

1 Introduction
Campylobacter is the most common bacterial cause of gastroenteritis, worldwide. Since the first description of the disease in the 1970s (Skirrow, 1977), the incidence of human campylobacteriosis in the UK, measured in terms of laboratory reports, has risen steadily, peaking at 57,674 reports in the year 2000; with 46,603 reports in 2006 (http://www.hpa.org.uk). Although generally self limiting, this disease has an important economic impact (Skirrow and Blaser, 1992). More serious complications, such as motor neurone paralysis, arise in 1–2 cases per 100,000 people in the UK and USA (Nachamkin et al., 1998). The disease also has an appreciable, yet less defined, impact in developing countries. Approximately 90% of human infection is caused by C. jejuni, with C. coli accounting for much of the rest (Gillespie et al., 2002).
C. jejuni, and C. coli are Gram-negative curved rods with polar flagella and are highly motile (Ketley, 1997). In common with other members of the genus Campylobacter they are fastidious bacteria that are best isolated in a microaerophilic atmosphere using specific complex media, since they lack many of the genes needed to degrade carbohydrates or amino acids. Unlike most other Campylobacter species, they show optimum growth at 42°C, perhaps as a consequence of their association with avian species. It is thought that where campylobacters become environmentally stressed they enter a viable non-culturable state, in which case pre-incubation in an enrichment broth may help to recover the organism (Humphrey, 1989; Ketley, 1997).
Campylobacter are ubiquitous, being commensal members of the gastrointestinal microbiota of poultry and other farm, animals, as well as many wild species. Such animal infection is rarely symptomatic, but provides sources of contamination, both directly and via the consumption of the afflicted animal. Run-off from farmland leads to the contamination of ground-water sources, acting as a source of infection for animals, birds and humans, if consumed (Hopkins et al., 1984; Peabody et al., 1997). However, human campylobacteriosis is most commonly associated with the consumption of chicken or chicken products (Adak et al., 2005). Ingestion of Campylobacter by humans may cause disease resulting from the invasion of the intestinal epithelial layer, leading to localized inflammation and diarrhea (Young et al., 2007).
The multiple potential reservoirs for human infection, together with the high levels of genetic diversity of these bacteria, have combined to complicate understanding of the relative contributions of different infection sources to the human disease burden – an essential prerequisite to effective disease control. Here, we illustrate how molecular epidemiological techniques, especially those based on nucleotide sequence determination, have contributed to unravelling the epidemiology of these important human pathogens.
2 Campylobacter Typing and Population Structure
Multi-locus sequence typing (MLST) is a method of unambiguously indexing genetic variation among bacterial isolates (Maiden, 2006). For every isolate investigated, nucleotide sequence data are obtained for an internal fragment of each of seven housekeeping loci. Genetic variation in housekeeping genes is indexed as the variation is present in all isolates and is under stabilising selection for the conservation of metabolic function. This allows for the monitoring of long-term evolutionary events (Dingle et al., 2001). Nucleotide sequence lengths of ~500 bp are usually employed, since accurate data can be readily obtained by the use of a single primer extension for each DNA strand. For each locus, every unique gene fragment sequence (or MLST allele) is assigned a unique but arbitrary number, regardless of whether allele differences have occurred as a result of a single or multiple base changes – an important criterion when analyzing highly recombinogenic bacteria (Maiden, 2006). Consequently, each isolate investigated has an “allelic profile” or “sequence type” (ST), consisting of seven integers which, in the case of the Campylobacter scheme, represents 3,309 bp of unique nucleotide sequence from multiple loci around the genome. Genetic relationships between isolates can be determined from these data since closely related isolates have identical STs or allelic profiles differing at few loci, while the profiles of unrelated isolates are different.
Examination of C. jejuni and C. coli collections by MLST has confirmed these organisms to be genetically diverse, with a semi-clonal population structure (Dingle et al., 2001, 2005). Such bacterial populations contain clusters of related isolates but, as in other highly recombinogenic bacteria, phylogenetic relationships among clusters cannot be determined, due to the reassortment of alleles by frequent recombination (Holmes et al., 1999). These clusters are referred to as clonal complexes and in the case of Campylobacter are pragmatically defined as those isolates sharing four or more alleles with a central genotype, after which the complex is named (Dingle et al., 2001). As observed in other bacteria (Maiden, 2006), the central genotypes of Campylobacter clonal complexes have a higher prevalence than other STs in population samples and are stable during global spread over time (Dingle et al., 2002). An example of this is the ST-45 clonal complex, in which the central genotype is the most abundant; with the majority of other STs observed much less frequently, in many cases only once. Clonal complexes have become the main unit of analysis of Campylobacter genotypes for epidemiological investigations (Dingle et al., 2002).
3 Epidemiology of Human Infection
Prior to the development of MLST, immunological typing methods, predominantly targeted to lipooligosacharide (LOS) variants and capsular antigens, were used for the characterization of Campylobacter isolates (Penner et al., 1983) but these methods failed to advance understanding of Campylobacter epidemiology. The reasons for this failure became clear on comparison of serotyping and MLST data. Firstly, particular serotypes can be associated with more than one ST or clonal complex; furthermore, a given genotype can contain isolates expressing various serotypes (Dingle et al., 2002; Wareing et al., 2003). This lack of association of serotype with genotype is due both to recombinational reassortment of antigen genes among genotypes and to phase variation, so that the same isolate may express very different serotypes at different times (Parkhill et al., 2000).
A major advantage of MLST is that the results are highly reproducible and the technique is portable, enabling data collected in different laboratories to be readily compared. MLST data available via the Internet at the PubMLST database website (http://pubmlst.org/Campylobacter) show that while Campylobacter genotyopes have a global distribution, their prevalence among human disease is not uniform worldwide (Dingle et al., 2008). The distribution of clonal complexes among disease isolates from two regions of the UK might be similar, for example (Dingle et al., 2002; Sopwith et al., 2006), but might differ from those seen among disease isolates collected in Australia (Mickan et al., 2007). Intriguingly, the disease isolates from the UK and Australia are more similar to each other than they are to isolates obtained from cases in the Dutch West Indies (Duim et al., 2003) (Fig. 1). These findings probably reflect different dietary infection sources in different countries. Of particular note is the similarity of the genotypes recovered from poultry meat and human disease, which is consistent with contaminated poultry being a major source of human infection (Dingle et al., 2002; Colles et al., 2008).
Fig. 1.

Human clinical isolates in geographically distinct locations. The relative abundance of the clonal complexes detected in Oxfordshire, UK during a one-year study (Dingle et al., 2008). Comparison with the clonal complexes detected in three other studies of human Campylobacter infections in NW England, UK (Sopwith et al., 2006) Curaçao, (Duim et al., 2003) and New South Wales, Australia (Mickan et al., 2007)
Outbreaks of human Campylobacter infection are identified infrequently due to the relatively long incubation period, lack of accurate epidemiological information, and the high incidence and wide distribution of human-disease related central genotypes. The application of a ten-locus typing scheme that combines MLST data and nucleotide sequences of flaA, flaB and porA antigen genes, enhances discrimination among isolates. In a study of 620 isolates collected over a one-year period from Oxfordshire, UK the discriminatory index (DI) was increased from 0.976 achieved by MLST alone to 0.992 by achieved with ten-locus typing. This greatly enhanced the ability to detect outbreaks, although the contribution of such outbreaks to overall disease burden does, indeed, appear to be low (Dingle et al., 2008).
In addition to being useful in the study of human disease epidemiology, clonal complexes have provided information about the association of particular Campylobacter genotypes with particular animal hosts. In an analysis of 814 Campylobacter isolates from diverse sources, including farm and wild animals, as well as human disease, the ST-21 complex was isolated from all the sources; ST-45 and ST-257 complex predominantly from human disease or poultry; ST-61 and ST-48 complexes mainly from human disease, cattle and sheep, whereas ST-177 and ST-179 complexes were exclusively isolated from beach sand, presumably ultimately coming from wild birds. These findings suggest that the frequency distribution of some clonal complexes is in part associated with the environment from which isolates are obtained and that their persistence in particular hosts may be a result of niche adaptation (Dingle et al., 2002).
4 Host Association Studies of Chickens, Geese and Starlings
Understanding patterns of host association of different Campylobacter genotypes is important in elucidating the ecology of this food-borne zoonotic infection. If particular genotypes are associated with particular host species, it becomes possible to use genetic data to attribute cases of human infection to particular host sources. Initial investigations with MLST suggested that Campylobacter genotypes associated with human disease are found in the farm environment (Colles et al., 2003; French et al., 2005), indicating as obvious transmission routes to humans contaminated food that has been inadequately prepared.
Analysis of Campylobacter isolates obtained from 975 individual chickens representing 64 free-range broiler flocks, 331 wild geese and 964 wild starlings at the University farm in Oxfordshire between 2002 and 2005 enabled host association and transmission in a single farm to be investigated (Colles et al., 2008). There were some significant differences in the carriage rate and species distribution of the organism between the broiler chickens and wild birds. The average shedding rate was much higher amongst the broiler chickens (90.4%) compared to the wild geese (50.5%) and wild starlings (30.4%). In addition, C. coli was isolated much more commonly from the broiler chickens (50.9% of isolates) compared to the geese (0.6% of isolates) and starlings (1.7% of isolates). The results were consistent with those from other studies suggesting that C. coli is a later coloniser of chickens than C. jejuni and is thus commonly isolated from free-range fowl and poultry organically reared over a longer period of time (El-Shibiny et al., 2005).
Further comparison of the Campylobacter genotypes recovered revealed strong host association, with little overlap between the different host sources. Only five clonal complexes and four STs overlapped amongst the chickens and wild birds, and only two Campylobacter isolates with identical ST and flaA SVR type were isolated from the chickens and starlings, although the time period was 6 months apart. Both Fisher’s statistic, used to compare the gene flow between Campylobacter populations, and analysis using the software package Structure (Falush et al., 2003) to predict the host source from an allelic profile employing probabilistic methods, supported a strong association of particular Campylobacter genotypes with certain hosts (Fig. 2). This finding was consistent with mapping genealogical data to host source, which shows an overrepresentation of the same genotypes in farm – specifically poultry – sources and human disease (Fig. 3). This association of genotypes with hosts and human disease supports the use of genetic methods to determine major sources of human infection.
Fig. 2.
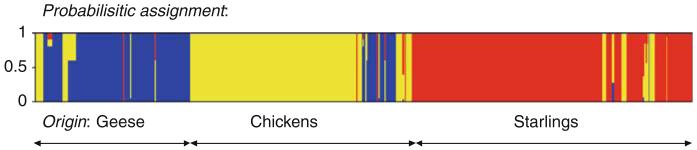
Probabilistic assignment of the Campylobacter jejuni allelic profiles isolated from geese (blue), chickens (yellow) and starlings (red), using the software package Structure. Each vertical bar represents an allelic profile and gives the estimated probability that it comes from each of the sources
Fig. 3.

Comparison of human Campylobacter jejuni isolates with those from poultry, ovine/bovine and environmental sources. The phylogenetic trees were constructed using ClonalFrame software and the ST-based clonal complexes are indicated. The shaded area of the pie charts is proportional to the frequency that each clonal complex occurs in comparison of scaled data sets (n = 765)
Molecular epidemiology can also be used to track infection on-farm. The free-range poultry flocks examined in the Oxfordshire study were on a rolling production system using 16 different plots at two different farm sites. Plots were fallow for 7 weeks between successive flocks and a total of four flocks was raised per plot over the study period. The number of occasions in which an identical Campylobacter genotype was identified on the same plot was unpredictable and no different to that which might expected by chance, despite the inevitable contamination that would result from the previous flocks. Similarly, the extent of ranging behaviour showed no correlation with the shedding rate of Campylobacter or the diversity of genotypes. Higher rates of genotypic diversity were, however, linked with lower growth rates and better hock health. Genotypes were clustered over time, independently of farm site and plot, suggesting that the flocks were contaminated from a common source over a period of time. Taken together these data suggest that wild birds are not a common source of contamination for the free-range poultry but imply that Campylobacter genotypes may be circulating within the poultry industry (Colles et al., 2008).
5 Genetics of Host Association and Speciation
A further application of MLST data has been the investigation of the evolutionary processes underlying host adaptation. A particular conundrum is how distinct genotypes can emerge in the face of the high rates of recombination in Campylobacter populations. Specifically, the persistence of different host-adapted genotypes, and indeed the two species, seem to be inconsistent with the apparently high rates of genetic exchange (Fraser et al., 2007). The existence of distinct groups suggests the presence of effective barriers to gene flow (Lawrence, 2002). This can be (i) mechanistic – imposed by homology dependence of recombination (Fraser et al., 2007) or other factors promoting DNA specificity (Eggleston and West, 1997) such as restriction/modification systems; (ii) ecological – a consequence of physical separation of bacterial populations in distinct niches; (iii) adaptive – implying selection against hybrid genotypes (Zhu et al., 2001). For many bacteria, observed levels of genetic exchange should prevent divergence of subpopulations (Gupta and Maiden, 2001; Cohan, 2002; Hanage et al., 2006).
In Campylobacter there is evidence of the mechanisms of adaptation and it has been suggested that C. jejuni and C. coli are despeciating as a result of expansion into an agricultural niche (Sheppard et al., 2008). As livestock farming became an increasingly important part of human food production, natural barriers to bacterial recombination became less rigid. Animals with undomesticated predecessors, from whom they were separated in the wild, were brought together at high stocking densities with rapid generation times. The convergence of Campylobacter species in this new niche demonstrates that bacterial evolution can occur by mechanisms analogous to those in sexual eukaryotic populations, such as Darwin’s Finches, where ecological factors can generate and maintain incipient species associated with distinct niches (Grant and Grant, 1992; Mallet, 2007).
6 Conclusion – Impact of Molecular Epidemiology on Control of Human Infection
Molecular epidemiological studies have contributed to our understanding of the biology of Campylobacter infection, and indeed help to elucidate where Campylobacter comes from, in a number of important ways. MLST and related typing techniques have provided a robust typing tool that has enabled data from multiple laboratories to be compared. The clonal complex has emerged as a practical, yet biologically based, unit of epidemiological analysis. Enhancement of these data with additional loci encoding more variable genes has provided robust tools for contact tracing and outbreak investigation. MLST has also enabled the association of particular genotypes with particular host species, further confirming the important role of poultry as a source of human disease and indicating that the animals, themselves, have Campylobacter populations distinct from those found among wild birds such as geese and starlings. Finally the data have helped us to understand the evolutionary processes whereby distinct genotypes arise and are maintained.
In practical terms, much emphasis has been placed on reducing the number of Campylobacter organisms reaching the consumer, since is directly proportional to the risk of infection (Newell and Fearnley, 2003; Lindqvist and Lindblad, 2008). In order to achieve this reduction, research has largely concentrated on three main areas; on-farm biosecurity, hygiene during the slaughter process and hygiene during food preparation. Although some progress has been made, further improvement is needed to meet the target set by the UK Food Standards Agency in 2005 to achieve a 50% reduction in Campylobacter-infected UK-produced chickens by 2010. The results of the molecular epidemiological studies suggest that two additional areas could be targeted in the future: improved animal welfare and the impact of intensive farming (Fig. 4) (Humphrey, 2006; Colles et al., 2008; Sheppard et al., 2008). This illustrates how molecular epidemiological and population genetic studies are important in shedding light on the best means to reduce the incidence of this major pathogen.
Fig. 4.
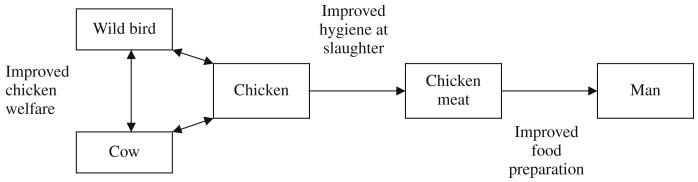
Schematic representation of intervention points for reducing rates of human infection, suggested by molecular epidemiology investigations
References
- Adak GK, Meakins SM, Yip H, Lopman BA, O’Brien SJ. Disease risks from foods, England and Wales, 1996-2000. Emerg Infect Dis. 2005;(11):365–372. doi: 10.3201/eid1103.040191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan FM. What are bacterial species? Annu Rev Microbiol. 2002;(56):457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- Colles FM, Jones K, Harding RM, Maiden MC. Genetic diversity of Campylobacter jejuni isolates from farm animals and the farm environment. Appl Environ Microbiol. 2003;(69):7409–7413. doi: 10.1128/AEM.69.12.7409-7413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colles FM, Jones TA, McCarthy ND, Sheppard SK, Cody AJ, Dingle KE, Dawkins MS, Maiden MC. Campylobacter infection of broiler chickens in a free-range environment. Environ Microbiol. 2008;(10):2042–2050. doi: 10.1111/j.1462-2920.2008.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, Colles FM, Falush D, Maiden MC. Sequence typing and comparison of population biology of Campylobacter coli and Campylobacter jejuni. J Clin Microbiol. 2005;(43):340–347. doi: 10.1128/JCM.43.1.340-347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, McCarthy ND, Cody AJ, Peto TE, Maiden MC. Extended sequence typing of Campylobacter spp. Emerg Infect Dis. 2008;(14):1620–1622. doi: 10.3201/eid1410.071109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, Colles FM, Ure R, Wagenaar J, Duim B, Bolton FJ, Fox AJ, Wareing DRA, Maiden MCJ. Molecular characterization of Campylobacter jejuni clones: a rational basis for epidemiological investigations. Emerg Infect Dis. 2002;(8):949–955. doi: 10.3201/eid0809.02-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, Colles FM, Wareing DRA, Ure R, Fox AJ, Bolton FJ, Bootsma HJ, Willems RJL, Urwin R, Maiden MCJ. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. 2001;(39):14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duim B, Godschalk PC, van den Braak N, et al. Molecular evidence for dissemination of unique Campylobacter jejuni clones in Curacao, Netherlands Antilles. J Clin Microbiol. 2003;(41):5593–5597. doi: 10.1128/JCM.41.12.5593-5597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggleston AK, West SC. Recombination initiation: easy as A, B, C, D… chi? Curr Biol. 1997;(7):R745–R749. doi: 10.1016/s0960-9822(06)00394-0. [DOI] [PubMed] [Google Scholar]
- El-Shibiny A, Connerton PL, Connerton IF. Enumeration and diversity of campylobacters and bacteriophages isolated during the rearing cycles of free-range and organic chickens. Appl Environ Microbiol. 2005;(71):1259–1266. doi: 10.1128/AEM.71.3.1259-1266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;(164):1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C, Hanage WP, Spratt BG. Recombination and the nature of bacterial speciation. Science. 2007;(315):476–480. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French N, Barrigas M, Brown P, Ribiero P, Williams N, Leatherbarrow H, Birtles R, Bolton E, Fearnhead P, Fox A. Spatial epidemiology and natural population structure of Campylobacter jejuni colonizing a farmland ecosystem. Environ Microbiol. 2005;(7):1116–1126. doi: 10.1111/j.1462-2920.2005.00782.x. [DOI] [PubMed] [Google Scholar]
- Gillespie IA, O’Brien SJ, Frost JA, Adak GK, Horby P, Swan AV, Painter MJ, Neal KR, Collaborators C.S.S.S. A case-case comparison of Campylobacter coli and Campylobacter jejuni infection: a tool for generating hypotheses. Emerg Infect Dis. 2002;(8):937–942. doi: 10.3201/eid0809.10.3201/eid0809.010187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PR, Grant BR. Hybridization of bird species. Science. 1992;(256):193–197. doi: 10.1126/science.256.5054.193. [DOI] [PubMed] [Google Scholar]
- Gupta S, Maiden MCJ. Exploring the evolution of diversity in pathogen populations. Trends Microbiol. 2001;(9):181–192. doi: 10.1016/s0966-842x(01)01986-2. [DOI] [PubMed] [Google Scholar]
- Hanage WP, Spratt BG, Turner KM, Fraser C. Modelling bacterial speciation. Phil Trans Roy Soc Lond B Biol Sci. 2006;(361):2039–2044. doi: 10.1098/rstb.2006.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Urwin R, Maiden MCJ. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol. 1999;(16):741–749. doi: 10.1093/oxfordjournals.molbev.a026159. [DOI] [PubMed] [Google Scholar]
- Hopkins RS, Olmsted R, Istre GR. Endemic Campylobacter jejuni infection in Colorado: identified risk factors. Am J Pub Health. 1984;(74):249–250. doi: 10.2105/ajph.74.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey T. Are happy chickens safer chickens? Poultry welfare and disease susceptibility. Br Poult Sci. 2006;(47):379–391. doi: 10.1080/00071660600829084. [DOI] [PubMed] [Google Scholar]
- Humphrey TJ. An appraisal of the efficacy of pre-enrichment for the isolation of Campylobacter jejuni from water and food. J Appl Bacteriol. 1989;(66):119–126. doi: 10.1111/j.1365-2672.1989.tb02461.x. [DOI] [PubMed] [Google Scholar]
- Ketley JM. Pathogenesis of enteric infection by Campylobacter. Microbiology. 1997;(143):5–21. doi: 10.1099/00221287-143-1-5. [DOI] [PubMed] [Google Scholar]
- Lawrence JG. Gene transfer in bacteria: speciation without species? Theor Popul Biol. 2002;(61):449–460. doi: 10.1006/tpbi.2002.1587. [DOI] [PubMed] [Google Scholar]
- Lindqvist R, Lindblad M. Quantitative risk assessment of thermophilic Campylobacter spp. and cross-contamination during handling of raw broiler chickens evaluating strategies at the producer level to reduce human campylobacteriosis in Sweden. Int J Food Microbiol. 2008;(121):41–52. doi: 10.1016/j.ijfoodmicro.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;(60):561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybrid speciation. Nature. 2007;(446):279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- Mickan L, Doyle R, Valcanis M, Dingle KE, Unicomb L, Lanser J. Multilocus sequence typing of Campylobacter jejuni isolates from New South Wales, Australia. J Appl Microbiol. 2007;(102):144–152. doi: 10.1111/j.1365-2672.2006.03049.x. [DOI] [PubMed] [Google Scholar]
- Nachamkin I, Allos BM, Ho T. Campylobacter species and Guillain-Barré syndrome. Clin Microbiol Rev. 1998;(11):555–567. doi: 10.1128/cmr.11.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell DG, Fearnley C. Sources of Campylobacter colonization in broiler chickens. Appl Environ Microbiol. 2003;(69):4343–4351. doi: 10.1128/AEM.69.8.4343-4351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;(403):665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- Peabody R, Ryan MJ, Wall PG. Outbreaks of Campylobacter infection: rare events for a common pathogen. CDR. 1997;(7):R33–R37. [PubMed] [Google Scholar]
- Penner JL, Hennessy JN, Congi RV. Serotyping of Campylobacter jejuni andCampylobacter coli on the basis of thermostable antigens. Eur J Clin Microbiol. 1983;(2):378–383. doi: 10.1007/BF02019474. [DOI] [PubMed] [Google Scholar]
- Sheppard SK, McCarthy ND, Falush D, Maiden MC. Convergence of Campylobacter species: implications for bacterial evolution. Science. 2008;(320):237–239. doi: 10.1126/science.1155532. [DOI] [PubMed] [Google Scholar]
- Skirrow MB. Campylobacter enteritis: a “new” disease. BMJ. 1977;(2):9–11. doi: 10.1136/bmj.2.6078.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirrow MB, Blaser MJ. In: Clinical and Epidemiological Considerations. Tompkins LS, editor. American Society for Microbiology; Washington, DC: 1992. pp. 3–8. [Google Scholar]
- Sopwith W, Birtles A, Matthews M, Fox A, Gee S, Painter M, Regan M, Syed Q, Bolton E. Campylobacter jejuni multilocus sequence types in humans, northwest England, 2003-2004. Emerg Infect Dis. 2006;(12):1500–1507. doi: 10.3201/eid1210.060048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareing DR, Ure R, Colles FM, Bolton FJ, Fox AJ, Maiden MC, Dingle KE. Reference isolates for the clonal complexes of Campylobacter jejuni. Lett Appl Microbiol. 2003;(36):106–110. doi: 10.1046/j.1472-765x.2003.01270.x. [DOI] [PubMed] [Google Scholar]
- Young KT, Davis LM, Dirita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol. 2007;(5):665–679. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- Zhu P, van der Ende A, Falush D, et al. Fit genotypes and escape variants of subgroup III Neisseria meningitidis during three pandemics of epidemic meningitis. Proc Natl Acad Sci USA. 2001;(98):5234–5239. doi: 10.1073/pnas.061386098. [DOI] [PMC free article] [PubMed] [Google Scholar]