

ABSTRACT
The human microbiome plays important roles in health, but when disrupted, these same indigenous microbes can cause disease. The composition of the microbiome changes during the transition from health to disease; however, these changes are often not conserved among patients. Since microbiome-associated diseases like periodontitis cause similar patient symptoms despite interpatient variability in microbial community composition, we hypothesized that human-associated microbial communities undergo conserved changes in metabolism during disease. Here, we used patient-matched healthy and diseased samples to compare gene expression of 160,000 genes in healthy and diseased periodontal communities. We show that health- and disease-associated communities exhibit defined differences in metabolism that are conserved between patients. In contrast, the metabolic gene expression of individual species was highly variable between patients. These results demonstrate that despite high interpatient variability in microbial composition, disease-associated communities display conserved metabolic profiles that are generally accomplished by a patient-specific cohort of microbes.
IMPORTANCE
The human microbiome project has shown that shifts in our microbiota are associated with many diseases, including obesity, Crohn’s disease, diabetes, and periodontitis. While changes in microbial populations are apparent during these diseases, the species associated with each disease can vary from patient to patient. Taking into account this interpatient variability, we hypothesized that specific microbiota-associated diseases would be marked by conserved microbial community behaviors. Here, we use gene expression analyses of patient-matched healthy and diseased human periodontal plaque to show that microbial communities have highly conserved metabolic gene expression profiles, whereas individual species within the community do not. Furthermore, disease-associated communities exhibit conserved changes in metabolic and virulence gene expression.
INTRODUCTION
The human body is an excellent culture vessel, providing nutrients and a hospitable environment that support the growth of countless microbes. Collectively, these microbial species constitute the human microbiota. Nearly 500 years ago, Leeuwenhoek observed these tiny “animalcules” under his microscope and recorded the great diversity in cell size and shape in human dental plaque. In recent years, researchers have begun using marker gene surveys to catalog the species that colonize different regions of our bodies, including the oral cavity (1–3). These studies have primarily used high-throughput sequencing of the highly conserved rRNA gene to identify and quantify the numerous species constituting the microbiota (1, 2). Among the best-characterized human-associated microbial communities are the extremely diverse gut and oral microbiota. It is now appreciated that our indigenous microbiota are tightly linked to health. Studies using germfree mice have shown that key members of the microbiota promote normal immune system development (4–6). However, several human diseases, including diabetes, Crohn’s disease, and periodontitis, are linked to disruptions in the gut and oral microbial populations (3, 7–10). In light of these results, microbiota-associated diseases such as periodontitis are increasingly examined through an ecological lens.
Microbiota-associated diseases are characterized by changes in the relative abundances of different species during disease. Periodontal disease is one such “microbial shift” disease associated with massive reorganization of the microbiota residing in the subgingival crevice, the region between the tooth surface and the gingival epithelium (3, 11). While marked changes in microbial population structure are observed during periodontitis, the actual community members can differ greatly from person to person (12). In fact, both healthy and disease-associated oral microbial communities vary significantly among people, among locations in the mouth, and even on a daily basis at the same site within the mouth (12, 13).
One possible explanation for the variability observed in marker gene surveys is that a variety of organisms are capable of occupying the multitude of niches present in health- and disease-associated communities. Thus, the question arises to what extent changes in the ecosystem are attributable to alterations in the abundance of certain community members or changes in the activities of existing organisms. Furthermore, while the species that make up health- and disease-associated communities may change, are there conserved metabolic changes in the microbiota associated with the transition to disease? Transcriptional profiling provides an avenue to explore bacterial behavior and metabolism in complex communities (14, 15). In this study, we used massively parallel RNA sequencing to profile changes in both the composition and gene expression of the human oral microbiota in health and in periodontitis.
RESULTS
Disease-associated periodontal microbiota are more similar than are health-associated communities.
Patient-matched healthy and diseased periodontal samples were collected from 10 patients with aggressive periodontitis (AgP) (Table 1). Each healthy and diseased periodontal plaque sample was a pool of populations from three healthy or diseased teeth from each patient. Thus, our study encompassed 30 total health-associated and 30 total disease-associated microbial periodontal populations. This collection technique was important for two main reasons. First, microbial periodontal plaque populations are very small, especially those populating healthy teeth; therefore, our pooled collection methods allowed us to obtain enough microbial cells to isolate RNA for population-wide diversity and gene expression analyses. Second, periodontal microbial populations have been shown to differ widely from one tooth to another, and our approach allowed us to capture the mean microbial population composition for healthy and disease-associated plaque for each patient.
TABLE 1 .
Aggressive periodontitis patient data
| Sample | Age (yr) | Genderc | PDa (full) | CALb (full) | Plaque indexd | Bleeding (%) | Smoking | Mean PDa per sampling site |
|
|---|---|---|---|---|---|---|---|---|---|
| Diseased | Healthy | ||||||||
| 1 | 36 | M | 5 | 6 | 2 | 90 | Yes | 5 | 2 |
| 2 | 40 | F | 5 | 5 | 1 | 60 | No | 5 | 3 |
| 3 | 33 | F | 6 | 6 | 2 | 70 | Yes | 6 | 2 |
| 4 | 34 | F | 6.5 | 6.5 | 1 | 60 | No | 5 | 3 |
| 5 | 36 | M | 6 | 7 | 2 | 80 | Yes | 5 | 3 |
| 6 | 30 | F | 5 | 5 | 2 | 70 | Yes | 6 | 2 |
| 7 | 34 | M | 5 | 6.5 | 1 | 70 | No | 5 | 2 |
| 8 | 34 | M | 5.5 | 5.5 | 1 | 70 | No | 7 | 2 |
| 9 | 36 | F | 5.5 | 5.5 | 2 | 80 | No | 5 | 2 |
| 10 | 37 | F | 5 | 5 | 1 | 60 | No | 5 | 3 |
PD, probing depth of the subgingival crevice (mm).
CAL, clinical attachment loss of gingival epithelium (mm).
M, male; F, female.
1, plaque detected by probe; 2, plaque visible to the naked eye.
High-throughput sequencing of rRNA genes and of rRNA has been used to identify and quantify species in microbial communities (16). In this study, we elected to use rRNA sequencing, because rRNA reflects organisms’ capacities to produce proteins and alter community activity (16). Using rRNA sequencing (see Table S1 in the supplemental material), we found that many bacteria are present in both health- and disease-associated communities; however, many of the most ribosome-rich microbes in disease samples were those previously associated with infection, including Tannerella sp., Prevotella sp., Treponema sp., and Porphyromonas sp. (see Fig. S1A). Alpha diversity analyses of rRNA content in health- and disease-associated populations showed that disease-associated communities were significantly less diverse than health-associated populations: they contained fewer overall species (Fig. 1A) and were less species rich (Fig. 1B). Comparing rRNA gene abundance to rRNA abundance for a subset of our samples revealed a stronger correlation between these two measures for disease-associated populations than for health-associated populations, suggesting that a larger fraction of the disease-associated population is ribosome rich and thus can contribute to overall community activity (see Table S2). In contrast, rRNA gene abundance correlated less well with rRNA abundance in healthy communities, indicating that many members of this community have low ribosome content. Beta diversity analysis comparing the relatedness of disease- and health-associated populations from multiple individuals showed that disease- and health-associated populations segregated into distinct groups (PERMANOVA, P = 0.01), and diseased populations were less dispersed than healthy populations (PERMDISP, P = 0.008) (Fig. 1C). Additionally, disease-associated populations were more related to the average disease state than to paired health-associated populations from the same individual (Fig. 1D). These data show that health-associated periodontal populations are highly diverse and patient specific, while a few commonly found, ribosome-rich organisms overwhelm health-associated microbiota during aggressive periodontitis. Previous studies have shown that the oral microbiota can vary from site to site within individuals (13), yet our data suggest that common features are seen in microbial communities associated with aggressive periodontitis.
FIG 1 .

Ribosome quantification reveals that disease-associated periodontal microbiota are less diverse and contain fewer low-abundance species than do health-associated populations. (A) Number of distinct 16S rRNA sequences (OTUs) observed in healthy (blue) and diseased (red) samples with increasing numbers of sequences sampled from each population. Error bars indicate standard errors of the means (n = 10). (B) Shannon indices show that health-associated populations are more species rich than diseased populations (*, P = 0.03, paired two-tailed Student t test). (C) Beta diversity was measured using the unweighted Unifrac method to calculate relatedness of paired health-associated (blue) and disease-associated (red) microbial populations by assessment of shared and unique species in each community. Principal coordinates 1 and 2 are plotted. Mean diseased and healthy centroids (mean ± standard deviation) are indicated by ellipses. Distances between samples and corresponding centroids are shown as blue and red lines, respectively. Black lines show distances between paired populations from the same patient. (D) Mean Euclidean distance (mean ± standard deviation) from each sample to corresponding centroids and corresponding paired sample from same patient (**, P = 0.0005, paired two-tailed Student t test).
Community gene expression analysis.
Changes in the composition of the microbiota have previously been associated with numerous diseases, including periodontitis, and the results of our rRNA sequencing show that a few members of the community produce a majority of the rRNA during periodontal disease. Despite these findings, it is unclear how specific activities of different members of the community impact disease. To address this question, we used high-resolution community transcriptional profiling. We were able to obtain sufficient quantities of total RNA from three patient-matched healthy and diseased samples representing 9 health- and 9 disease-associated periodontal plaque populations, which, following depletion of highly abundant human and bacterial rRNA, were sequenced on an Illumina HiSeq system. In total, 1.5 billion RNA sequencing (RNA-seq) reads were obtained (see Table S3 in the supplemental material). Prior to microbial gene expression analyses, we aligned the reads to the Human Oral Microbiome Database consisting of complete and draft genomes (HOMD; 4.4 billion bp), the RefSeq human RNA database (huRNA; 135 million bp), and the RefSeq viral genome database (virusDB; 121 million bp). For each sample, between 55 and 65% of the total reads aligned to these reference databases, and the majority (>99%) of these reads were prokaryotic (see Table S4).
To quantify gene expression, reads were aligned to a 60-organism “metagenome” comprised of completed and draft genomes representing microbes comprising 60 to 90% of total healthy or diseased rRNA (see Supplementary File 1 at http://web.biosci.utexas.edu/whiteley_lab/pages/resources.html). Since we characterized both health- and disease-associated communities with RNA-seq, we could analyze differential expression of the >160,000 bacterial genes represented in our metagenome simultaneously between healthy and diseased sites in the same individual. For each sample, 28 to 85 million RNA-seq reads mapped to the 60-organism metagenome, including 17.3 ± 2.05 million mRNA reads per sample. This sequencing depth provided sufficient data for differential expression analysis at the community and organismal levels (for raw read counts per gene, see Supplementary File 2 at http://web.biosci.utexas.edu/whiteley_lab/pages/resources.html; median, 12 to 21 reads per mRNA; mean, 75 to 156 reads per mRNA). In total, 66 to 91% of reads that mapped to the HOMD, huRNA, and virusDB databases mapped to the 60-species metagenome, suggesting that our reference metagenome sufficiently represents the oral microbiome as determined by shotgun metagenomic sequencing data.
Disease-associated communities change metabolic gene expression.
Previous studies have used genomic information to predict disease-associated shifts in metabolism; however, these models are based on the genetic capacity of the population rather than microbial community metabolic gene expression (17, 18). Our RNA-seq approach allows modeling of the metabolism of the microbiota during health and disease based solely on gene expression. In this approach, we used Enzyme Commission (EC) numbers to assign biochemical function to the >160,000 genes present in the 60-organism metagenome. EC numbers classify enzymes based on the reaction that they catalyze (i.e., enzymes catalyzing the same reaction will have the same EC number). This allowed us to calculate changes in expression of metabolic enzymes for the entire community during health and disease, resulting in a quantitative, high-resolution view of metabolism. Among ~1,100 unique enzyme-encoding gene families in the oral metagenome, ~18% were differentially expressed (P < 0.05) at the microbiome level during disease (Fig. 2; see also Supplementary File 3 at http://web.biosci.utexas.edu/whiteley_lab/pages/resources.html). Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (19) metabolic pathway database, we were able to reveal enzymatic steps whose genes were upregulated, downregulated, or unchanged in the microbiome during disease (Fig. 3). These results revealed that within each individual, disease-associated populations showed defined changes in expression of metabolic genes, suggesting that specific metabolic shifts are occurring in disease-associated communities. Specific pathways that showed enhanced gene expression in all diseased sites included lysine fermentation to butyrate, histidine catabolism, nucleotide biosynthesis, and pyruvate fermentation. The observation that these pathways were observed in all three patients strongly suggests that they are important for stability of disease-associated populations and likely contribute to the disease process. In support of this, butyrate levels have been shown to increase during periodontitis (20) and likely contribute to disease by preventing human cell proliferation (21). These data provide the first metabolic reconstruction (from gene expression data) of the microbial population in healthy and diseased periodontal pockets and identified numerous pathways not previously associated with disease along with one pathway (lysine fermentation to butyrate) previously proposed to be important.
FIG 2 .

Differential expression of enzyme gene families in health and disease. Log2 fold change during disease is plotted against the log2 mean read counts per million total reads for each EC enzyme-encoding gene family. Gene families upregulated in health are shown in blue, while gene families upregulated in disease are shown in red.
FIG 3 .

Differential metabolic gene expression in the diseased periodontal microbiome. Metabolic network reconstruction. Black lines indicate enzyme-encoding genes that were expressed and unchanged in health and disease, red lines indicate genes upregulated during disease, and blue lines indicate genes upregulated during health. Colored regions identify different sections of the metabolic pathway map. Those highlighted in yellow represent important pathways that were upregulated in disease. Complete data showing all differentially regulated genes are available in supplementary files 2 and 3 at http://web.biosci.utexas.edu/whiteley_lab/pages/resources.html. THF, tetrahydrofolate metabolism; TCA, tricarboxylic acid.
In addition to defining community-level metabolic gene expression, our high-resolution RNA-seq analyses allow for identification of the individual microbes mediating shifts in metabolic gene expression during disease. For example, while several oral microbes have the capacity to produce butyrate, the Gram-negative bacterium Fusobacterium nucleatum is the sole bacterium responsible for community lysine degradation to butyrate in all patients (Fig. 4A). In contrast, several bacteria were responsible for enhanced expression of genes involved in histidine degradation and pyruvate fermentation during disease (Fig. 4B and C), and the bacteria differed between patients. Collectively, these data provide two novel insights into this microbiota-mediated disease. (i) We propose that F. nucleatum is a keystone species during periodontitis, functioning in all patients by shifting its gene expression to produce a metabolite (butyrate) that establishes a hospitable growth environment for the disease-associated community. Notably, the rRNA of F. nucleatum in health- and disease-associated communities is proportionally identical (see Fig. S1 in the supplemental material), indicating that the changes in gene expression observed are not due to an increase in abundance in disease-associated populations. While F. nucleatum and other bacteria have been proposed as keystone species in the past, even the most convincing data supporting these theories have arisen from defined model communities containing few species grown in animal models (22). Because the ability to serve as a keystone species is dependent on the constituents of the community (which vary between patients), our data provide the first evidence for the role of this bacterium as a keystone species in a naturally occurring microbial community during human infection. We also pinpoint lysine fermentation as the key metabolic pathway contributing to its keystone role, which was previously unappreciated. (ii) We also show that, while metabolism is conserved at the community level, for pathways like histidine degradation and pyruvate fermentation multiple microbes contribute to gene expression changes (Fig. 4B and C). The interchangeability of community members in each patient provides insight into why metagenomic analyses of the oral microbiome have displayed little conservation. Remarkably, we observed that the bacteria contributing to expression of known extracellular virulence factors vary between patients (Fig. 4D), suggesting that in addition to metabolism, distinct microbes produce conserved virulence determinants in each individual.
FIG 4 .

Metabolic niche dynamics in diseased populations. (A) Production of butyrate is primarily due to F. nucleatum lysine fermentation. (B) Multiple species that vary among patients fill histidine degradation and tetrahydrofolate (THF) metabolic niches. (C) Multiple species that vary among patients carry out pyruvate fermentation. For panels A to C, community fold changes of EC enzyme-encoding gene expression are indicated at each arrow. (D) Different organisms fill virulence niches in diseased periodontal communities. In patient 1, Tannerella forsythia is the major source of collagenase expression, whereas collagenase expression is augmented by Prevotella tannerae in patient 2 and by Porphyromonas gingivalis in patient 3. Protease production follows similar patterns, whereby combinations of different species express proteases in each patient. In panels A to D, heat maps indicate relative normalized expression (log2 reads per million reads in each sample) of different enzyme-encoding genes or virulence genes by species in each patient. Abbreviations of species names and the color scale for heat maps are indicated. CoA, coenzyme A.
Community metabolic gene expression is highly conserved relative to individual species.
Our data suggest that metabolic pathways are well conserved in health- and disease-associated communities, while the organisms carrying out these processes often vary between communities. If this were true, we hypothesized that variance of gene expression at the individual gene level (i.e., a gene within a single species) would be high, while variance at the EC-binned level (orthologous genes from all species in the community) would be low. In support of this hypothesis, variance estimations show that as expression increases, dispersion increases at the individual gene level while it decreases at the community EC expression level (Fig. 5A and B). Since the decreased variance observed at the EC-binned level could potentially be due to a condensation of the data to fewer total data points, we randomly binned genes into 1,137 groups (equal to the number of ECs) and performed the variance analyses on this artificial data set. As expression increases in the randomly grouped gene set, variance increases (Fig. 5C), demonstrating that the decreased variance observed in EC-binned genes is indeed biological and not due to compression of the data. These data indicate that an individual gene (e.g., pyruvate formate lyase in a single microbe) displays high variability in expression between communities (i.e., patients); however expression of orthologous genes (e.g., pyruvate formate lyase from all microbes) is highly conserved.
FIG 5 .
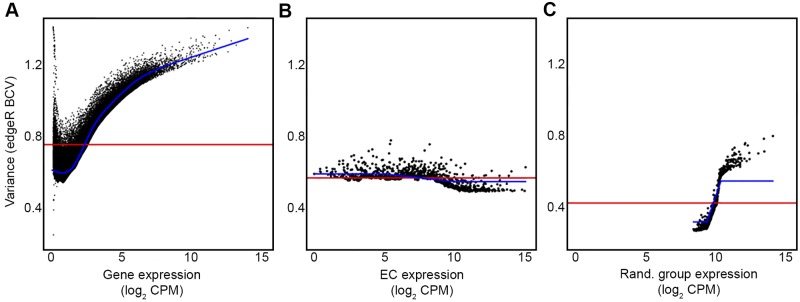
EC expression is less variable than individual gene expression. (A) Variance estimations for genes in the metagenome determined in edgeR analyses. (B) Variance estimations for EC expression determined in edgeR analyses. (C) Variance estimations for genes randomly binned into 1,137 gene groups determined in edgeR analyses. Blue lines in panels A to C indicate the tagwise dispersion, while red lines show the common dispersions calculated with edgeR. BCV, biological coefficient of variance.
DISCUSSION
Previous studies of the human microbiota have indicated that microbial diversity is high between individuals and can vary significantly over time and between different locations on the same individual. Thus, in studies focusing on the microbiota, it is important to ensure that sufficient samples are examined to capture the breadth of this variability. Previous metatranscriptomics studies examining the human microbiota associated with bacterial vaginosis, in feces treated with different drugs, and in feces from people with various diets have successfully revealed conserved community gene expression responses with as few as two biological replicates per condition (14, 15, 23). Here, we provided a complete transcriptome-based reconstruction of microbial metabolism in nine patient-matched health- and disease-associated periodontal plaque populations from three patients. Importantly, the results of this reconstruction recapitulated a key phenomenon consistently found during the transition to periodontal disease: the increased production of short-chain fatty acids such as butyrate (20). This indicates that we were able to identify conserved changes in microbial community metabolism in the face of high interpatient variability in microbiota composition.
Our study represents an important advance in several ways. First, we captured gene expression at an extremely high resolution, examining expression of 160,000 genes simultaneously. To comprehensively analyze expression of such a large gene set, it was important to achieve sufficient sequencing depth to accurately determine differences among plaque communities. Therefore, we focused our study on a relatively small patient group and were able to compare gene expression of numerous high- and low-abundance organisms in these communities. Also, our EC-based computational approach allowed us to take a more global view of microbial metabolism than previously appreciated. Other studies have used KEGG orthologs to study metabolism (14, 15, 23). While this approach is similar to ours, it is complicated by the fact that many KEGG orthologs can encode proteins with the same catalytic activity, whereas each enzyme is assigned only one EC number for its specific catalytic activity. Therefore, our approach truly distilled genes into functional rather than orthologous groups, allowing us to accurately look at whole-community metabolism in an ancestry-independent manner.
While population composition varied among plaque populations, we found that enzyme expression was well conserved. This suggests that multiple organisms that vary among populations are capable of filling conserved metabolic niches. This study is an important step toward characterizing the influence of human microbiota on health and disease. While mRNA is not an exact prediction of metabolic activity, it is a closer approximation of metabolism than 16S rRNA or genome-based predictions that have been reported previously. RNA-seq is especially well suited for studying human microbiota because it is highly sensitive, and therefore, experiments can be performed with small amounts of starting material, like dental plaque. Other approaches, including proteomics or metabolomics, may aid in the understanding of human-associated microbial metabolism but are technically challenging due to the sample sizes required for these techniques.
A major question that remains with all microbial shift diseases is whether changes in microbiota composition and behavior cause disease or are a consequence of disease. Here, we found that differential expression of metabolic genes in certain pathways was associated with the periodontal disease state. For instance, expression of butyrate production genes by F. nucleatum increased during disease. Increased butyrate levels have been measured in diseased periodontal pockets, and studies in cell culture have shown that these butyrate concentrations can arrest human cell growth, potentially delaying the healing process (20, 21). In combination with the findings presented here, this suggests that F. nucleatum butyrate production likely promotes disease. However, carefully designed future studies will be necessary to better support this hypothesis.
Several different metatranscriptomic approaches will help elucidate the answer to the “chicken or egg” question in microbial shift disease research. One potential approach is to carefully examine composition and behavior of the microbiota throughout disease progression. In this approach, one could simultaneously analyze the host symptoms and microbiota to determine whether changes occur first at the host level or at the microbial level at disease onset. An alternate approach would be to examine a large number of healthy and disease-associated microbial communities. This would allow the characterization of common features of health and disease, and one would expect to find intermediate compositional and expression states that might get to the root of the question. In addition, results from metatranscriptomics studies will help guide more directed studies to utilize mRNAs as markers for disease.
MATERIALS AND METHODS
Study population.
A total of 10 individuals seeking dental treatment in the School of Dentistry, Ege University, Izmir, Turkey, were involved in the present study. Ten systemically healthy, untreated patients with generalized aggressive periodontitis (AgP) were recruited from September 2011 to August 2012 (Table 1). The study was conducted in full accordance with ethical principles, including the World Medical Association’s Declaration of Helsinki, as revised in 2000. The study protocol was explained, and written informed consent was received from each individual before clinical periodontal examinations and subgingival plaque sampling. Medical and dental histories were obtained, and smoking habits were recorded. Individuals with medical disorders, such as diabetes mellitus or immunological disorders, and those who had antibiotic or periodontal treatment in the last 6 months were excluded from the study.
Individuals with AgP were diagnosed in accordance with the clinical criteria stated in the consensus report of the World Workshop in Periodontitis. Individuals had at least 6 permanent teeth, including incisors and/or first molars, with at least one site with probing depth (PD) and clinical attachment loss (CAL) of ≥5 mm and 6 teeth other than first molars and incisors with similar PD and CAL measurements, and familial aggregation (all individuals were asked if they had any family member with current severe periodontal disease or a history of such).
Subgingival plaque sampling.
For the diseased samples, the deepest 3 pockets were selected and pooled in a single Eppendorf tube. Supragingival plaque was first removed from the sample teeth with sterilized Gracey curettes and sterilized gauze. The site was then cleaned and isolated using cotton rolls and air dried gently. Another sterilized Gracey curette was inserted into the deepest part of the pocket and removed by applying a slight force toward the root surface. The tip of the curette was then inserted in the Eppendorf tube containing RNALater and shaken until the plaque was removed from the curette. For the healthy subgingival plaque samples, in the same patient 3 healthy sites that did not show any sign of inflammation and bleeding on probing were chosen and pooled in an Eppendorf tube. The same procedures were followed for the subgingival sampling. After 24 h, the samples were frozen and stored at −40°C until the sample collection period was completed.
Clinical periodontal measurements.
Subsequent to saliva and serum sampling, clinical periodontal recordings, including plaque index, PD, CAL, and bleeding on probing (BOP) (+/−), were performed at 6 sites (mesiobuccal, midbuccal, distobuccal, mesiolingual, midlingual, and distolingual locations) on each tooth present, except the third molars, using a Williams periodontal probe. CAL was assessed from the cement enamel junction to the base of the probable pocket. BOP (deemed positive if it occurred within 15 s after periodontal probing) was recorded dichotomously by visual examination. All measurements were performed by two precalibrated examiners (P.G. and N.N.). Interexaminer and intraexaminer calibration was analyzed using the kappa-Cohen test. The initial intraexaminer kappa values were 0.96 (PD) and 0.86 (CAL) for P.G. and 0.93 (PD) and 0.79 (CAL) for N.N. The interexaminer values were 0.92 (PD) and 0.75 (CAL).
Total RNA isolation.
Subgingival plaque samples stored in RNALater were centrifuged at 16,100 × g to collect whole cells. Cell pellets were resuspended in 1 ml RNA Bee and transferred to a bead-beating tube. Cells were lysed by bead-beating 3 times for 60 s and incubated on ice for 1 min between bead beatings. Lysed cell solutions were transferred to new microcentrifuge tubes, and 200 µl chloroform was added. Tubes were shaken vigorously for 1 min to mix and incubated for 5 min in an ice bath. Samples were centrifuged at 13,100 × g for 30 min at 4°C to separate aqueous and organic phases. The aqueous phase from each sample was transferred to a new microcentrifuge tube, and RNA was precipitated with an equal volume of isopropanol and 2 µg linear acrylamide for 16 h at −80°C. Samples were thawed in an ice bath and centrifuged for 30 min at 13,100 × g at 4°C. Supernatants were removed, and RNA pellets were washed with twice with ice-cold 75% ethanol by resuspension and centrifugation for 10 min at 16,100 × g at 25°C. Following the second ethanol wash, RNA pellets were air dried for 5 min at 25°C and resuspended in 22 µl RNase-free water. RNA concentrations for each sample were determined with a Nanodrop spectrophotometer (Thermo Scientific).
rRNA sequencing.
rRNA sequencing was carried out by modifying 2 protocols from previous studies which sequenced bacterial 16S rRNA genes (1, 2). Total subgingival plaque RNA for all 10 healthy and diseased samples was used to reverse transcribe 16S cDNA with SSII reverse transcriptase (RT) (Invitrogen) and the universal bacterial 16S 926 RT gene-specific primer (see Table S5 in the supplemental material), which anneals immediately downstream of the 16S rRNA V5 variable region. Negative-control reactions with reaction mixtures lacking SSII were conducted on all RNA samples in parallel to ensure that DNA was not copurified with the total RNA. From each RT reaction, including negative-control reactions, 2 µl was removed, and cDNA was used as the template to minimally PCR amplify the 16S rRNA V4/V5 variable region using indexed sample-specific primers 16SV5926R-BC0 through 16SV5926R-BC19 and the common primer 16SV4515F (see Table S5). All RT-PCR products were separated by agarose gel electrophoresis, stained with ethidium bromide, and viewed with a GBox imaging system. Distinct cDNA bands were visible for all positive-control reactions, while negative-control reactions with reaction mixtures lacking RT showed no product, verifying the absence of DNA contamination in the original RNA preparations. Paired-end 250-bp sequencing was performed on the 16S cDNA libraries using an Illumina MiSeq system at the University of Texas Genomic Sequencing and Analysis Facility (UTGSAF) with custom MiSeq16SV4515F forward, MiSeq16SV5926R reverse, and MiSeq16SV4V5Index index sequencing primers (see Table S5).
Total DNA isolation and 16S rRNA gene library preparation.
To organic phases from RNA isolations from healthy and diseased samples from patients 1, 2, and 3, 500 µl Tris-EDTA (TE) buffer (pH 8.0) was added, and samples were mixed by rotation for 10 min at 25°C to elute DNA from organic phases. Samples were centrifuged for 30 min at 16,100 × g at 4°C, and the aqueous phase was transferred to a new tube. To each sample, 750 µl ice-cold 100% ethanol, 25 µl 3 M sodium acetate (pH 5.5), and 1 µl 1-mg/ml linear acrylamide were added and inverted to mix, and samples were incubated for 4 h at −80°C. Samples were centrifuged for 15 min at 16,100 × g at 4°C, supernatants were discarded, and DNA pellets were washed with 750 µl ice-cold 70% ethanol. Samples were centrifuged for 5 min at 16,100 × g at 25°C, supernatants were discarded, and DNA pellets were washed one more time with 750 µl ice-cold 70% ethanol. Samples were centrifuged for 5 min at 16,100 × g at 25°C, supernatants were discarded, and DNA pellets were dried for 5 min at 25°C. DNA was resuspended in 22 µl TE buffer, pH 8.8. Sequencing libraries were prepared by PCRs with 5 µl DNA, the 16SV4f-515F forward primer, and unique bar-coded reverse primers for each sample (primers BC8 to BC13; see Table S5 in the supplemental material). Paired-end 250-bp sequencing was performed on the 16S cDNA libraries using an Illumina MiSeq system at the University of Texas Genomic Sequencing and Analysis Facility (UTGSAF) with custom MiSeq16SV4515F forward, MiSeq16SV5926R reverse, and MiSeq16SV4V5Index index sequencing primers (see Table S5).
Bacterial population analyses.
The paired 250-bp forward and reverse MiSeq sequencing reads were assembled using fastq-join (24). Unassembled reads were discarded, improving read accuracy. Qiime (25) was used to search the assembled 16S cDNA sequences from all 10 healthy and 10 diseased samples with Uclust against the 97% Greengenes reference database (26) for species-level identification of operational taxonomic units (OTUs) using the Qiime python script pick_otus_through_otu_table.py (25). Prior to alpha diversity analyses, samples were rarefied, or subsampled, 10 times at each step from 500 to 5,000 sequences with a 500-sequence step-size. Mean alpha diversity, or within-sample diversity, was calculated using the Qiime python scripts alpha_diversity.py and collate_alpha.py to determine the number of observed species at each subsampling depth in the rarefication analysis as well as the Shannon indices, which reflect species richness within samples. Significant differences in mean Shannon indices for diseased and healthy samples were determined with a paired Student t test. Jackknifed beta diversity, or between-sample diversity, was determined for 5,000 sequences per sample using the Qiime python script jackknifed_beta_diversity.py. A multidimensional scaling (MDS) analysis plot was generated from the average of 10 distance matrices determined by unweighted Unifrac analysis (27) calculated by the jackknifed_beta_diversity.py script and was used to determine the similarity between sample populations. Briefly, Unifrac analysis takes into account the number of shared and unique species between two populations and provides a distance metric that represents the overall similarity of the two populations (27). Healthy and disease centroids on the MDS plot were determined from the mean positions of the respective samples on the plot. PERMANOVA and PERMDISP analyses were calculated using the Qiime script compare_categories.py to determine whether healthy and diseased samples formed distinct groups and if the two groups had unequal dispersions. Euclidean distances were calculated to determine relatedness between paired healthy and diseased samples, and Euclidean distances from samples to their respective healthy or disease centroids were calculated. Significant differences between Euclidean distances to centroids versus pairs for diseased and healthy samples were determined with a paired Student t test.
Comparing 16S rRNA gene and 16S rRNA sequencing.
16S rRNA gene and rRNA sequencing reads from patients 1, 2, and 3 were assembled and assigned OTUs using Qiime (25), as described above for rRNA sequencing. To determine relatedness of 16S rRNA and rRNA gene sequencing, Spearman rank correlation analysis was performed using the core R package to compare rRNA and rRNA gene sequencing OTU abundances for healthy and diseased samples from each patient.
RNA-seq.
Patients 1, 2, and 3 were selected for total RNA sequencing to analyze microbial population gene expression in periodontal health and disease because they demonstrated OTU patterns that were representative of the average healthy and diseased populations (see Fig. S1A and B in the supplemental material) and there was sufficient RNA to make RNA-seq libraries. Total RNA samples were treated with the RiboZero Epidemiology kit (Epicentre) to deplete bacterial and eukaryotic rRNA and purified by ethanol precipitation using 20 µg linear acrylamide to precipitate the RNA. Depleted RNA was fragmented with NEB RNA fragmentation buffer, according to the manufacturer’s protocol. Fragmented RNA was ethanol precipitated with linear acrylamide and eluted in RNase-free water. RNA-seq libraries were prepared using the NEB Next Multiplex Small RNA Library Prep Set for Illumina, according to the manufacturer’s protocol. The resulting strand-specific cDNA libraries were stained with SYBR gold nucleic acid stain (Invitrogen) and visualized on a GBox imaging system, and cDNA between ~150 and 300 bp was extracted, corresponding to fragmented RNA between nucleotides (nt) 31 and 181. Gel extracted cDNA was eluted in NEB polyacrylamide gel elution buffer, ethanol precipitated, and resuspended in TE buffer (NEB). Libraries were quantified and analyzed using a Nanodrop spectrophotometer (Thermo Scientific) and a Bioanalyzer (Agilent). Single-end 50-bp sequencing was conducted at the UTGSAF on an Illumina HiSeq2000 system producing ~1.5 billion sequencing reads (see Table S3).
RNA-seq fastq read processing.
HiSeq reads were trimmed with Flexbar (28), as described previously (29), to remove contaminating adapter sequences from the cDNA library preparation. Flexbar was run with settings to collect reads 15 to 50 bp following adapter trimming for further analysis, because these reads are specific: 15-bp sequences are predicted to occur randomly only once per ~1 billion bp. Because our reference metagenome contained 161 million bp, the chance of a 15-bp read mapping randomly to the genome was 10% and therefore should not skew our results.
Determining origin of metatranscriptome sequencing reads.
All metatranscriptome data analysis was conducted on the Texas Advanced Computing Center Stampede supercomputer. Human oral bacterial genome sequences (oral_microbiome.na.zip, >4 billion bp) were downloaded from the Human Oral Microbiome Database (30) (HOMD) available on the World Wide Web via ftp://ftp.homd.org/human_oral_microbial_genomic_sequences/20130520/, the human RNA database (human.rna.fna.gz) was downloaded on the World Wide Web through NCBI RefSeq via ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/mRNA_Prot/, and the viral genome database (viral.1.1.genomic.fna.gz, ~121 million bp) consisting of sequenced viruses and bacteriophage available was downloaded through NCBI RefSeq via ftp://ftp.ncbi.nih.gov/refseq/release/viral/. Reference sequences were indexed with Bowtie 2.0 (31). Since the HOMD sequences exceeded the size limit for Bowtie 2.0 (31), the sequences were split into two files with the custom Perl script FastaSplit.pl (http://github.com/khturner/metaRNA-seq), and then each file was indexed. Trimmed fastq sequencing reads were split into chunks of 10 million reads using the UNIX split command. Each read chunk was mapped separately to the four indexed reference sequences using Bowtie 2.0 (31), keeping only 1 match for each fastq read for each reference. Unmapped reads were discarded, and mapped reads were labeled to indicate whether they mapped to either of the 2 human oral microbiome indexed databases (HOMD1 and HOMD2), the indexed human RNA database, or the indexed viral RNA database. The resulting labeled mapped reads in sam file format for each read chunk in each sample were concatenated and sorted by read name using the UNIX cat and sort commands. Since initially we were interested in whether a read was of bacterial origin, human origin, or viral origin, if a single fastq read mapped to both HOMD1 and HOMD2, the read mapped to HOMD2 was discarded; however, if a read mapped to multiple references (e.g., HOMD and human), it was labeled in the file using the custom Perl script MatchMarker.pl (http://github.com/khturner/metaRNA-seq). The numbers of uniquely mapping reads and reads mapping to multiple reference databases for each sample were determined using the UNIX uniq and pattern-matching grep commands.
Generating a reference metagenome for differential gene expression analysis.
Genomes for differential gene expression analyses were selected using 16S rRNA sequencing for patients 1, 2, and 3. Reference genome sequences and annotations were downloaded in Fasta and GFF formats, respectively. Genomes were downloaded, concatenated, and processed to include only protein-encoding genes using the custom Perl scripts GenomeMerge.pl and HOMDpull.sh (http://github.com/khturner/metaRNA-seq) to generate an annotated metagenome to serve as a reference. Individual genome sequences and annotations were obtained from NCBI Genbank (ftp://ftp.ncbi.nih.gov/genbank) and HOMD (http://www.homd.org/index.php?&name=seqDownload&type=G). When available, EC numbers for genes were downloaded from KEGG (19) using the custom Perl scripts PullEC.pl and HOMD_GenomeMerge.pl (http://github.com/khturner/metaRNA-seq).
Differential gene expression analyses.
Trimmed RNA-seq reads produced by Flexbar (28) were mapped against the indexed reference metagenome, and reads mapping to each gene were counted using the custom UNIX shell script MapCount_RNASeq.sh (http://github.com/khturner/metaRNA-seq), which depends on Bowtie 2.0 (31) and the Python package HTSeq (https://pypi.python.org/pypi/HTSeq). The trimmed sequencing reads were read into the script and mapped to the metagenome, and the number of reads in each sample mapping to each annotated gene in the metagenome was counted. Paired differential gene expression was determined using the custom UNIX shell script calcRNASeqPaired.sh (http://github.com/khturner/metaRNA-seq), which depends on the R package edgeR (32) and the supporting R script Pairwise_edgeR.r (http://github.com/khturner/metaRNA-seq). This analysis normalizes read counts between samples, fits the data to a negative binomial distribution, and determines pairwise differential expression using the patient-matched samples.
Differential expression analysis of EC enzymes.
EC numbers obtained from the HOMD and KEGG databases were added to the table containing raw read counts per gene produced by MapCount_RNASeq.sh (above). Genes lacking EC numbers were removed from the table, and the table was sorted by the EC numbers. The total number of reads mapping to each EC number was calculated using the custom Perl script ECcounter.pl (http://github.com/khturner/metaRNA-seq), to produce a table containing the number of reads mapping to each EC number in each sample. Differential expression of EC enzymes was determined using the custom UNIX shell script Pairwise_edgeR.sh (http://github.com/khturner/metaRNA-seq), which depends on the R package edgeR (32) and the supporting R script Pairwise_edgeR.r (http://github.com/khturner/metaRNA-seq).
Nucleotide sequence accession numbers.
rRNA sequencing data are available at http://datadryad.org/ at doi:10.5061/dryad.d41v4, and RNA-seq sequencing data are available at NCBI in the sequence read archive under BioProject accession number SRP033605.
SUPPLEMENTAL MATERIAL
Average bacterial species abundances in healthy and diseased periodontal samples determined by rRNA-seq. (A) rRNA-seq was used to determine OTUs present in health and disease. Shown are average abundances for healthy (blue) and diseased (red) samples. Error bars indicate standard errors of the means (n = 10). (B) OTU abundances for healthy and diseased samples from patients 1, 2, and 3, which were selected for RNA-seq. Download
rRNA-seq sequencing and analysis information.
Spearman correlations of 16S rRNA gene and 16S rRNA sequencing.
RNA-seq sequencing information.
Percent specific RNA-seq reads aligned to the HOMD database, human mRNA database, and viral genome database.
Primer information.
ACKNOWLEDGMENTS
We thank the Whiteley lab members for critical discussions of the manuscript.
This work was supported by grants from the NIH (1R01DE020100 to M.W. and 5F31DE021633-02 to P.J. and M.W.). M.W. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Citation Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M. 2014. Metatranscriptomics of the human oral microbiome during health and disease. mBio 5(2):e01012-14. doi:10.1128/mBio.01012-14.
REFERENCES
- 1. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6:1621–1624. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O’Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. 10.1093/nar/gkq873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6:1176–1185. 10.1038/ismej.2011.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wesemann DR, Portuguese AJ, Meyers RM, Gallagher MP, Cluff-Jones K, Magee JM, Panchakshari RA, Rodig SJ, Kepler TB, Alt FW. 2013. Microbial colonization influences early B-lineage development in the gut lamina. Nature 501:112–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hooper LV, Littman DR, Macpherson AJ. 2012. Interactions between the microbiota and the immune system. Science 336:1268–1273. 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. 2011. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331:337–341. 10.1126/science.1198469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. 2013. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498:99–103. 10.1038/nature12198 [DOI] [PubMed] [Google Scholar]
- 8. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023. 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- 9. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J. 2006. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55:205–211. 10.1136/gut.2005.073817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. 2013. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339:1084–1088. 10.1126/science.1233521 [DOI] [PubMed] [Google Scholar]
- 11. Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 7:1016–1025. 10.1038/ismej.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ge X, Rodriguez R, Trinh M, Gunsolley J, Xu P. 2013. Oral microbiome of deep and shallow dental pockets in chronic periodontitis. PLoS One 8:e65520. 10.1371/journal.pone.0065520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simón-Soro A, Tomás I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. 2013. Microbial geography of the oral cavity. J. Dent. Res. 92:616–621. 10.1177/0022034513488119 [DOI] [PubMed] [Google Scholar]
- 14. Macklaim JM, Fernandes AD, Di Bella JM, Hammond JA, Reid G, Gloor GB. 2013. Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 1:12. 10.1186/2049-2618-1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maurice CF, Haiser HJ, Turnbaugh PJ. 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152:39–50. 10.1016/j.cell.2012.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. 2013. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7:2061–2068. 10.1038/ismej.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kastenmüller G, Schenk ME, Gasteiger J, Mewes HW. 2009. Uncovering metabolic pathways relevant to phenotypic traits of microbial genomes. Genome Biol. 10:R28. 10.1186/gb-2009-10-3-r28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31:814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27:29–34. 10.1093/nar/27.20.e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niederman R, Buyle-Bodin Y, Lu BY, Robinson P, Naleway C. 1997. Short-chain carboxylic acid concentration in human gingival crevicular fluid. J. Dent. Res. 76:575–579. 10.1177/00220345970760010801 [DOI] [PubMed] [Google Scholar]
- 21. Chang MC, Tsai YL, Chen YW, Chan CP, Huang CF, Lan WC, Lin CC, Lan WH, Jeng JH. 2013. Butyrate induces reactive oxygen species production and affects cell cycle progression in human gingival fibroblasts. J. Periodontal Res. 48:66–73. 10.1111/j.1600-0765.2012.01504.x [DOI] [PubMed] [Google Scholar]
- 22. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aronesty E. 2013. Comparisons of sequencing utility programs. Open Bioinforma. J. 7:1–8. 10.2174/1875036201307010001 [DOI] [Google Scholar]
- 25. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dodt M, Roehr J, Ahmed R, Dieterich C. 2012. FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1:895–905. 10.3390/biology1030895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jorth P, Trivedi U, Rumbaugh K, Whiteley M. 2013. Probing bacterial metabolism during infection using high-resolution transcriptomics. J. Bacteriol. 195:4991–4998. 10.1128/JB.00875-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. 2010. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010:baq013. 10.1093/database/baq013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson MD, Smyth GK. 2007. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23:2881–2887. 10.1093/bioinformatics/btm453 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Average bacterial species abundances in healthy and diseased periodontal samples determined by rRNA-seq. (A) rRNA-seq was used to determine OTUs present in health and disease. Shown are average abundances for healthy (blue) and diseased (red) samples. Error bars indicate standard errors of the means (n = 10). (B) OTU abundances for healthy and diseased samples from patients 1, 2, and 3, which were selected for RNA-seq. Download
rRNA-seq sequencing and analysis information.
Spearman correlations of 16S rRNA gene and 16S rRNA sequencing.
RNA-seq sequencing information.
Percent specific RNA-seq reads aligned to the HOMD database, human mRNA database, and viral genome database.
Primer information.