prot.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denisov VP, Jonsson BH, Halle B. Hydration of denatured and molten globule proteins. Nat Struct Biol. 1999;6(3):253–260. doi: 10.1038/6692. [DOI] [PubMed] [Google Scholar]
- 41.Baldwin RL. Properties of hydrophobic free energy found by gas-liquid transfer. Proc Natl Acad Sci USA. 2013;110(5):1670–1673. doi: 10.1073/pnas.1220825110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hibbard LS, Tulinsky A. Expression of functionality of alpha-chymotrypsin. Effects of guanidine hydrochloride and urea in the onset of denaturation. Biochemistry. 1978;17(25):5460–5468. doi: 10.1021/bi00618a021. [DOI] [PubMed] [Google Scholar]
- 43.Behe MJ, Lattman EE, Rose GD. The protein-folding problem: The native fold determines packing, but does packing determine the native fold? Proc Natl Acad Sci USA. 1991;88(10):4195–4199. doi: 10.1073/pnas.88.10.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bromberg S, Dill KA. Side-chain entropy and packing in proteins. Protein Sci. 1994;3(7):997–1009. doi: 10.1002/pro.5560030702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhattacharyya S, Varadarajan R. Packing in molten globules and native states. Curr Opin Struct Biol. 2013;23(1):11–21. doi: 10.1016/j.sbi.2012.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.