

ABSTRACT
“Candidatus Synechococcus spongiarum” is a cyanobacterial symbiont widely distributed in sponges, but its functions at the genome level remain unknown. Here, we obtained the draft genome (1.66 Mbp, 90% estimated genome recovery) of “Ca. Synechococcus spongiarum” strain SH4 inhabiting the Red Sea sponge Carteriospongia foliascens. Phylogenomic analysis revealed a high dissimilarity between SH4 and free-living cyanobacterial strains. Essential functions, such as photosynthesis, the citric acid cycle, and DNA replication, were detected in SH4. Eukaryoticlike domains that play important roles in sponge-symbiont interactions were identified exclusively in the symbiont. However, SH4 could not biosynthesize methionine and polyamines and had lost partial genes encoding low-molecular-weight peptides of the photosynthesis complex, antioxidant enzymes, DNA repair enzymes, and proteins involved in resistance to environmental toxins and in biosynthesis of capsular and extracellular polysaccharides. These genetic modifications imply that “Ca. Synechococcus spongiarum” SH4 represents a low-light-adapted cyanobacterial symbiont and has undergone genome streamlining to adapt to the sponge’s mild intercellular environment.
IMPORTANCE
Although the diversity of sponge-associated microbes has been widely studied, genome-level research on sponge symbionts and their symbiotic mechanisms is rare because they are unculturable. “Candidatus Synechococcus spongiarum” is a widely distributed uncultivated cyanobacterial sponge symbiont. The genome of this symbiont will help to characterize its evolutionary relationship and functional dissimilarity to closely related free-living cyanobacterial strains. Knowledge of its adaptive mechanism to the sponge host also depends on the genome-level research. The data presented here provided an alternative strategy to obtain the draft genome of “Ca. Synechococcus spongiarum” strain SH4 and provide insight into its evolutionary and functional features.
INTRODUCTION
As one of the oldest, most primitive metazoans, sponges are distributed globally and play important ecological roles (1–3). The association of symbiotic microbes with the sponge was identified several decades ago (4). Since then, sponge-associated microbial communities and their diversity have been studied extensively (5). Pyrosequencing techniques further facilitated the investigation of sponge-associated microbes (6, 7). A recent study reported up to 32 bacterial phyla and candidate phyla in sponges (7). To some extent, sponge-associated microbial communities showed sponge-species specificity and tropical-subtropical dissimilarity (6, 7). In addition, dissimilarity in the composition of microbial communities between sponges with high and low microbial abundance has been identified (8).
Diverse symbiotic microbes in sponges function in nitrogen fixation, nitrification, photosynthesis, and sulfate reduction and affect the health, ecological distribution, and evolutionary processes of the host (5, 9). However, most sponge symbionts are unculturable and some fall into sponge-specific clusters (10), which makes it difficult to understand their functions and symbiotic mechanisms at the genome level. To date, researchers have only obtained one complete genome of sponge symbiotic microbes that belong to the psychrophilic crenarcheon Cenarchaeum symbiosum (11). On the other hand, a draft genome of an uncultured deltaproteobacterium in association with the sponge Cymbastela concentrica was extracted from metagenomic data using the tetranucleotide frequency method (12). The single-cell method has also been used to study the genomes of poribacterial symbionts in marine sponges (13). However, our knowledge of sponge symbionts at the genome level remains limited.
Cyanobacteria represent one of the most common members of the sponge-associated microbial communities and are considered to play important roles in photosynthesis, nitrogen fixation, UV protection, and defensive toxin production (5, 14). Identified cyanobacterial sponge symbionts belong to Synechocystis, Aphanocapsa, Anabaena, Oscillatoria, and “Candidatus Synechococcus spongiarum” (5). “Ca. Synechococcus spongiarum,” proposed by Usher et al. (15), was found in at least 40 sponge species and represents the largest sponge-specific cluster to date (10). Electron and fluorescence micrographs of “Ca. Synechococcus spongiarum” symbionts revealed their presence in the intercellular environment of host sponges and provided evidence for their interaction with sponge amebocytes (16). In addition, vertical transmission of “Ca. Synechococcus spongiarum” from parents to offspring has been reported (17). Although the genetic differentiation of “Ca. Synechococcus spongiarum” is considered to be very low among populations from different host species or geographical regions according to the similarity of the 16S rRNA genes, their internal transcribed spacer (ITS) region displays high variations (18). The functional properties of this highly prevalent sponge symbiont and its symbiotic interaction with sponges remain unclear. Marine picocyanobacteria of the genera Prochlorococcus and Synechococcus overwhelmingly dominate the picophytoplankton of the world ocean and contribute vitally to global primary production (19, 20). The features that distinguish “Ca. Synechococcus spongiarum” from free-living picocyanobacteria and the mechanism underlying its ability to adapt to the symbiotic partnership are still unclear. Genomic analyses may provide answers to these questions.
According to our previous data, “Ca. Synechococcus spongiarum” is highly abundant in the sponge Carteriospongia foliascens, collected from the Red Sea, and represents the dominant cyanobacterial symbiont (see Fig. S1 in the supplemental material), thus permitting the extraction of its genome from the microbial community. The development of bioinformatics also makes it feasible to obtain genomic sequences of uncultured bacteria from multiple metagenomes by genome binning based on differential coverage and tetranucleotide frequency (12, 21). Here, we report a draft genome of “Ca. Synechococcus spongiarum” strain SH4 extracted from metagenomic data. Using the extracted genome, we examined the evolutionary relationship and functional dissimilarity of “Ca. Synechococcus spongiarum” SH4 with closely related free-living cyanobacterial strains and so pave the way to understand its adaptive mechanism to the sponge-symbiont partnership.
RESULTS AND DISCUSSION
Genome binning.
Metagenomic DNA of the microbial community in the Red Sea sponge C. foliascens was subjected to 454 pyrosequencing, and metagenomic reads were assembled using GS De Novo Assembler (Newbler). A full-length cyanobacterial 16S rRNA gene was predicted from the assembled metagenomic contigs and completely matched with the dominant cyanobacteria (represented by operational taxonomic unit 1691 [OTU1691]; see Fig. S1 in the supplemental material) in the sponge C. foliascens. A phylogenetic tree based on the predicted 16S rRNA gene indicated that this symbiont is distantly related to free-living Synechococcus and Prochlorococcus species and that it groups together with sequences from “Ca. Synechococcus spongiarum” derived from various sponge species (15, 18) and shares more than 99% identity with them (Fig. 1). This cyanobacterial symbiont in the sponge C. foliascens was designated “Candidatus Synechococcus spongiarum” strain SH4. Analysis of 16S rRNA genes in 454 metagenomic reads revealed a consistently high abundance of the symbiont “Ca. Synechococcus spongiarum” SH4 (see Fig. S2 in the supplemental material). Among the 16S rRNA reads, 67% (158/236) were assigned to Cyanobacteria, of which 155 reads matched the 16S rRNA gene of SH4 with more than 99% identity. These reads were thus sorted to “Ca. Synechococcus spongiarum” SH4. The high abundance of SH4 in the metagenome data implied that the genome coverage of SH4 in the assembled contigs (average coverage, ~28×) was much higher than those of other sponge-associated microbes, which facilitated distinguishing contigs of SH4 from the others.
FIG 1 .

Neighbor-joining phylogenetic tree of strain SH4 and closely related cyanobacterial strains based on partial 16S rRNA genes (1,100 bp). CSS, cluster of “Ca. Synechococcus spongiarum.” Bootstrap values based on 1,000 replications are shown as percentages at branch angles. Scale bar indicates 1% estimated sequence divergence.
Using a combination of genome coverage and tetranucleotide frequency patterns and taking the GC content and essential genes into account (21), we extracted the genome of “Ca. Synechococcus spongiarum” SH4 from the assembled metagenomic contigs (Table 1; see Fig. S3 in the supplemental material). The draft genome of SH4 was 1.6 Mbp in length, with a GC content of 63.4%. A total of 96 out of 106 single-copy, essential genes were identified in the draft genome, assuming a genome recovery of 90%. Although the genome recovery was not effectively complete, the draft genome is good enough to permit analysis of the functional properties of the sponge symbiont “Ca. Synechococcus spongiarum.” To validate the occurrence of genome reduction, the genes of interest that were lacking in the SH4 draft genome were checked again in the remaining assembled metagenomic contigs with lengths longer than 500 bp and 454 metagenomic coverage higher than 8×.
TABLE 1 .
General features and functional comparison of “Candidatus Synechococcus spongiarum” SH4 and related picocyanobacterial strains
| Genome feature or gene function | Value for indicated taxona |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Genome recovery | ~90% | F | D | D | F | F | D | F |
| Genome size (Mbp) | 1.66 | 2.22 | 2.58 | 3.04 | 2.61 | 1.75 | 2.83 | 3.34 |
| No. of essential genes | 96 | 106 | 104 | 106 | 105 | 106 | 105 | 106 |
| % GC content | 63.4 | 60.8 | 64.5 | 65.4 | 52.5 | 36.4 | 68.7 | 68.7 |
| % Coding density | 87.5 | 94.2 | 92.7 | 88.4 | 87.2 | 89.3 | 87.6 | 89.5 |
| No. of genes in KEGG pathways | ||||||||
| Photosynthesis | 47 | 60 | 64 | 61 | 64 | 53 | 57 | 59 |
| Antenna proteins | 24 | 28 | 18 | 23 | 28 | 8 | 21 | 17 |
| TCA cycles | 10 | 9 | 8 | 11 | 8 | 11 | 11 | 11 |
| DNA replication | 13 | 13 | 13 | 14 | 13 | 13 | 15 | 14 |
| DNA repair | 31 | 38 | 41 | 47 | 38 | 36 | 40 | 41 |
| Cysteine/methionine metabolism | 10 | 21 | 22 | 24 | 19 | 17 | 21 | 24 |
| No. of genes in SEED/subsystems | ||||||||
| Response to oxidative stress | 12 | 28 | 26 | 32 | 26 | 21 | 29 | 31 |
| Resistance to antibiotics and toxins | 8 | 22 | 22 | 28 | 23 | 9 | 30 | 36 |
| CPS and EPS biosynthesis | 3 | 13 | 13 | 16 | 10 | 8 | 19 | 17 |
| Gram-negative cell wall | 7 | 14 | 12 | 14 | 9 | 10 | 20 | 17 |
| Peptidoglycan biosynthesis | 16 | 15 | 15 | 17 | 15 | 15 | 17 | 16 |
| Eukaryotic-like domain | ||||||||
| Fibronectin type III domain | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| AR | 17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TPR | 3 | 9 | 7 | 17 | 1 | 6 | 12 | 14 |
| LRR | 9 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. strain RCC307; 3, Synechococcus sp. strain RS9917; 4, Synechococcus sp. strain WH 5701; 5, Synechococcus sp. strain CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. strain PCC 7001; 8, Cyanobium gracile PCC 6307. Genome recovery: F, finished genome; D, draft genome.
Phylogenomic inference.
The explosive growth of genomic data has provided conserved marker genes as alternatives to 16S rRNA genes for phylogenetic inference (22). Here, a phylogenomic tree based on 31 concatenated marker genes (Fig. 2a) revealed the evolutionary distinction of SH4 from picocyanobacteria of the genera Synechococcus, Prochlorococcus, and Cyanobium cluster Synechococcus and supported its evolutionary divergence from free-living cyanobacteria (18). The bipartition point where SH4 branched from other picocyanobacteria suggested that SH4 was an independent cyanobacterial lineage that had adapted to the symbiotic lifestyle for a long period of time. This is in accord with previous findings demonstrating that these symbionts are vertically inherited from the parent sponges (17). Average nucleotide identity (ANI) and tetranucleotide frequency are powerful tools for comparison analysis of genome composition (23). SH4 showed higher ANIs and tetranucleotide frequency similarity with Synechococcus and Cyanobium cluster Synechococcus than to Prochlorococcus (Fig. 2b). In addition, SH4 represented one of the cyanobacterial strains with the highest GC contents (>60%) and was more similar to Synechococcus and Cyanobium cluster Synechococcus than to Prochlorococcus (Fig. 2c). These results indicated that SH4 was more closely related to free-living Synechococcus than to Prochlorococcus. Accordingly, closely related strains affiliated with Synechococcus and Cyanobium cluster Synechococcus were selected for the genome-level functional comparison with SH4 (Table 1). The low-light-adapted cyanobacterium Prochlorococcus marinus strain SS120 (CCMP1375 in Table 1), with a nearly minimal oxyphototrophic genome, was also included in the comparison analysis.
FIG 2 .

Phylogenomic inference of “Ca. Synechococcus spongiarum” SH4. (a) Maximum-likelihood phylogenomic tree using 31 concatenated conserved proteins of SH4 and cyanobacterial strains. The branches were colored according to the bootstrap values, ranging from purple (80%) to red (100%). Scale bar indicates 10% estimated sequence divergence. (b) ANI values plus correlation indexes of tetranucleotide frequency between SH4 and closely related Synechococcus, Prochlorococcus, Cyanobium cluster Synechococcus, and other cyanobacterial strains revealed differences in their genome composition. (c) GC content and genome size of SH4 and other cyanobacterial strains.
Functional features at the genomic level.
The numbers of genes present in selected pathways of “Ca. Synechococcus spongiarum” SH4 and free-living picocyanobacteria strains are presented in Table 1; these data suggest the near completeness of key functional pathways, including photosynthesis, the citric acid cycle (tricarboxylic acid [TCA] cycle), DNA replication, and peptidoglycan biosynthesis. As a sponge symbiont, SH4 also displayed unique symbiotic features, with the highlight being genome streamlining following the loss of unnecessary genes in several pathways (Table 1). Previous studies have revealed the distribution and evolutionary divergence of the sponge symbiont “Ca. Synechococcus spongiarum” using molecular ecology techniques (18). Although “Ca. Synechococcus spongiarum” was thought to enhance host metabolism and ecological fitness by providing a photosynthesis-derived carbon source, its functional properties at the genome level remain unclear. Here, the extracted genome provided direct insights into the carbon and energy metabolism of “Ca. Synechococcus spongiarum” SH4 and its symbiotic adaptation to the sponge host.
Host-symbiont interaction.
Proteins with ankyrin repeats (ARs), leucine-rich repeats (LRRs), and fibronectin type III domains were enriched in SH4 but rare in free-living cyanobacteria (Table 1). Proteins with eukaryoticlike domains, such as ARs, tetratricopeptide repeats (TPRs), LRRs, NHL repeats (PF01436), and fibronectin type III, have been reported to be enriched in sponge symbiotic microbes (12, 24) and were suggested to modulate host behavior by interfering with eukaryotic protein-protein interactions. Specially, AR proteins from sponge symbionts modulate amoebal phagocytosis and might help symbionts escape digestion by the sponge host (25). The enrichment of these domains in SH4 is consistent with the role of “Ca. Synechococcus spongiarum” as a sponge symbiont. Interestingly, the number of TPR proteins was lower in SH4 than in several free-living cyanobacterial strains (Table 1). The TPR motif was originally identified in yeast (26) but was recently found in a wide variety of prokaryotes and suggested to be involved in numerous cell processes (27). The hypothesis that TPR functions as a symbiotic factor in the sponge-symbiont interaction requires further careful evaluation.
Amino acid metabolism.
Although obligatory symbiotic bacteria tend to lose essential metabolic pathways that are required for free-living organisms, especially those responsible for amino acid metabolism (28, 29), the numbers of genes in most of the amino acid metabolism pathways of “Ca. Synechococcus spongiarum” SH4 were similar to the numbers in free-living cyanobacterial strains (see Fig. S4 in the supplemental material). Surprisingly, the cysteine and methionine metabolism pathway was dramatically reduced (Table 1). In-depth analysis showed that there was no enzyme for the de novo biosynthesis of the methionine precursor (homocysteine), although methionine synthase (metH) was present in the SH4 draft genome (Fig. 3). In addition to de novo biosynthesis, the methionine salvage pathway is an important metabolic pathway for maintaining the concentration of l-methionine in bacteria (30). Key genes in the methionine salvage pathway, including S-adenosylmethionine decarboxylase (speD), spermidine synthase (speE), methylthioribose-1-phosphate isomerase (mtnA), and 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase (mtnD), were lost (Fig. 3). The lack of de novo biosynthesis and salvage of methionine suggested that the essential methionine might be provided by exogenous sources. In the methionine salvage pathway, SpeD and SpeE are two enzymes responsible for the biosynthesis of spermidine, a prevalent polyamine found in bacteria (31). Previous studies have shown that polyamines in bacteria play important roles in optimal cell growth, signaling cell differentiation, DNA protection, biofilm formation, and antibiotic resistance (31). High-performance liquid chromatograph analysis revealed that polyamines were widely distributed in cyanobacteria, which indicates their important roles for the cyanobacteria (32). SH4 lives in the sponge intercellular environment (18) and likely acquires exogenous polyamines therein. “Ca. Synechococcus spongiarum” has been shown to interact with host amebocytes (18), a mobile cell responsible for food digestion. Amebocytes might digest food and release nutrients to satisfy the necessities, such as polyamines and methionine, for the cyanobacterial symbiont. Since other symbiotic bacteria were also found in the sponge host (Fig. S1), they might be an alternative source of these essential chemicals for “Ca. Synechococcus spongiarum.”
FIG 3 .

Schematic overview of the methionine metabolism pathway in “Ca. Synechococcus spongiarum” SH4. Red and green labels indicate the absence and presence, respectively, of that enzyme in SH4.
Photosynthetic system.
“Ca. Synechococcus spongiarum” has been reported to contain phycocyanin and phycoerythrin (15). Here, we showed that “Ca. Synechococcus spongiarum” SH4 contains genes encoding all three types of antenna proteins (phycocyanin, phycoerythrin, and allophycocyanin) (Fig. 4), which suggests that this symbiont absorbs a wide spectrum of light for photosynthesis. The eight subunits of F-type ATPase were identified. Compared to free-living cyanobacterial strains, however, genes in photosystem II (PSII), including psbP, psbI, psbK, psbM, and psbY, were missing (Fig. 4). In further analysis, these genes could not be detected in the entire assemblage of metagenomic contigs or raw pyrosequencing reads. PsbP, together with PsbO, PsbQ, PsbU, and PsbV, form the oxygen-evolving complex (OEC) of PSII in cyanobacteria. This complex oxidizes water to provide protons for photosystem I (PSI). Synechocystis sp. strain PCC 6803 mutants with inactive PsbP exhibit reduced photoautotrophic growth (33). PsbI, PsbK, PsbM, and PsbY are low-molecular-weight peptides involved in the assembly, stabilization, dimerization, and photoprotection of the photosynthetic center of PSII (34). The loss of PsbP and low-molecular-weight peptides implied that the PSII complex of “Ca. Synechococcus spongiarum” SH4 is less stable than those of free-living strains and may represent a low-light-adapted photosynthetic system (35). Several genes encoding low-molecular-weight peptides in the PSI complex and cytochrome b6/f were also not found in SH4 (Fig. 4). The abnormalities observed in the photosynthetic system of “Ca. Synechococcus spongiarum” SH4 might represent a protective mechanism against damage caused by a high dosage of photosynthesis-derived oxygen and/or oxidative stress to the sponge host (36).
FIG 4 .
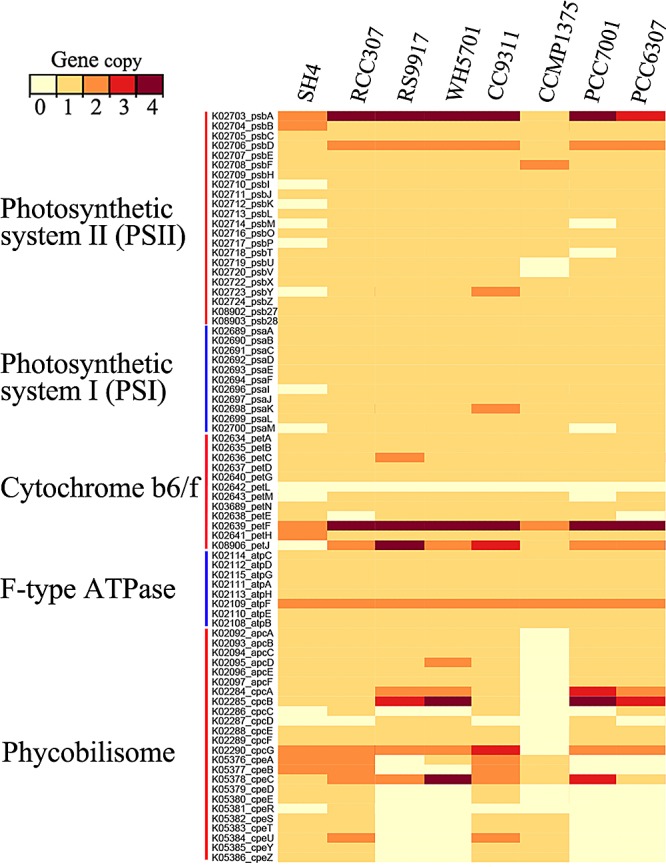
Abundance of photosynthetic genes in “Ca. Synechococcus spongiarum” SH4 and related cyanobacteria strains based on KEGG orthology annotation. The identities of the cyanobacterial strains are described in Table 1.
Resistance to oxidative stress.
Reactive oxygen species (ROS) are by-products of aerobic metabolism and can cause intracellular oxidative damage. ROS generated by the photosynthetic electron transport chain pose a significant threat to photosynthetic organisms, such as cyanobacteria (36). The ability to rapidly perceive ROS and initiate antioxidant defense is crucial for the survival of these organisms. In cyanobacterial strains, antioxidant enzymes play important roles in resistance to oxidative stress (36). However, “Ca. Synechococcus spongiarum” SH4, as a photosynthetic microbe, has lost several antioxidant enzymes, including superoxide dismutase (SOD), glutathione peroxidase (GPX), and DNA-binding protein (Dps) (Fig. 5; see Table S1 in the supplemental material). P. marinus strain CCMP1375, a low-light-adapted picocyanobacteria with a nearly minimal oxyphototrophic genome (35), also lacks SOD and Dps (Fig. 5) and escapes from oxidative damage through living at the bottom of the illuminated layer (35). The observed loss of antioxidant enzymes consistently confirmed that SH4 is a low-light-adapted organism. However, different from the antioxidant mechanism used by P. marinus CCMP1375 (35), “Ca. Synechococcus spongiarum” SH4 lives in the mild intercellular environment of the sponge host (16), which provides the barrier of the sponge body to forbid too much sunlight arriving at the cyanobacterial symbiont, and thus it avoids the oxidative damage caused by a highly efficient photosynthesis process.
FIG 5 .

Genes of “Ca. Synechococcus spongiarum” SH4 and related cyanobacterial strains important for resistance to oxidative stress, antibiotics, and environmental toxins based on SEED/Subsystems annotation. See details in Tables S1 and S2. The identities of the cyanobacterial strains are described in Table 1.
Resistance to antibiotics and toxic compounds.
In addition to the loss of genes encoding antioxidant enzymes, there was also a dramatic reduction of genes involved in resistance to antibiotics and environmental toxins in “Ca. Synechococcus spongiarum” SH4 (Fig. 5; see Table S2 in the supplemental material). There was a depletion of genes encoding proteins involved in arsenic resistance, multidrug resistance efflux pumps, integrase, beta-lactamase, and the negative regulator of beta-lactamase. Genes encoding cobalt-zinc-cadmium and methicillin resistance were also dramatically reduced in “Ca. Synechococcus spongiarum” SH4 compared to their occurrence in other free-living cyanobacterial strains. Interestingly, a large fraction of these genes were also lost in the genome of Prochlorococcus marinus CCMP1375. Cyanobacteria are a large and highly diverse group of photosynthetic prokaryotes which can adapt to various habitats, including those containing natural and artificial antibiotics and heavy metals (37). Resistance to these toxins in open water is important for the survival of these organisms (38). However, “Ca. Synechococcus spongiarum,” which inhabits the mild intercellular environment of the sponge host (16), can evade these toxins via the barriers of the sponge host. Accordingly, genes involved in resistance to environmental factors are not required and were lost during the evolutionary development of this symbiont.
Cell wall and capsule composition.
Similar to the case for closely related free-living cyanobacterial strains, most of the genes responsible for the biosynthesis of peptidoglycan and lipopolysaccharide (LPS) could be found in SH4 (Table 1). The presence of these genes allows the formation of a rigid cell wall and is consistent with the characteristic spiral thylakoid membrane of “Ca. Synechococcus spongiarum” observed by transmission electron microscopy (16). Interestingly, genes responsible for the biosynthesis of capsular polysaccharide (CPS) and extracellular polysaccharides (EPS) were almost completely lost (Table 1). CPS and EPS are extracellular products of a wide range of microorganisms, including cyanobacteria (39), and play important roles, such as protection against environmental stresses (40), biofilm formation (41), and survival against phagocytosis or antibiotics (42, 43). The absence of CPS and EPS further indicated that SH4 has a low resistance to environmental stresses. However, this characteristic is likely to diminish the barrier between the symbiont and sponge cells, thus benefiting sponge-symbiont interactions and nutrient exchange.
DNA replication and repair.
Just like other reported bacterial symbionts (28), “Ca. Synechococcus spongiarum” SH4 retained the same set of genes for DNA replication as are found in closely related free-living cyanobacterial strains, but genes for DNA repair capabilities are limited (Table 1). Although the base excision repair and nucleotide repair pathway were found in SH4, the exonuclease Exo VII complex in the mismatch repair pathway, the exonuclease V complex (RecBCD) in the homologous recombination pathway, and ATP-dependent DNA ligase were not detected (Table 1; see Table S3 in the supplemental material). There are also reports of a reduction of DNA repair genes in the symbiotic cyanobacterium UCYN-A (44) and in the cyanobacterium-originating inclusions termed chromatophores (28). The absence of DNA repair genes in “Ca. Synechococcus spongiarum” SH4, similar to many cases of bacterial symbionts with extraordinarily small genomes (45), likely facilitates the evolution of the genome to adapt to the symbiotic partnership.
Overview of the possible lifestyle of “Ca. Synechococcus spongiarum” SH4.
Based on the analysis of the extracted draft genome and its comparison with those of free-living picocyanobacteria, we proposed schematic functional features and adaptive schemes of “Ca. Synechococcus spongiarum” SH4 to the sponge-symbiont partnership (Fig. 6). Proteins with eukaryoticlike domains were identified in SH4, which is consistent with the role of “Ca. Synechococcus spongiarum” as a sponge symbiont. Genes involved in the biosynthesis of methionine and spermidine were lost, suggesting that “Ca. Synechococcus spongiarum” depends on the sponge host and/or the other sponge symbionts for essential nutrients and chemical factors. The presence of a functional photosynthesis pathway in this symbiont guarantees a steady carbon supply to the host and ensures its ecological success (5). However, the photosynthetic system in SH4 might be unstable and has a low efficiency due to the reduction of PsbP in the OEC complex and the loss of several low-molecular-weight peptides. Furthermore, SH4 should have a low resistance to oxidative stress because of the loss of several antioxidant enzymes. These features indicate that “Ca. Synechococcus spongiarum,” similar to Prochlorococcus marinus SS120, should be a low-light-adapted picocyanobacterium but uses the alternative strategy of symbiosis for adaptation to low light (35). “Ca. Synechococcus spongiarum” also had a low resistance to environmental antibiotics and toxins, which may be further compromised by the defect in the biosynthesis of CPS and EPS. These features force the symbiont to inhabit the mild intercellular environment of the host. However, the defect in the biosynthesis of CPS and EPS may represent a mechanism used to diminish the barrier between symbiont and sponge cells to benefit sponge-symbiont interactions and nutrient exchange. In addition, the loss of DNA repair genes may play roles in facilitating the genome evolution of “Ca. Synechococcus spongiarum” SH4 to adapt to the sponge-symbiont partnership.
FIG 6 .

Schematic of mode of life of the sponge symbiont “Ca. Synechococcus spongiarum.” The schematic figure was deduced from the genomic analysis of the draft genome of strain SH4.
Summary.
Picocyanobacteria in the genera Prochlorococcus and Synechococcus numerically dominate the picophytoplankton communities of the world’s ocean (19, 20). During their adaptation to open ocean and coastal environments, these organisms have overcome various environmental stresses (35, 46). In contrast to free-living picocyanobacteria, “Ca. Synechococcus spongiarum” is a sponge symbiont. The exclusive detection of “Ca. Synechococcus spongiarum” in sponges (18), their vertical transmission between generations (17), and their large phylogenomic dissimilarity to free-living picocyanobacteria (Fig. 2) suggest an intimate symbiotic relationship between “Ca. Synechococcus spongiarum” and the sponge host. The draft genome of “Ca. Synechococcus spongiarum” SH4 provided further insight into the adaptive mechanism of this intercellular symbiont to live in the sponge host (Fig. 6).
Although the draft genome is estimated to have a recovery of 90% and is not effectively completed, the absence of certain genes has been confirmed through searching against the entire assemblage of metagenomic contigs. However, the incomplete genome precludes the detection of several other potentially symbiotic features, such as transposase-driven genome rearrangement and horizontal gene transfer (45). The recovery of an effectively complete genome by combining a metagenome and a single-cell-derived genome, perhaps even using the bacterial artificial chromosome (BAC) library method, should further elucidate this symbiotic partnership. According to the observed intimate interdependence of the symbiont with the sponge and the phylogenomic dissimilarity with free-living picocyanobacteria, our study suggests that the symbiotic partnership between “Ca. Synechococcus spongiarum” and the sponge has been established for a long time. However, this conclusion is inconsistent with the widespread distribution but low genetic differentiation of “Ca. Synechococcus spongiarum” (47). Although cryptic diversity of “Ca. Synechococcus spongiarum” among sponges has been suggested based on the variation in ITS regions, which supports the potential cryptic genetic differentiation (18), additional evidence is required to confirm the intraspecies diversity and divergence of this sponge symbiont. Due to the high abundance of “Ca. Synechococcus spongiarum” in the sponge C. foliascens, binning genomes of these symbionts from multiple individuals of this sponge species located in a single and/or different geographical sites will improve our knowledge about their genetic diversity and differentiation.
MATERIALS AND METHODS
Sample collection and DNA extraction.
Sponge tissues collected from site RB4 (22°44′56″N, 38°59′35″E), located in the Rabigh Bay of Saudi Arabia along the Red Sea coast, in April 2012, were placed into separate sterile plastic bags and immediately transported back to the laboratory. Sponge tissues were flushed using 0.22-µm-membrane-filtered seawater to remove loosely attached microbes and debris. Ten-milliliter amounts of the flushed sponge tissues were preserved in 70% ethanol for identification of the sponge species. The tissues (0.5 ml) were dissected, cut into small pieces with a sterile razor blade, and frozen in 0.8 ml of extraction buffer (100 mM Tris-HCl, 100 mM EDTA, 100 mM Na2HPO4, 1.5 M NaCl, 1% cetyltrimethylammonium bromide [CTAB], pH 8.0) for DNA extraction. Total genomic DNA was extracted using the modified sodium dodecyl sulfate-based method described previously by Lee et al. (6) and purified using the Mo Bio soil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, USA) according to the manufacturer’s manual. The products were qualified and quantified using a NanoDrop ND-100 device (Thermo Fisher, USA) and stored at −20°C until use.
Metagenome sequencing and assembly.
Metagenomic DNA was sequenced on a 454 FLX platform using Titanium chemistry. This produced a total of 315,119 reads with a total length of 160.2 Mbp. Raw reads were subjected to quality filtering by using the 454QC.pl script in the NGS QC (next-generation sequencing quality control) Toolkit with default parameters (48), and reads smaller than 100 bp and containing more than 5 ambiguous nucleotides were removed. Qualified reads with a total length of 157.5 Mbp were assembled using GS De Novo Assembler (Newbler) with a threshold of 100-bp overlap and 98% identity, which produced 7,203 contigs longer than 100 bp with a total length of 9.8 Mbp. A total of 1,114 contigs longer than 2,000 bp with a total length of 4.2 Mbp were used for genome binning.
Genome binning.
Draft genome binning was carried out mainly based on genome coverage and tetranucleotide frequency patterns according to a previously described method (21), but several modifications were made. The 454 pyrosequencing reads were mapped to the assembled contigs using Bowtie2 (49), and the genome coverage was calculated with SAMtools (50) and Perl scripts. Another sample that contained highly abundant sponge cells was subjected to DNA extraction and Illumina sequencing. The Illumina reads were mapped to the assembled metagenomic contigs, and a secondary genome coverage was obtained. The tetranucleotide frequency of the assembled contigs was calculated using Perl scripts written by Albertsen et al. (21). Principal component analysis of the tetranucleotide frequency was performed using the Vegan package 2.0-5. The open reading frames (ORFs) of the assembled contigs were predicted using Prodigal (51). A set of 107 hidden Markov models (HMM) of essential proteins (21) were searched against the predicted ORFs with default cutoff values in the HMM datasets. The essential proteins identified were searched against the NCBI NR database with BLASTP (E value of 1e-05) and taxonomically assigned using MEGAN 4.0 (52). Contigs were labeled according to the phylum-level taxonomic affiliation of the essential proteins.
Using the previously described R pipelines (21), the genome of “Ca. Synechococcus spongiarum” SH4 was extracted from the assembled contigs (see Fig. S3 in the supplemental material). A group of contigs with 454 metagenomic coverage of greater than 20× showed high GC content and included most of the labeled cyanobacterial contigs. From these contigs, a core set of cyanobacterial contigs were extracted (Fig. S3, dataset 1). Because several contigs that were assigned to the Cyanobacteria fell into a region with lower coverage than the core set, a binning was carried out on a larger region (Fig. S3, dataset 2) and another set of contigs were obtained (Fig. S3, dataset 3). The ORFs in each contig were taxonomically assigned using the BLASTP and MEGAN programs according to the method described above. If more than 50% of the ORFs of a contig in dataset 2 were assigned to the Cyanobacteria, that contig was also included in the SH4 genome. Finally, a draft genome of “Ca. Synechococcus spongiarum” SH4, composed of 273 contigs, was obtained (Table 1).
Genome analysis.
For the KEGG annotation, predicted amino acid sequences of the SH4 draft genome and other reference genomes downloaded from the NCBI Genome database were searched against the KEGG database (53) by using BLASTP with a maximum E value cutoff of 1e-05. Amino acid sequences were also searched against the GenBank NR database, and the output xml file was imported into MEGAN for taxonomic affiliation and SEED/Subsystems annotation (54). KEGG and SEED/Subsystems annotations of the SH4 and closely related cyanobacterial relatives were compared to evaluate the genome reduction of the sponge symbiont “Ca. Synechococcus spongiarum.” Genes of interest that could not be found in the SH4 draft genome were rechecked in the remaining assembled metagenomic contigs with lengths longer than 500 bp and 454 metagenomic coverage greater than 8×. Low-molecular-weight peptides in photosynthetic systems were also searched through the entire assemblage of metagenomic contigs and raw pyrosequencing reads. If the overlooked gene was detected and assigned to the Cyanobacteria, it was considered to be affiliated with “Ca. Synechococcus spongiarum” SH4. For the phylogenomic analysis, 31 proteins encoding phylogenetic markers were predicted from the SH4 draft genome and cyanobacterial genomes in the JGI database using AMPHORA (22). The sequences of each marker gene were aligned individually using ClustalW (55). The aligned sequences were concatenated, and a maximum-likelihood phylogenetic tree was constructed using PhyML (56) according to a previously described method (22). Bootstrap values were calculated with 100 replications. Average nucleotide identity values (ANIs) and z scores of the tetranucleotide frequency between the extracted genome of SH4 and closely related cyanobacterial genomes were calculated using JSpecies (23). Eukaryoticlike domains, including ARs, TPRs, LRRs, NHL repeats, fibronectin type III, and cadherins, were annotated by using the pfam_scan.pl script to search against the PFAM database (57) according to a previously described method (58).
16S rRNA prediction and phylogenetic tree construction.
The 16s rRNA genes in the qualified pyrosequencing reads and the metagenomic assembled contigs were predicted using Meta-RNA (59). Predicted 16S rRNA gene fragments that were longer than 100 bp were loaded into the online RDP classifier for taxonomic assignment. For phylogenetic analysis, a full-length cyanobacterial 16S rRNA gene predicted from the assembled contigs was searched against the NCBI GenBank database using BLASTN to detect closely related relatives. A neighbor-joining tree was constructed using MEGA5.1 software (60). Multiple alignment was performed using ClustalW (55). Distance matrices were calculated using Kimura’s two-parameter correction model (61). Bootstrap values were determined with 1,000 replications.
Nucleotide sequence accession numbers.
The 16S rRNA gene sequence of “Ca. Synechococcus spongiarum” SH4 was deposited in the NCBI GenBank database under accession number KJ174471. All the metagenomic sequences are available in the NCBI Sequence Reads Archive (SRA) database under accession number SRP035516. The draft genome of the “Ca. Synechococcus spongiarum” has been deposited at GenBank under accession number JENA00000000. The version described in this paper is version JENA01000000.
SUPPLEMENTAL MATERIAL
High abundance of “Ca. Synechococcus spongiarum” (represented by OTU1691) in the sponge C. foliascens according to the percentage of 16S rRNA gene amplicons analyzed by tag pyrosequencing. Download
Microbial community compositions in the sponge C. foliascens revealed by 16S rRNA genes predicted from the metagenome in this study and (a) and tag pyrosequencing of 16S rRNA gene amplicons (b). Download
Genome binning of “Ca. Synechococcus spongiarum” SH4. Each circle represents a metagenomic contig with size proportional to length and colored by GC content (a) or taxonomic affiliation (b). Only contigs greater than 2 kbp in length are shown. (c) Sequence composition-independent binning of metagenomic contigs based on the coverage. (d) Principal component analysis clustering on tetranucleotide frequencies for further genome binning. Download
Abundance of genes involved in amino acid metabolism pathways in “Ca. Synechococcus spongiarum” SH4 and closely related free-living cyanobacterial strains. Functional pathways are referred to the KEGG database. Download
Number of genes important for resistance to oxidative stress in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, Cyanobium gracile PCC 6307.
Number of genes important for resistance to antibiotics and environmental toxins in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, C. gracile PCC 6307.
Number of genes important for DNA replication and repair in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, C. gracile PCC 6307.
ACKNOWLEDGMENTS
We thank WP Zhang and G. Zhang from Hong Kong University of Science and Technology (HKUST) and the technical team from King Abdullah University of Science and Technology (KAUST) for their assistance during sample collection. We are also grateful to Rob von Soest, Zoological Museum, University of Amsterdam, for sponge identification.
This study was supported by the Nature Science Foundation of China (grant U1301232), the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (grants XDB06010100 and XDB06010200), and awards from the Sanya Institute of Deep Sea Science and Engineering, CAS (grants SIDSSE-201206, SIDSSE-BR-201303, and SIDSSE-201305), and KAUST (grant SA-C0040/UK-C0016).
Footnotes
Citation Gao Z-M, Wang Y, Tian R-M, Wong YH, Batang ZB, Al-Suwailem AM, Bajic VB, Qian P-Y. 2014. Symbiotic adaptation drives genome streamlining of the cyanobacterial sponge symbiont “Candidatus Synechococcus spongiarum.” mBio 5(2):e00079-14. doi:10.1128/mBio.00079-14.
REFERENCES
- 1. Li S, Dumdei EJ, Blunt JW, Munro MH, Robinson WT, Pannell LK. 1998. Theonellapeptolide IIIe, a new cyclic peptolide from the New Zealand deep water sponge, Lamellomorpha strongylata. J. Nat. Prod. 61:724–728. 10.1021/np970417r [DOI] [PubMed] [Google Scholar]
- 2. Hedges SB, Blair JE, Venturi ML, Shoe JL. 2004. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol. Biol. 4:2. 10.1186/1471-2148-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Soest RW, Boury-Esnault N, Vacelet J, Dohrmann M, Erpenbeck D, De Voogd NJ, Santodomingo N, Vanhoorne B, Kelly M, Hooper JN. 2012. Global diversity of sponges (Porifera). PLoS One 7:e35105. 10.1371/journal.pone.0035105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vacelet J, Donadey C. 1977. Electron microscope study of the association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 30:301–314. 10.1016/0022-0981(77)90038-7 [DOI] [Google Scholar]
- 5. Taylor MW, Radax R, Steger D, Wagner M. 2007. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71:295–347. 10.1128/MMBR.00040-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee OO, Wang Y, Yang J, Lafi FF, Al-Suwailem A, Qian PY. 2011. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 5:650–664. 10.1038/ismej.2010.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW. 2012. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6:564–576. 10.1038/ismej.2011.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, Ravasi T, Hentschel U. 20 August 2013. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 10.1111/mec.12365 [DOI] [PubMed] [Google Scholar]
- 9. Weisz JB, Hentschel U, Lindquist N, Martens CS. 2007. Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar. Biol. 152:475–483. 10.1007/s00227-007-0708-y [DOI] [Google Scholar]
- 10. Simister RL, Deines P, Botté ES, Webster NS, Taylor MW. 2012. Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14:517–524. 10.1111/j.1462-2920.2011.02664.x [DOI] [PubMed] [Google Scholar]
- 11. Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y, Sugahara J, Preston C, de la Torre J, Richardson PM, DeLong EF. 2006. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc. Natl. Acad. Sci. U. S. A. 103:18296–18301. 10.1073/pnas.0608549103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu MY, Kjelleberg S, Thomas T. 2011. Functional genomic analysis of an uncultured δ-proteobacterium in the sponge Cymbastela concentrica. ISME J. 5:427–435. 10.1038/ismej.2010.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamke J, Sczyrba A, Ivanova N, Schwientek P, Rinke C, Mavromatis K, Woyke T, Hentschel U. 2013. Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 7:2287–2300. 10.1038/ismej.2013.111. PubMed. PubMed. [DOI] [PMC free article] [PubMed]
- 14. Webster NS, Taylor MW. 2012. Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. 14:335–346. 10.1111/j.1462-2920.2011.02460.x [DOI] [PubMed] [Google Scholar]
- 15. Usher KM, Fromont J, Sutton DC, Toze S. 2004. The biogeography and phylogeny of unicellular cyanobacterial symbionts in sponges from Australia and the Mediterranean. Microb. Ecol. 48:167–177. 10.1007/s00248-003-1062-3 [DOI] [PubMed] [Google Scholar]
- 16. Erwin PM, López-Legentil S, Turon X. 2012. Ultrastructure, molecular phylogenetics, and chlorophyll a content of novel cyanobacterial symbionts in temperate sponges. Microb. Ecol. 64:771–783. 10.1007/s00248-012-0047-5 [DOI] [PubMed] [Google Scholar]
- 17. Usher KM, Sutton DC, Toze S, Kuo J, Fromont J. 2005. Inter-generational transmission of microbial symbionts in the marine sponge Chondrilla australiensis (Demospongiae). Mar. Freshw. Res. 56:125–131. 10.1071/MF04304 [DOI] [Google Scholar]
- 18. Erwin PM, Thacker RW. 2008. Cryptic diversity of the symbiotic cyanobacterium Synechococcus spongiarum among sponge hosts. Mol. Ecol. 17:2937–2947. 10.1111/j.1365-294X.2008.03808.x [DOI] [PubMed] [Google Scholar]
- 19. Flombaum P, Gallegos JL, Gordillo RA, Rincón J, Zabala LL, Jiao N, Karl DM, Li WK, Lomas MW, Veneziano D, Vera CS, Vrugt JA, Martiny AC. 2013. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. U. S. A. 110:9824–9829. 10.1073/pnas.1307701110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR, Post AF, Hagemann M, Paulsen I, Partensky F. 2009. Ecological genomics of marine picocyanobacteria. Microbiol. Mol. Biol. Rev. 73:249–299. 10.1128/MMBR.00035-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31:533–538. 10.1038/nbt.2579 [DOI] [PubMed] [Google Scholar]
- 22. Wu M, Eisen JA. 2008. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9:R151. 10.1186/gb-2008-9-10-r151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106:19126–19131. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Siegl A, Kamke J, Hochmuth T, Piel J, Richter M, Liang C, Dandekar T, Hentschel U. 2011. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5:61–70. 10.1038/ismej.2010.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen MT, Liu M, Thomas T. 26 August 2013. Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 10.1111/mec.12384 [DOI] [PubMed] [Google Scholar]
- 26. Sikorski RS, Boguski MS, Goebl M, Hieter P. 1990. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell 60:307–317. 10.1016/0092-8674(90)90745-Z [DOI] [PubMed] [Google Scholar]
- 27. Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, Stulik J. 2013. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect. Immun. 81:629–635. 10.1128/IAI.01035-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nowack EC, Melkonian M, Glöckner G. 2008. Chromatophore genome sequence of Paulinella sheds light on acquisition of photosynthesis by eukaryotes. Curr. Biol. 18:410–418. 10.1016/j.cub.2008.02.073 [DOI] [PubMed] [Google Scholar]
- 29. Ramsey JS, MacDonald SJ, Jander G, Nakabachi A, Thomas GH, Douglas AE. 2010. Genomic evidence for complementary purine metabolism in the pea aphid, Acyrthosiphon pisum, and its symbiotic bacterium Buchnera aphidicola. Insect Mol. Biol. 19(Suppl 2):241–248. 10.1111/j.1365-2583.2009.00945.x [DOI] [PubMed] [Google Scholar]
- 30. Albers E. 2009. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB Life 61:1132–1142. 10.1002/iub.278 [DOI] [PubMed] [Google Scholar]
- 31. Wortham BW, Patel CN, Oliveira MA. 2007. Polyamines in bacteria: pleiotropic effects yet specific mechanisms. Adv. Exp. Med. Biol. 603:106–115. 10.1007/978-0-387-72124-8_9 [DOI] [PubMed] [Google Scholar]
- 32. Hosoya R, Hamana K, Isobe M, Yokota A. 2005. Polyamine distribution profiles within Cyanobacteria. Microbiol. Cult. Collect. 21:3–8 [Google Scholar]
- 33. Thornton LE, Ohkawa H, Roose JL, Kashino Y, Keren N, Pakrasi HB. 2004. Homologs of plant PsbP and PsbQ proteins are necessary for regulation of photosystem II activity in the cyanobacterium Synechocystis 6803. Plant Cell 16:2164–2175. 10.1105/tpc.104.023515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi LX, Schröder WP. 2004. The low molecular mass subunits of the photosynthetic supracomplex, photosystem II. Biochim. Biophys. Acta 1608:75–96. 10.1016/j.bbabio.2003.12.004 [DOI] [PubMed] [Google Scholar]
- 35. Dufresne A, Salanoubat M, Partensky F, Artiguenave F, Axmann IM, Barbe V, Duprat S, Galperin MY, Koonin EV, Le Gall F, Makarova KS, Ostrowski M, Oztas S, Robert C, Rogozin IB, Scanlan DJ, Tandeau de Marsac N, Weissenbach J, Wincker P, Wolf YI, Hess WR. 2003. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc. Natl. Acad. Sci. U. S. A. 100:10020–10025. 10.1073/pnas.1733211100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Latifi A, Ruiz M, Zhang CC. 2009. Oxidative stress in cyanobacteria. FEMS Microbiol. Rev. 33:258–278. 10.1111/j.1574-6976.2008.00134.x [DOI] [PubMed] [Google Scholar]
- 37. El-Sorogy AS, Mohamed MA, Nour HE. 2012. Heavy metals contamination of the Quaternary coral reefs, Red Sea coast, Egypt. Environ. Earth Sci. 67:777–785. 10.1007/s12665-012-1535-0 [DOI] [Google Scholar]
- 38. Budd K, Craig SR. 1981. Resistance to arsenate toxicity in the blue-green alga Synechococcus leopoliensis. Can. J. Bot. 59:1518–1521. 10.1139/b81-207 [DOI] [Google Scholar]
- 39. Bertocchi C, Navarini L, Cesàro A, Anastasio M. 1990. Polysaccharides from Cyanobacteria. Carbohydr. Polym. 12:127–153. 10.1016/0144-8617(90)90015-K [DOI] [Google Scholar]
- 40. Ophir T, Gutnick DL. 1994. A role for exopolysaccharides in the protection of microorganisms from desiccation. Appl. Environ. Microbiol. 60:740–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Houdt R, Michiels CW. 2005. Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res. Microbiol. 156:626–633. 10.1016/j.resmic.2005.02.005 [DOI] [PubMed] [Google Scholar]
- 42. Campos MA, Vargas MA, Regueiro V, Llompart CM, Albertí S, Bengoechea JA. 2004. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 72:7107–7114. 10.1128/IAI.72.12.7107-7114.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith HE, Damman M, van der Velde J, Wagenaar F, Wisselink HJ, Stockhofe-Zurwieden N, Smits MA. 1999. Identification and characterization of the cps locus of Streptococcus suis serotype 2: the capsule protects against phagocytosis and is an important virulence factor. Infect. Immun. 67:1750–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thompson AW, Foster RA, Krupke A, Carter BJ, Musat N, Vaulot D, Kuypers MM, Zehr JP. 2012. Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science 337:1546–1550. 10.1126/science.1222700 [DOI] [PubMed] [Google Scholar]
- 45. McCutcheon JP, Moran NA. 2011. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10:13–26. 10.1038/nrmicro2670 [DOI] [PubMed] [Google Scholar]
- 46. Palenik B, Ren Q, Dupont CL, Myers GS, Heidelberg JF, Badger JH, Madupu R, Nelson WC, Brinkac LM, Dodson RJ, Durkin AS, Daugherty SC, Sullivan SA, Khouri H, Mohamoud Y, Halpin R, Paulsen IT. 2006. Genome sequence of Synechococcus CC9311: insights into adaptation to a coastal environment. Proc. Natl. Acad. Sci. U. S. A. 103:13555–13559. 10.1073/pnas.0602963103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steindler L, Huchon D, Avni A, Ilan M. 2005. 16S rRNA phylogeny of sponge-associated Cyanobacteria. Appl. Environ. Microbiol. 71:4127–4131. 10.1128/AEM.71.7.4127-4131.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357-359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21:1552–1560. 10.1101/gr.120618.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114. 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33:5691–5702. 10.1093/nar/gki866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML. Syst. Biol. 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 57. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, Thomas T. 2012. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U. S. A. 109:E1878–E1887. 10.1073/pnas.1203287109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang Y, Gilna P, Li W. 2009. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25:1338–1340. 10.1093/bioinformatics/btp161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16:111–120. 10.1007/BF01731581 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
High abundance of “Ca. Synechococcus spongiarum” (represented by OTU1691) in the sponge C. foliascens according to the percentage of 16S rRNA gene amplicons analyzed by tag pyrosequencing. Download
Microbial community compositions in the sponge C. foliascens revealed by 16S rRNA genes predicted from the metagenome in this study and (a) and tag pyrosequencing of 16S rRNA gene amplicons (b). Download
Genome binning of “Ca. Synechococcus spongiarum” SH4. Each circle represents a metagenomic contig with size proportional to length and colored by GC content (a) or taxonomic affiliation (b). Only contigs greater than 2 kbp in length are shown. (c) Sequence composition-independent binning of metagenomic contigs based on the coverage. (d) Principal component analysis clustering on tetranucleotide frequencies for further genome binning. Download
Abundance of genes involved in amino acid metabolism pathways in “Ca. Synechococcus spongiarum” SH4 and closely related free-living cyanobacterial strains. Functional pathways are referred to the KEGG database. Download
Number of genes important for resistance to oxidative stress in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, Cyanobium gracile PCC 6307.
Number of genes important for resistance to antibiotics and environmental toxins in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, C. gracile PCC 6307.
Number of genes important for DNA replication and repair in SH4 and related cyanobacterial strains. Taxa: 1, “Ca. Synechococcus spongiarum” SH4; 2, Synechococcus sp. RCC307; 3, Synechococcus sp. RS9917; 4, Synechococcus sp. WH 5701; 5, Synechococcus sp. CC9311; 6, P. marinus CCMP137; 7, Cyanobium sp. PCC 7001; 8, C. gracile PCC 6307.