

Abstract
Autosomal recessive limb-girdle muscular dystrophies (AR-LGMDs) are characterized by clinical and genetic heterogeneity. LGMD type 2C, or γ-sarcoglycanopathy, is the most frequent in North African populations as a result of the founder c.525delT mutation in the SGCG gene. Its epidemiology is poorly known in Morocco, and its prevalence among the Moroccan population has never been evaluated. This study screened 26 patients with a LGMD2C and 45 patients with an AR-LGMD phenotype for the c.525delT mutation. DNA extracted from umbilical cord blood samples of 250 newborns was tested for the same mutation. Molecular epidemiologic methods were used to calculate the frequency of heterozygotes for this mutation in Moroccan newborns and to estimate the prevalence of LGMD2C in the Moroccan population. The carrier frequency was estimated to be 1/250, which would imply that the prevalence of LGMD2C would be approximately 1/20,492 considering the effect of consanguinity. The homozygous c.525delT mutation was found in 65% of all patients with AR-LGMDs. These findings suggest that AR-LGMDs are prevalent in the Moroccan population and LGMD2C is one of the most common forms. This information might be useful for the development of diagnostic strategies on a large scale for better management of patients with AR-LGMD and genetic counseling of families.
Introduction
Limb-girdle muscular dystrophies (LGMDs) are heterogeneous inherited muscle disorders characterized by progressive weakness and muscle wasting. The most common clinical forms are autosomal recessive (AR), characterized by great clinical and genetic variability (Bönnemann 2005). They typically show degeneration/regeneration (dystrophic changes) on muscle biopsy, which is usually associated with elevated serum creatine kinase concentrations. Immunohistochemical studies with specific antibodies on muscle biopsies are very useful for classifying the different types prior to genetic analysis (Anderson 1996; Boito et al., 2003). The relative frequency of the different types of AR-LGMD varies from one ethnic population to another (Trabelsi et al., 2008). However, previous studies indicated that LGMD type 2C (Online Mendelian Inheritance in Man #253700) is the most frequent in North African populations, as a result of the founder mutation c.525delT (El Kerch et al., 1994; Ben Othmane et al., 1995). LGMD2C is caused by mutations in the SGCG gene on 13q12, which encodes γ-sarcoglycan. It is characterized by a childhood onset of progressive muscular dystrophy. The mean age of onset is between 5 and 6 years, and half of these patients lose ambulation by age 12 years. Calf hypertrophy and lumbar lordosis are common (Bönnemann and Finkel, 2002). On the basis of these clinical findings, LGMD2C is referred to as a severe childhood autosomal recessive muscular dystrophy or as a Duchenne-like autosomal recessive muscular dystrophy (Ozawa et al., 1998).
In Morocco, the epidemiology of LGMD2C is poorly known. This study aimed to estimate the frequency of the recurrent mutation c.525delT in Moroccan patients and controls in order to deduce, by a molecular epidemiology approach, the prevalence of LGMD type 2C in the Moroccan population.
Materials and Methods
Patients
We enrolled two groups of patients: The first group consisted of 26 patients with LGMD type 2C based on an immunohistochemical study. They were 12 males and 14 females, ranging in age from 5.5 to 13 years. Eleven of them were consanguineous, and 19 were familial cases with at least one sibling pair. In addition to clinical, biological, and histologic signs of LGMD, none had staining for γ-sarcoglycan.
The second group comprised 45 patients (9 males and 36 females) aged 6–12 years, with a tentative clinical autosomal recessive LGMD diagnosis, based on the following conditions: slowly progressive mild to moderate limb girdle weakness, elevated serum creatine kinase level, and various degrees of dystrophic changes on muscle biopsy. No patients had access to immunofluorescence studies. Twenty-eight were sporadic cases and 17 were familial cases, with at least one sibling pair. Twenty-nine of these families were consanguineous (Fig. 1). The autosomal recessive condition of the LGMD was validated by the presence of at least one affected female. For the 9 families with only an affected male, the diagnosis of dystrophinopathy was first excluded by immunohistochemical stain for dystrophin.
FIG. 1.

Siblings in 45 families with AR-LGMD phenotype (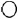 normal girl, ● affected girl,
normal girl, ● affected girl,  normal boy,
normal boy,  affected boy,
affected boy, 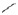 deceased child,
deceased child, 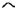 twin).
twin).
Controls
Umbilical cord blood samples of 250 unrelated newborns were investigated. They originated from different regions of Morocco, and the Moroccan origin of their parents and grandparents was confirmed. Two controls with homozygous and heterozygous deletion c.525delT were included in the study. The study was approved by the local ethics committee, and parents provided informed consent for DNA analysis.
Data collection
Genomic DNA of patients and controls was extracted from leukocytes isolated from ethylenediaminetetraacetic acid–anticoagulated blood using the salting-out method (Sambrook et al., 1989). The quality and quantity of the DNA were controlled by A260/A280 using a Nanodrop spectrophotometer (Fisher Scientific, Wilmington, DE) and aliquots (50–100 μL) of packed blood cells were stored at 4°C until analysis. First, we searched for c. 525delT mutation on exon 6 of the SGCG gene in the two groups of patients by direct sequencing (sequencer ABI PRISM 310; Applied Biosystems, Carlsbad, CA) using specific primers as previously reported (El Kerch et al., 2001). Next, we screened for the c.525delT mutation in the 250 controls by using real-time PCR (Applied Biosystems 7500 Fast Real-Time PCR Systems) using TaqMan probes (PE Applied Biosystems). It is simpler and faster than DNA fragment analysis. This method combines PCR and mutation detection in a single step. A hybridization probe is cleaved by the 5′ nuclease activity of Taq DNA polymerase only if the specific sequence is successfully amplified. Two TaqMan probes are used, one for each allele. TaqMan probe consists of an 18- to 22-bp oligonucleotide probe, which is labeled with a reporter fluorophore at the 5′ end and a quencher fluorophore at the 3′ end. Heterozygote samples for the c.525delT mutation detected by real-time PCR were confirmed by direct sequencing.
Data management
Assuming that the Moroccan population is in Hardy–Weinberg equilibrium, we estimated the frequency of the c.525delT mutation in a homozygous state. The Wright formula was used to calculate the frequency of homozygotes for a given mutation (M) in a population (P) (Wright 1921):
 |
where p is the prevalence of the mutation and F the mean inbreeding coefficient of the population (0.0065) (Jaouad et al., 2009). From the observed c.525delT mutation frequency in our newborns' sample, an estimate of the expected prevalence of LGMD2C in the Moroccan population was obtained by using the following formula:
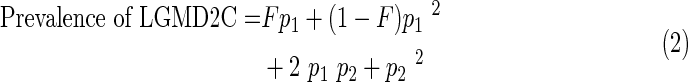 |
where F is the inbreeding coefficient, p1 the prevalence of allele c.525delT in our sample, and p2 the estimated prevalence of all non-c.525delT LGMD2C alleles in the population. An estimate of p2 can be obtained from p1 and the proportion Pc.525delT of c.525delT alleles among all LGMD2C alleles found in the first group of patients:
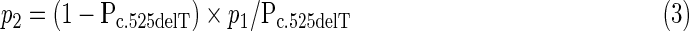 |
Pc.525delT is obtained by c.525delT allele counting among the total of alleles in the first group of patients.
Results
Nineteen patients of the 26 from the first group with a confirmed γ-sarcoglycanopathy were homozygous for c.525delT mutation (Fig. 2), and 2 were be heterozygous. It means that Pc.525delT=40 alleles c.525delT/52 alleles tested (77%). In the second group of 45 patients strongly suspected of having AR-LGMD, 22 females and 7 males were homozygous for the c.525delT mutation, which represents 58 alleles c.525delT among 90 alleles tested (65%). In this same group, 2 females were heterozygous, whereas 12 females and 2 males were not carriers for the c.525delT mutation. In the control group, 1 heterozygote was identified among the 250 cord blood samples tested for the c.525delT mutation (Fig. 3). The carrier frequency for c.525delT mutation was then 1/250 (i.e., 0.4%). Assuming Hardy–Weinberg balance and on the basis of the c.525delT allelic prevalence of 1/500 and a coefficient of inbreeding of 0.0065, the expected frequency of homozygosity for this mutation using formula (1) would thus be 1/250,000+0.0065×1/ 250×499/500=1/58,914. By using formulas (2) and (3) detailed in the Materials and Methods section, the birth prevalence of LGMD2C was then calculated and estimated to be 1/ 20,492.
FIG. 2.

Electropherogram showing the c.525delT mutation in the SGCG gene. (A) Normal sequence. (B) Homozygous state.
FIG. 3.

Curve of amplification of the study of c.525delT mutation. (A) Normal profile. (B) Heterozygous profile.
Discussion
AR-LGMD represents a heterogeneous group of muscular diseases with a wide spectrum of clinical disability ranging from very mild to severe forms. To date, at least 17 loci have been identified (Rosales et al., 2012). The different subtypes can be distinguished by immunohistochemical analysis in a muscle biopsy specimen in order to guide and abbreviate the molecular genetic investigations. There is a geographical difference in their incidence (Bushby and Beckmann, 1995). LGMD2C is one of the most severe forms, with the highest incidence reported in North Africa as a result of a founder mutation in the SGCG gene (McNally et al., 1996). In Morocco, the epidemiology of AR-LGMD is poorly known, but we expect that the prevalence of these disorders would be higher in the Moroccan population than others because of the phenomenon of consanguineous marriages, which reaches up to 15% (Jaouad et al., 2009). However, the scarcity of specialized centers and difficulties that can prevent patients from benefitting from immunohistochemical analysis complicate their diagnosis, management, and genetic counseling.
To bypass these difficulties, we therefore performed a low-cost molecular screening strategy to look for the most prevalent mutation encountered as a cause of LGM2C, the c.525delT mutation. Not surprisingly, 19 of 26 patients (73%), with no staining for γ-sarcoglycan, were homozygous for the c.525delT mutation, as previously reported in other North-African populations (Noguchi et al., 1995). The 2 patients found to be heterozygotes from this group suggest that there might be other LGMD2C mutations in the Moroccan population, which would be interesting to investigate. In the second group of patients with a strongly suggestive diagnosis of AR-LGMD but with no access to immunohistochemical analysis, we showed that nearly 65% of them were homozygous for c.525delT. Finally, on the basis of the identification of 1 heterozygote among 250 newborns (assuming that this cohort was fairly representative of the overall Moroccan population), we estimated that prevalence of LGMD-2C would be 1/20,492. This prevalence is nearly of 1/51,020 in Europeans (Les Cahiers d'Orphanet 2011). The observed difference between our population and others can be explained by the high rate of consanguinity (15.25%) and the increased prevalence of autosomal recessive diseases in Morocco in consanguineous marriages (60%) (Jaouad et al., 2009).
We therefore propose, in our context and on the basis of our preliminary results, that screening for c.525delT could be the first test for AR-LGMD, with a good cost/benefit ratio in public health strategies, until access to immunohistochemical analysis is generalized. This might be useful not only for management of patients, genetic counseling, and prenatal diagnosis in families but also for novel therapeutic approaches and future clinical trials.
Acknowledgments
We thank the patients and their families. We are grateful to all of the staff of the Department of Medical Genetics of the National Institute of Health for their continuous support.
Author Disclosure Statement
No competing financial interests exist.
References
- Anderson LV. (1996)) Optimized protein diagnosis in the autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 6:443–446 [DOI] [PubMed] [Google Scholar]
- Ben Othmane K, Speer MC, Stauffer J, et al. (1995) Evidence for linkage disequilibrium in chromosome 13-linked Duchenne-like muscular dystrophy (LGMD2C). Am J Hum Genet 57:732–734 [PMC free article] [PubMed] [Google Scholar]
- Boito C, Fanin M, Siciliano G, et al. (2003) Novel sarcoglycan gene mutations in a large cohort of Italian patients. J Med Genet 40:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönnemann CG, Finkel RS. (2002) Sarcolemmal proteins and the spectrum of limb-girdle muscular dystrophies. Semin Pediatr Neurol 9:81–99 [DOI] [PubMed] [Google Scholar]
- Bönnemann CG. (2005) Limb-girdle muscular dystrophy in childhood. Pediatr Ann 34:569–577 [DOI] [PubMed] [Google Scholar]
- Bushby KM, Beckmann JS. (1995) The limb-girdle muscular dystrophies: proposal for a new nomenclature. Neuromuscul Disord 5:337e43. [DOI] [PubMed] [Google Scholar]
- El Kerch F, Sbiti A, Azibi K, et al. (2001) La gamma-sarcoglycanopathie par la mutation del521T au Maroc. à propos de 20 cas. Rev Magh Pédiat 11:189–192 [Google Scholar]
- El Kerch F, Sefiani A, Azibi K, et al. (1994) Linkage analysis of families with severe childhood autosomal recessive muscular dystrophy in Morocco indicates genetic homogeneity of the disease in North Africa. J Med Genet 31:342–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaouad IC, Elalaoui SC, Sbiti A, et al. (2009) Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci 41:575–581 [DOI] [PubMed] [Google Scholar]
- Prévalence des maladies rares: Données bibliographiques Les Cahiers d'Orphanet, 2011. Novembre, Numéro 1 Available at: http://www.orpha.net/orphacom/cahiers/docs/FR/Prevalence_des_maladies_rares_par_ordre_alphabetique.pdf (Accessed June17, 2013)
- McNally EM, Passos-Bueno MR, Bönnemann CG, et al. (1996) Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. Am J Hum Genet 59:1040–1047 [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, McNally EM, Ben Othmane K, et al. (1995) Mutations in the dystrophin-associated protein y-sarcoglycan in chromosome 13 muscular dystrophy. Science 270:819–822 [DOI] [PubMed] [Google Scholar]
- Ozawa E, Noguchi S, Mizuno Y, et al. (1998) From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve 21:421–438 [DOI] [PubMed] [Google Scholar]
- Rosales XQ, Tsao CY. (2012) Childhood onset of limb-girdle muscular dystrophy. Pediatr Neurol 46:13–23 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. (1989) Isolation of DNA from mammalian cells. In: Sambrook J, Fritsch EF, Maniatis T. (eds). Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press: pp 917–919 [Google Scholar]
- Trabelsi M, Kavian N, Daoud F, et al. (2008) Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum Genet 16:793–803 [DOI] [PubMed] [Google Scholar]
- Wright S. (1921) Systems of mating. Genetics 6:111–178 [DOI] [PMC free article] [PubMed] [Google Scholar]