

ABSTRACT
Most fungal genomes are poorly annotated, and many fungal traits of industrial and biomedical relevance are not well suited to classical genetic screens. Assigning genes to phenotypes on a genomic scale thus remains an urgent need in the field. We developed an approach to infer gene function from expression profiles of wild fungal isolates, and we applied our strategy to the filamentous fungus Neurospora crassa. Using transcriptome measurements in 70 strains from two well-defined clades of this microbe, we first identified 2,247 cases in which the expression of an unannotated gene rose and fell across N. crassa strains in parallel with the expression of well-characterized genes. We then used image analysis of hyphal morphologies, quantitative growth assays, and expression profiling to test the functions of four genes predicted from our population analyses. The results revealed two factors that influenced regulation of metabolism of nonpreferred carbon and nitrogen sources, a gene that governed hyphal architecture, and a gene that mediated amino acid starvation resistance. These findings validate the power of our population-transcriptomic approach for inference of novel gene function, and we suggest that this strategy will be of broad utility for genome-scale annotation in many fungal systems.
IMPORTANCE
Some fungal species cause deadly infections in humans or crop plants, and other fungi are workhorses of industrial chemistry, including the production of biofuels. Advances in medical and industrial mycology require an understanding of the genes that control fungal traits. We developed a method to infer functions of uncharacterized genes by observing correlated expression of their mRNAs with those of known genes across wild fungal isolates. We applied this strategy to a filamentous fungus and predicted functions for thousands of unknown genes. In four cases, we experimentally validated the predictions from our method, discovering novel genes involved in the metabolism of nutrient sources relevant for biofuel production, as well as colony morphology and starvation resistance. Our strategy is straightforward, inexpensive, and applicable for predicting gene function in many fungal species.
INTRODUCTION
Fungi are estimated to account for 25% of the world’s biomass (1) and to comprise as many as 5 million species (2). Almost all fungal species can grow as filaments that invade the substrate as they feed. However, most of what we know about the genetic basis of fungal growth and the coordination of nutrient acquisition, transport, and metabolism has come from research on Saccharomyces cerevisiae, a unicellular yeast that feeds by diffusion (3). Recently, this situation has changed due to the remarkable suitability of small (~30-Mb), haploid, easily cultivatable and essentially immortal filamentous fungi as subjects for whole-genome sequencing, such that more than one-third of all eukaryotic, whole-genome sequences are fungal (4). This sequencing effort has led to a rich collection of genomes of many filamentous fungi but one that is poorly annotated because filamentous fungi harbor thousands of genes absent from unicellular yeast (5). In the filamentous fungus Neurospora crassa, a flagship model organism, ~40% of the predicted genes remain annotated with no known function.
Traditionally, functions of uncharacterized genes have often been discovered in screens of deletion mutants engineered in an isogenic background (6, 7). A powerful complementary approach instead exploits the genetic changes that have arisen naturally in wild populations. When variation across outbred individuals affects the regulation of genes of common function (8), the biological role of an unannotated gene falling into such a regulon can be inferred by reference to the annotations of the rest of the group (9). Unlike S. cerevisiae, where a heterogeneous population structure combined with the small number of available wild isolates has made it difficult to perform genome-wide association studies (10), N. crassa is particularly well suited for population analyses owing to the detailed understanding of population structure (11, 12) and the large and growing culture collection of wild strains in this species (13–17). Here, we report on the use of expression as a genome-scale screening tool in fewer than 150 wild individuals, far fewer than the >8,000 mutants of predicted nonessential genes in N. crassa that would be screened for phenotype in a library of deletion mutants.
We set out to survey transcriptional variation in wild N. crassa, both intrapopulation differences between strains isolated in Louisiana (18) and differences between the Louisiana population and one from the southern Caribbean (11). From these analyses, we formulated hypotheses about genes of unknown function that might mediate growth, metabolic, and regulatory traits in N. crassa, and we focused on several as case studies for validation of our methods. Our experiments targeted the roles of these genes with respect to three fundamental aspects of filamentous fungal biology: colony development, regulation of iron acquisition, and global regulation of nitrogen and carbon metabolism. The results led to the discovery of novel phenotypes for four previously uncharacterized genes.
RESULTS
Inferring multigene regulons from expression variation across an N. crassa population.
To survey variation in N. crassa gene expression, we made use of our transcriptional profiles recently generated from wild isolates of this fungus collected in Louisiana (19). Of the 9,733 predicted N. crassa genes, 8,876 had mapped reads in at least 24 of the wild isolates, and we considered the latter set of genes to represent the core active transcriptional program of N. crassa under the standard growth conditions of our cultured colonies.
To harness regulatory variation across strains to infer gene function, we first used our expression profiles to define coexpressed gene clusters, applying a resampling strategy to assess the significance of cluster sizes (see Materials and Methods). At a cluster size of nine genes, we identified 188 clusters whose gene expression was correlated across wild strains with a correlation coefficient of 0.4 or greater, whereas no such clusters were detected in permuted data sets (see Table S1 in the supplemental material). The majority of clusters (92%) contained at least three genes that have been annotated in functional categories according to the Functional Catalogue (FunCat) (20). In 72% of these clusters, we detected functional category enrichment at a P value of ≤0.05 (Benjamini-Hochberg-corrected hypergeometric test; see Data Set S1), thereby highlighting the potential of our clustering data set as a resource for the inference of function of uncharacterized genes.
Molecular validation of a novel hyphal morphology gene.
To investigate, at the molecular level, the inferred function of uncharacterized N. crassa genes that underlie growth traits, we first focused on a coexpressed cluster of genes (cluster 48 in Data Set S1 in the supplemental material; see also Table S2) encoding proteins that (i) are localized to septa and cell walls (RHO4, COT2, and ACW11), (ii) act as signaling molecules and transcription factors (RHO1, ADA6, BEK2, and CHM1), and (iii) have suspected roles in hyphal branching and septa formation (RHO4, RHO1, and CHM1). Also among the genes in this cluster was NCU04826, which encodes a hypothetical protein lacking any annotated function or protein domains. Sequence searches revealed homologs of this gene in species of filamentous fungi within the Sordariomycetes and Leotiomycetes, with amino acid identity to the N. crassa sequence ranging from 28% (Glarea lozoyensis) to 97% (Neurospora tetrasperma). Given the coregulation of NCU04826 with known cell wall and morphology genes, we hypothesized that this gene would have a role in hyphal morphology. To test this notion, we obtained an N. crassa strain of the FGSC (Fungal Genetics Stock Center) 2489 background harboring a deletion in NCU04826 (21), crossed it to an isogenic wild-type strain, and examined progeny for colony morphology. Strikingly, inheritance of the deletion cassette conferred a distinct hyperbranching phenotype, as predicted from our coexpression analysis (Fig. 1). In assays of the branching phenotypes of strains bearing deletions of the other genes from the NCU04826 coexpression cluster (see Table S2), the colony and branching morphology of a strain carrying a deletion of NCU02978 most closely resembled that of the ΔNCU04826 strain (see Fig. S1). NCU02978 is predicted to encode a protein similar to Sla1p in S. cerevisiae, an adaptor protein for endocytosis involved in assembly of the actin cytoskeleton. Consistent with a potential role for NCU04826 in the cytoskeleton, we reevaluated the predicted protein product of NCU04826 and detected homology to PFAM “intermediate filament” and “tropomyosin-like” protein families (PF00038 [E = 0.055] and PF12718 [E = 0.0005], respectively), both of which comprise cytoskeleton components. We conclude that NCU04826 represents a previously uncharacterized determinant of colony morphology and hyphal branching, possibly by affecting activity of the cytoskeleton, underscoring the power of our coexpression clustering methods to infer functions of unknown genes. We propose the name hbc-1, hyperbranching and cytoskeleton 1, for this gene.
FIG 1 .
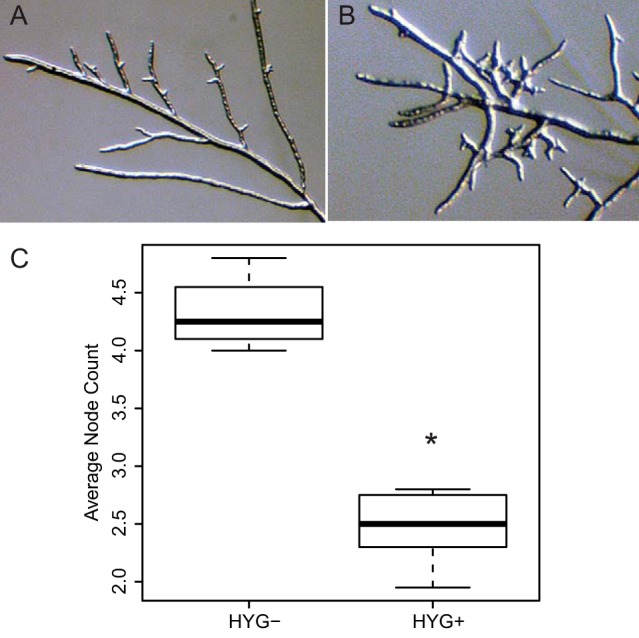
Deletion of the novel Neurospora crassa gene hbc-1 causes a hyperbranching phenotype. Micrographs show branching morphologies of a colony of the wild-type strain FGSC 2489 (A) or an isogenic strain bearing a deletion in hbc-1 (NCU04826) (B). (C) Each column reports the distribution of a quantitative measure of hyphal branching among progeny from a cross between the wild-type and the Δhbc-1 (hbc-1::HYG) strain, with the thick black horizontal bar reporting the median, 25% quartiles shown as a box, and whiskers extending to 1.5 times the interquartile range. The x axis reports genotype across the complete panel of cross progeny strains at the hbc-1 locus: HYG-, segregants inheriting the wild-type hbc-1 gene; HYG+, segregants inheriting the hbc-1 deletion. The y axis reports node count, determined by counting the number of branch junctions from the hyphal tip toward the colony center until a subtending branch was reached that was itself branching. *, the difference between node count in wild-type and hbc-1 mutant strains is significant at a Wilcoxon P value of 0.008.
Molecular validation of a novel amino acid metabolism gene.
We next set out to use our population genomics approach to investigate the regulatory functions of transcription factors. The majority of annotated transcription factors in N. crassa have no known function or target gene set (6), and we reasoned that our clusters of genes defined by coregulation across wild isolates could be harnessed to infer the pathways in which these transcription factors act. As a positive control for this approach, we first examined the 35 N. crassa transcription factors of known function (22) that fell into coexpression clusters enriched for one or more FunCat terms: of these, we identified 15 whose FunCat annotation overlapped with that of genes in their respective clusters (see Data Set S1 in the supplemental material; permutation P < 0.0001). For example, the known carbon catabolite repressor gene cre-1 was annotated in carbohydrate metabolism, as were the genes with which its expression was correlated (see cluster 176 in Data Set S1).
To evaluate this strategy in the context of an unannotated transcription factor, we focused on the gene NCU05257, which encodes a predicted zinc finger and homeobox DNA-binding protein and which fell into an expression cluster containing 58 other genes in our analysis of expression among wild N. crassa strains (cluster 15 in Data Set S1 in the supplemental material). This group was enriched for genes annotated in amino acid metabolism; see Data Set S1 and Table S2), and NCU05257 was previously reported to be a putative target of the N. crassa amino acid biosynthesis regulator CPC1 (23). To test the regulatory impact of NCU05257 directly, we used transcriptome sequencing (RNA-seq) to generate the transcriptional profile of a strain bearing a deletion in NCU05257, and we compared this profile to that of its isogenic wild-type control, identifying 43 genes affected by the NCU05257 mutation at a lenient statistical cutoff (Benjamini-Hochberg-corrected P value < 0.1; see Data Set S2). This expression signature was enriched for genes annotated in amino acid metabolism (see Table S3), a conclusion that was independent of the threshold used to call differential expression (data not shown). Likewise, the NCU05257 deletion signature was enriched for the members of the cluster of genes with which it was coregulated across wild strains (eight genes present in both sets; Table 1; Fisher’s exact test, P = 9.76 × 10−9), and in a quantitative analysis, this cluster was enriched for dramatic expression change upon NCU05257 deletion (resampling P = 0.007). Interestingly, in the NCU05257 mutant, we also noted upregulation of several iron scavenging genes (the aerobactin siderophore biosynthesis protein IUCB, the fatty acid coenzyme A [CoA] ligase NCU06063, which is involved in siderophore biosynthesis [24], and the siderophore iron transporter NCU06132; see Data Set S2).
TABLE 1 .
Genes coregulated with asi-1 (NCU05257) across wild strains are affected by asi-1 deletiona
| ID | Annotation | DE P value | Coexpression correlation |
|---|---|---|---|
| NCU00522 | Cystathionine beta-lyase | 2.3104e−05 | 0.78 |
| NCU00535 | Alanyl-tRNA synthetase | 0.0054 | 0.84 |
| NCU02019 | FAD-dependent oxidoreductase | 1.5469e-08 | 0.82 |
| NCU02543 | Aspartate aminotransferase | 0.0027 | 0.92 |
| NCU05045 | MFS monocarboxylate transporter | 1.8375e−09 | 0.90 |
| NCU05256 | Hypothetical | 0.0013 | 0.81 |
| NCU07126 | Acetyltransferase (GNAT) family | 0.0503 | 0.83 |
| NCU11365 | Aminotransferase | 0.0533 | 0.82 |
Each row shows analysis of one gene whose expression changed significantly upon deletion of asi-1 and was tightly correlated with expression of asi-1 across Louisiana strains of N. crassa. DE P value, Benjamini-Hochberg-corrected significance of differential expression between an engineered asi-1 deletion mutant and an isogenic wild-type-strain; the complete set of genes responsive to asi-1 deletion is given in Data Set S2 in the supplemental material. Coexpression correlation, Spearman’s rank correlation coefficient between the expression levels of the indicated gene and asi-1 across Louisiana isolates; the complete set of genes coregulated with asi-1 is given in Table S2. ID, identifier.
To investigate the phenotypic role of NCU05257, we used the histidine biosynthesis inhibitor 3-amino-1,2,4-triazole (3-AT), which induces growth defects in N. crassa amino acid biosynthesis regulator mutants (25, 26). We crossed the NCU05257 deletion strain to an isogenic wild-type strain and measured the growth rate of progeny in the presence of 3-AT. In this cross, strains inheriting ΔNCU05257 showed significantly compromised growth in 3-AT relative to that of wild-type progeny (Fig. 2), paralleling the behavior of a cpc-1 deletion strain (see Fig. S2 in the supplemental material) and confirming the importance of NCU05257 in the cellular response to amino acid starvation. We conclude that NCU05257 is a novel component of the amino acid metabolic control network in N. crassa with a link to iron scavenging, and we propose the name asi-1, for amino acid and siderophore regulation, for this gene.
FIG 2 .
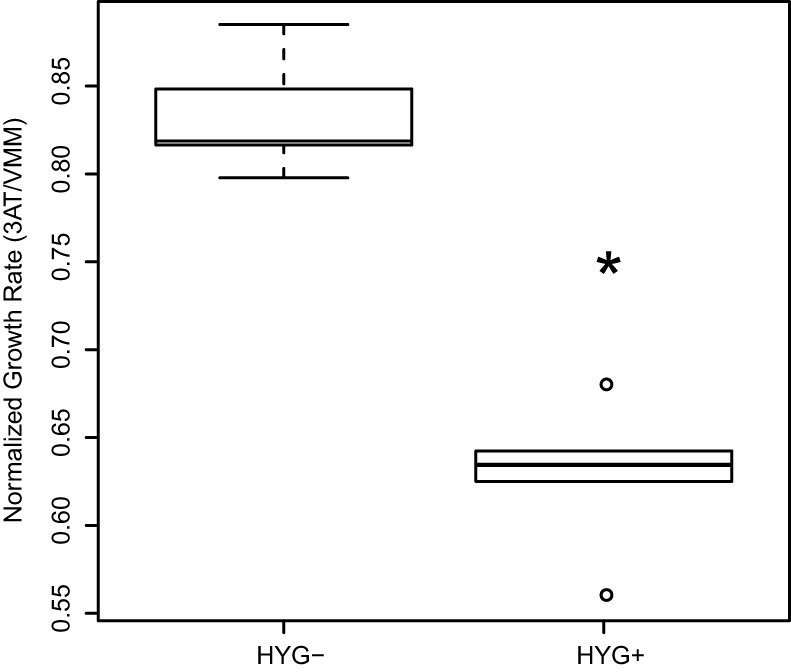
Deletion of asi-1 increases sensitivity to the histidine analogue 3-AT. Each column reports the distribution of growth rate among progeny from a cross between the wild-type strain, FGSC 4200, and an isogenic strain bearing a deletion in asi-1 (NCU05257; asi-1::HYG), with symbols as in Fig. 1 except that points >1.5-fold outside the interquartile range are shown as circles. The x axis reports genotype in cross progeny at the asi-1 locus: HYG-, segregants inheriting the wild-type asi-1 gene; HYG+, segregants inheriting the asi-1 deletion. The y axis reports the ratio between growth rate on Vogel’s minimal medium (VMM) containing 6 mM 3-amino-1,2,4-triazole (3-AT) and growth rate on VMM lacking 3-AT. *, the difference between the growth rate on 3-AT in the wild-type and Δasi-1 mutant strains is significant at a Wilcoxon P value of 0.008.
Regulatory variation between N. crassa populations and the nitrogen metabolite repression pathway.
Having explored the genetic differences among Louisiana strains of N. crassa, we next investigated the variation between two populations by incorporating transcriptional profiles previously reported for a southern Caribbean N. crassa population (11). To polarize changes with the use of an outgroup, we also added profiles of an N. crassa population from Panama (11). Genome-scale comparisons between Caribbean and Louisiana strains detected significant differential expression of 1,539 genes (Benjamini-Hochberg correction, P < 0.05; see Data Set S3 in the supplemental material).
To use expression divergence between populations as a test bed for inference of gene function, we began with the genes most strongly differentially expressed between Louisiana and Caribbean strains. At the top of the list was allantoicase (alc-1) (Fig. 3), a purine degradation gene canonically studied as part of the nitrogen metabolite repression (NMR) program, for its repression in the presence of preferred nitrogen sources (27, 28). Ammonium, at the concentrations used in our transcriptional profiling experiments, is sufficient to trigger NMR in N. crassa (27). This fact made it all the more striking that alc-1, a gene that ought to be under NMR, was highly expressed among wild Louisiana isolates of N. crassa (Fig. 3). This finding of unusual expression of an NMR target gene led us to further investigate interpopulation differences in metabolic repression programs. To analyze as many NMR targets as possible, we began with the set of NMR targets previously characterized in Neurospora, and we added to it Neurospora genes detected by a best-reciprocal-BLAST search of the N. crassa genome using known NMR targets in other fungi (see Data Set S4 in the supplemental material). Of these, we examined the 15 genes whose expression differed between the Louisiana and Caribbean populations and found 9 that were upregulated in Louisiana individuals relative to those of the Caribbean and Panamanian populations (Fig. 3, yellow-shaded panels). Finding such a large number of genes differentially upregulated in the same population constitutes a directional coherence significantly greater than that expected by chance from the genome as a whole (one-tailed Fisher’s exact test, P = 0.005). Thus, Louisiana isolates of N. crassa exhibited a program of high expression of NMR targets in rich medium that was unique among our study populations.
FIG 3 .

Targets of carbon catabolite and nitrogen metabolite repression are upregulated in the N. crassa Louisiana population. Each panel shows expression of one target gene of the nitrogen metabolite repression program (yellow shading; see Data Set S4 in the supplemental material) or of the carbon catabolite repression regulator CRE1 (orange shading [32]), measured in wild strains of N. crassa grown on standard rich medium. In a given panel, each column reports the distribution of expression of the indicated gene across the strains of one N. crassa population: LA, Louisiana; CARIB, Caribbean; PAN, Panama. Symbols are as in Fig. 2. NMR, nitrogen metabolite repression targets; CCR, carbon catabolite repression targets. Systematic gene identifiers, from top left to bottom right, are NCU01066, NCU01816, NCU02333, NCU03076, NCU03257, NCU05387, NCU07675, NCU08356, NCU10007, NCU00721, NCU00130, NCU01140, NCU01449, NCU02904, NCU03151, NCU04039, NCU04197, NCU04460, NCU04963, NCU08746, NCU10021, and NCU07363.
PP4 controls expression of nitrogen metabolite repression and carbon catabolite repression targets.
The NMR target genes differentially expressed between the Louisiana and Caribbean N. crassa clades also exhibited expression-level variation within the Louisiana population (Fig. 3). To complement our interpopulation comparison, we took a candidate-gene approach in hopes of identifying master regulators of metabolite expression programs, as follows. We identified N. crassa orthologs of the set of known NMR regulators in S. cerevisiae (29) (see Data Set S4 in the supplemental material) and examined their coding regions for sequence variation among Louisiana N. crassa strains. Among these genes, the only case of striking sequence difference between strains was in the putative protein phosphatase PP4 (NCU08301), a regulator of the circadian oscillator FRQ in N. crassa (30) whose homolog in S. cerevisiae activates the NMR transcription factor Gln3 (31); Louisiana individuals bore two derived amino acid variants in PP4 at high frequency relative to Caribbean strains (Fig. 4). To investigate the regulatory impact of pp4, we therefore transcriptionally profiled a strain harboring a deletion in this gene alongside an isogenic wild-type strain. Comparison between the two expression profiles identified 195 differentially expressed genes (Benjamini-Hochberg-corrected P value < 0.1; see Data Set S2), of which nearly all (187 genes) showed elevated expression upon pp4 deletion. The response to the deletion of pp4 was significantly more dramatic among NMR targets than expected under a null hypothesis based on genomic resampling (P < 0.0001), consistent with a role for PP4 in the repression of NMR target genes. In addition, we noted in the pp4 deletion signature a number of genes involved in the metabolism of alternative carbon sources, including the mannose metabolism genes NCU07067, NCU02322, NCU07269, and NCU07318 and the polysaccharide metabolism genes NCU08755, NCU04959, NCU09281, and NCU04431 (see Data Set S2). We thus suspected that in addition to the NMR genes we had originally analyzed (Fig. 3, yellow-shaded panels), PP4 also functioned as a repressor of genes subject to carbon catabolite repression (CCR) in glucose medium, which we call CCR targets. As an unbiased test for the role of pp4 in carbon catabolite repression, we considered the 75 genes whose expression increases upon deletion of the N. crassa carbon catabolite repressor CreA/CRE1 (32). Eight of these CCR (CRE1) targets were differentially expressed in the pp4 deletion, an overlap beyond that expected by chance (hypergeometric P = 0.0001). Furthermore, seven of the eight targets shared by CRE1 and PP4 showed elevated expression in the pp4 deletion strain, further supporting a model of PP4 as a repressor of CCR targets. We thus conclude that PP4 functions in the joint regulatory control of nitrogen and carbon metabolism genes in N. crassa.
FIG 4 .

A derived, high-frequency nonsynonymous variant in pp4 is associated with the expression level of a novel putative ammonium sensor. (A) The top panel shows an amino acid alignment of a region of the putative protein phosphatase PP4, in which the major allele among Louisiana strains of N. crassa (N_crassa_LA) encodes a glycine at residue 177, whereas the major allele among Caribbean isolates (N_crassa_CARIB) and the sequence in other Sordariomycetes encodes an aspartate. The inset shows allele frequencies of the aspartate and glycine alleles in the Louisiana and Caribbean N. crassa populations. (B) Each column reports the expression of Nc_nmr6 (NCU00789) across Louisiana N. crassa strains harboring the indicated allele at the PP4 variant in panel A. The Nc_nmr6 expression level associated at modest significance with the pp4 genotype across Louisiana strains (nominal P = 0.0001; Benjamini-Hochberg-corrected P = 0.12). Symbols are as in Fig. 2.
We next hypothesized that if the naturally occurring sequence variants among Louisiana strains in pp4 affected the function of this gene, inheritance at pp4 would be correlated with expression of nitrogen and carbon metabolism pathways across the Louisiana population. To test this notion, we examined both our directly inferred NMR targets and a broader curated set of nitrogen metabolism genes inferred from the nitrogen starvation transcriptional response of Magnaporthe grisea (33) (see Data Set S4 in the supplemental material; see also Materials and Methods) for association of their expression with inheritance at pp4 across Louisiana strains. We likewise tested for association between pp4 genotype and expression of CCR targets, again using the CRE1 target gene set as a reflection of the latter (32). Unexpectedly, the results were not consonant with natural variation in the pp4 sequence having a major impact on gene expression: expression of only one gene, NCU00789, showed modest association with the pp4 genotype across Louisiana strains (Benjamini-Hochberg-corrected P value = 0.12; Fig. 4). Thus, despite the dramatic impact of an engineered deletion in pp4, we concluded that natural polymorphisms in pp4 did not underscore most of the variation in metabolite repression genes within or between wild N. crassa populations. However, given that our exploration of pp4 led us to the uncharacterized gene NCU00789, we considered the latter in its own right as another candidate gene for inference of novel function in N. crassa.
Nitrogen and carbon metabolism gene expression associates with Nc_nmr6.
GenBank lists the protein encoded by NCU00789 (accession no. AY935520.1) as the N. crassa ortholog of the Hansenula polymorpha gene NMR6, an unpublished, 12-transmembrane ammonium sensor, and we refer to the Neurospora version of this gene as Nc_nmr6. The Nc_nmr6 locus harbored extensive variation at coding and silent sites across N. crassa isolates (Fig. 5A), which defined two haplotypes. One Nc_nmr6 haplotype was borne by most strains of the Caribbean population and a few strains from Louisiana, and a second Nc_nmr6 haplotype was apparent in the majority of Louisiana isolates. We thus hypothesized that Nc_nmr6 was a previously uncharacterized component of the nitrogen and carbon metabolism gene network in N. crassa and that polymorphisms at Nc_nmr6 served to tune the expression of its target genes even in medium containing preferred carbon and nitrogen sources.
FIG 5 .

A derived haplotype at the Nc_nmr6 locus (NCU00789) is associated with a gene expression program that mirrors the response to nitrogen starvation. (A) Alignment and insets are as in Fig. 4A, except that a polymorphic region of Nc_nmr6 is shown. (B and C) In a given panel, each row reports expression of one gene for which expression of the N. crassa ortholog was significantly associated with the genotype at Nc_nmr6 across Louisiana strains (nominal P < 0.05; see Materials and Methods for association test details), and expression of the M. grisea ortholog changed >2-fold upon nitrogen starvation in a laboratory strain (33). For a given row, the left column reports the log2 of the average expression level of the respective gene among Louisiana strains bearing the derived haplotype of Nc_nmr6, relative to the analogous average across strains with the ancestral haplotype; the right column reports the log2 of the expression of the respective gene in nitrogen-starved M. grisea, relative to the analogous measurement in untreated cells. (B) Genes for which the derived haplotype of Nc_nmr6 was associated with high expression relative to the ancestral haplotype. (C) Genes for which the derived haplotype of Nc_nmr6 was associated with low expression relative to the ancestral haplotype. For ease of visualization, all association effect sizes in panels B and C were normalized by a multiplicative factor of 5.
To validate our inference of Nc_nmr6 as a determinant of metabolic gene expression, we first investigated the relationship between inheritance at Nc_nmr6 and NMR target gene expression across Louisiana individuals. The results revealed a robust association signal, with the major haplotype of Nc_nmr6 associated with increased NMR target gene expression among Louisiana strains (one-tailed paired Wilcoxon P value = 0.006). We next evaluated the effect of the Nc_nmr6 genotype on the broader set of nitrogen starvation genes inferred from profiles of M. grisea (33). Again we observed a striking relationship (Fig. 5B and C): among the 62 genes for which the major allele of Nc_nmr6 among Louisiana individuals was associated with lower expression, largely components of the translation machinery, 50 were downregulated during nitrogen starvation (Fig. 5B) (binomial P = 9.204e−05). Likewise, of the 37 genes activated by the major allele of Nc_nmr6, 31 were upregulated during nitrogen starvation (Fig. 5C; binomial P = 8.197e−07). The latter included genes involved in the metabolism of alternative carbon sources (e.g., the xylanase NCU08189, the gluconate reductase NCU09519, and the rhamnose synthase NCU10683), as well as nitrogen metabolism genes (e.g., the uricase NCU07853 and the pyrimidine catabolism gene hydantoinase NCU00689). We thus suspected that Nc_NMR6 had a role in the expression of CCR targets as well as NMR targets. To evaluate the impact of Nc_nmr6 on carbon catabolite repression, we examined the association between the Nc_nmr6 genotype and expression of CRE1 target genes (32). Of the latter, 18 were differentially expressed between our Louisiana and Caribbean N. crassa populations, with 14 exhibiting a derived program of increased expression in Louisiana individuals relative to outgroups (one-tailed Fisher’s exact P value = 0.005; Fig. 3, orange-shaded panels). As predicted, among Louisiana individuals, the major Nc_nmr6 haplotype was associated with high expression of CRE1 targets (one-tailed paired Wilcoxon P value = 0.005). We conclude that the major allele of Nc_nmr6 among Louisiana strains is associated with an expression program in rich medium that mirrors the response to nitrogen starvation and the loss of carbon catabolite repression, strongly suggesting that Nc_NMR6, like PP4, functions in the regulation of genes that metabolize nonpreferred nutrient sources.
Nc_nmr6 is a novel determinant of metabolism gene expression.
As a direct test of the regulatory impact of Nc_nmr6, we constructed a strain bearing a deletion of this gene (see Materials and Methods), using as the genetic background FGSC 2489, which bears the major allele from the Louisiana population. We transcriptionally profiled this ΔNc_nmr6 strain and its wild-type isogenic progenitor after growth in rich medium, finding 80 genes that were differentially expressed between the strains (Benjamini-Hochberg-corrected P value < 0.1; see Data Set S2 in the supplemental material), enriched for annotation in a number of functions, including carbohydrate metabolism (see Table S3). Analysis of the Nc_nmr6 deletion signature bore out our prediction of a role for Nc_nmr6 in nitrogen metabolism regulation: the suite of NMR targets that we had originally ascertained based on upregulation in Louisiana strains relative to levels in other populations (Fig. 3, yellow-shaded panels) were expressed at low levels in the ΔNc_nmr6 mutant (resampling P value = 0.01; Fig. 6). Likewise, again conforming to our expectation, the engineered ΔNc_nmr6 mutant expressed CRE1 targets at lower levels than the wild-type control (Fig. 6). We conclude that Nc_NMR6 functions in the regulation of NMR and CCR gene targets and, in contrast to the behavior of the putative repressor PP4, is required for the high expression of these genes by the Louisiana strain FGSC 2489 in rich medium. Given the impact of both PP4 and Nc_NMR6 on multiple nutrient response pathways, our data thus implicate these proteins as two novel control points for expression of metabolite repression programs in N. crassa.
FIG 6 .

Nc_nmr6 (NCU00789) controls expression of target genes of the carbon catabolite and nitrogen metabolite repression programs. Each column reports the distribution, across the genes of the indicated set, of the change in expression between a strain harboring an engineered deletion in Nc_nmr6 (KO) and an isogenic wild-type (WT) strain. All genes, the complete set of genes expressed in the knockout experiment; DE between populations, the set of genes significantly differentially expressed between Louisiana and Caribbean N. crassa strains (see Data Set S3 in the supplemental material); NMR genes upregulated in LA, the set of inferred targets of the nitrogen metabolite repression program upregulated in Louisiana strains relative to other N. crassa populations (see yellow panels in Fig. 3); CCR genes upregulated in LA, the set of genes repressed by the carbon catabolite repression regulator CRE1 and upregulated in Louisiana strains relative to other N. crassa populations (see orange panels in Fig. 3). For the last three columns, the text at the top reports the resampling-based estimate of significance of the difference between the indicated regulon and the set of all genes with respect to the expression effects of Nc_nmr6 deletion. Symbols are as in Fig. 2, except that for ease of visualization, data points falling outside the box plot whiskers are not shown.
DISCUSSION
Transcripts that are up- and downregulated together across conditions may often encode proteins of similar functions. This idea has motivated studies of coregulation between unknown and well-characterized genes (34, 35), to shed light on novel, species- or condition-specific gene functions. A key roadblock in the field is that such highly specialized genes may be inactive under the experimental conditions used for standard analyses of gene coregulation, even in microbes (35–37). In contrast, when natural genetic variation impacts the expression of a wide range of genes, it can enable a broader survey of gene function than almost any other experimental design (36). To date, schemes using wild populations to predict gene function have been almost untested in the literature (9), and experimental follow-up of predictions from coexpression analyses has been at a premium. In this work, we established a pair of wild populations of N. crassa as a model system for the inference of gene function from expression profiles. This approach resulted in a diverse set of functional annotations enriched across 143 multigene coexpression clusters. Using the power of molecular genetics with N. crassa, we then experimentally validated the roles of unannotated genes from these clusters in filamentous fungus traits.
Hyphal branching plays a critical role in the interaction between a fungus and itself (formation of mycelial colonies), other organisms (nonself recognition), and its growth substrate (foraging) (38). Our analysis of the novel hyphal branching factor NCU04826 focused on a cluster of N. crassa genes whose products localize to cell or hyphal peripheries or have known functions as regulators of cell polarity and hyphal growth. The pattern of coexpression across strains indicates that expression of these upstream regulators is itself under joint control in the regulatory network, suggestive of feedback. Our results implicate NCU04826, which we named hbc-1, for hyphal branching and cytoskeleton, as a novel determinant of the polar growth machinery. The hyperbranching phenotype of the NCU04826 deletion strain resembles the effects of mutations in the cytoskeleton assembly control protein NCU02978 and those in the cot-1 and pod-6 kinases, which block hyphal extension and force lateral rather than directional growth (39, 40). In light of the apparent absence of NCU04826 homologs in unicellular yeasts, this gene likely contributes to the distinct biology of polar growth in the filamentous fungi (41), highlighting the utility of dedicated annotation efforts for these species.
Functional inference methods are in urgent demand for transcription factors, whose biological roles often remain unknown even in exhaustively studied model organisms like budding yeast (37). Our case study for characterization of a novel transcription factor was a coexpression cluster enriched for amino acid metabolism genes that contained NCU05257, a gene encoding a protein with predicted zinc finger and homeobox DNA-binding domains. Of the genes whose expression dropped when NCU05257 was deleted, many overlapped with the original coexpression gene cluster and/or were implicated in amino acid metabolism and iron scavenging via siderophores. Challenging the ∆NCU05257 strain with an amino acid biosynthesis inhibitor demonstrated the importance of this gene in the response to amino acid starvation. We named the NCU05275 gene asi-1, for amino acid and siderophore regulation; our results provide a first window onto the potential function of this gene in coordinating the joint regulation of fungal siderophore biosynthesis with amino acid supply (42, 43).
Besides analyzing expression variation across Louisiana strains of N. crassa, we also predicted gene function using the differences between two populations of this fungus, isolates from Louisiana and the southern Caribbean. The gene with the strongest interpopulation transcription difference, that encoding allantoicase, is part of the NMR program. This observation led us to a focus on patterns of coordinated expression between nitrogen and carbon metabolism genes and the discovery of a role in these metabolic networks for two putative upstream signaling factors. Our experiments implicated the putative protein phosphatase gene pp4 in the repression of genes involved in the metabolism of both nonpreferred nitrogen and carbon sources. In S. cerevisiae, where nutrient sensing networks have been well delineated, no protein phosphatase is known to participate in both carbon catabolite and nitrogen metabolite repression (44); it is thus tempting to speculate that the division of labor among protein phosphatases in nutrient sensing pathways has diverged between yeast and N. crassa. Likewise, given that PP4 has been well characterized in N. crassa as a regulator of the circadian clock machinery (30), we hypothesize that this protein may function as a novel component of the recently discovered gene network for circadian control of metabolism in N. crassa (45), plausibly linking the circadian clock with nitrogen and carbon scavenging under nutrient-poor conditions.
Our analysis of the putative transmembrane protein Nc_NMR6 identified a haplotype in Louisiana strains associated with high expression of genes involved in metabolism of nonpreferred carbon and nitrogen sources, even during growth in glucose- and ammonium-containing medium. The reduced expression of these nutrient-scavenging genes that we observed in an Nc_nmr6 deletion strain is consistent with either of two possible models for the function of the encoded protein. On the one hand, the ancestral haplotype of Nc_NMR6 could transduce a positive signal of the presence of preferred carbon and nitrogen sources, analogous to well-characterized glucose and ammonium receptors in many fungi (46–48). Under this model, the major allele of Nc_nmr6 in the Louisiana population would act as a dominant negative loss of function, failing to signal the availability of preferred nutrient sources in rich medium and leading to derepression of NMR and CCR targets. Upon deletion of this allele of Nc_nmr6 in the Louisiana genome, metabolite repression would be reinstated by CRE1, NMR1, and other NMR and CCR regulators in N. crassa (49, 50). Alternatively, the ancestral form of Nc_NMR6 could actively transduce a signal in the absence of preferred nutrient sources, activating the metabolic machinery for nutrient scavenging; the major haplotype in the Louisiana population would then act as a gain of function to elevate expression of NMR and CCR targets even in rich medium, and deletion of this allele would abrogate the expression effect. In either case, our results make clear that Nc_NMR6, like PP4, functions as part of a complex network for the joint control of carbon and nitrogen metabolism. The regulatory impact of both genes dovetails with that of the sugar sensor TPS1 in Magnaporthe oryzae, which signals to regulators of genes for metabolism of nonpreferred nitrogen and carbon sources (51–53). And our de novo inference of the functions of these previously uncharacterized genes underscores the power of the comparative transcriptomic approach for gene discovery.
Coexpression analysis can be a powerful approach for the prediction of gene function, though to date, relatively few genome-scale studies have experimentally validated the in silico inference of a gene’s role at the organismal level (54–59). By validating the regulatory and organismal impact of four novel genes, our work makes clear that expression differences between wild isolates can lead to biologically meaningful inferences of gene function, whether the loci of interest are involved in gross morphology or the regulation of metabolism. The coexpression clusters we have reported here contain 2,243 other unannotated genes well suited for a similar functional discovery pipeline, in which the deletion strain of any such gene can be assayed for traits in which its coregulated partners are known to play a role. We hope that by demonstrating the approach, our work will stimulate other researchers to use the collection of Louisiana and Caribbean N. crassa strains that have been transcriptionally profiled to determine functions for unknown genes in the pathways that interest them. Given the homology among genes in filamentous Ascomycota, the N. crassa collection will be of immediate use in inference of gene function in other species. And the strategies we have established for population-genomic transcriptional profiling and analysis in N. crassa will be relevant for future work in any fungus for which sufficient isolates can be collected.
MATERIALS AND METHODS
Strains, sequencing, and gene expression quantification.
The complete set of wild Louisiana isolates and engineered strains used in this study is listed in Table S4 in the supplemental material and was obtained from the Fungal Genetics Stock Center (FGSC) (60). Our analysis of Louisiana strain transcriptomes used 48 of the RNA-seq libraries from reference 19, which, for analysis on the same footing as data for other N. crassa populations (see below), we remapped as follows. For each RNA-seq data set, we mapped Illumina reads to the Neurospora crassa OR74A version 10 reference assembly (61) using the software program TopHat (62). We required reads to map uniquely with a maximum of two mismatches and minimum and maximum intron lengths of 40 and 200 bp, respectively, for mapping, coverage, and split-segment searches. We supplied TopHat with the Broad Institute’s February 2010 release of the N. crassa version 4 annotation (http://www.broadinstitute.org/annotation/genome/neurospora/) and also allowed the program to search for novel splice junctions; we then integrated all putative isoforms into a single expression measure per gene. We used a custom Perl script and the N. crassa annotation to calculate the number of raw reads overlapping each gene model and used the third-quartile method from reference 63 to normalize read counts between lanes. For expression measurements from wild strains, we excluded from further analysis any gene for which >50% of the individuals analyzed had an expression level of zero.
Expression clusters.
To generate expression clusters from transcriptional profiles of wild Louisiana strains of N. crassa, we first normalized read counts for each gene in each RNA-seq library by gene length. We next used the Statistics::RankCorrelation Perl module to calculate Spearman’s rank correlation coefficients between all pairwise comparisons of the 8,361 N. crassa genes using expression levels from the 48 Louisiana individuals. We used the absolute value of each correlation coefficient as input into the hclust package in the R software environment (64) and applied the method of reference 65 to define groups of coregulated genes. Briefly, given a correlation coefficient R, each cluster contains a set of genes among which any pair exhibit correlated expression across the strains with a coefficient of at least R. To establish a cutoff for R in Table S1 in the supplemental material, we permuted expression measurements between individuals and repeated the clustering analysis; then, given a cluster size s, in this null set we tabulated the number npermut of clusters containing at least s genes. Across 10 such permutations, for R = 0.4 and s = 9, npermut was zero (see Table S1); clusters of size 9 or larger in analysis of the real data are reported in Data Set S1.
Functional enrichment tests in clusters.
We tested each cluster of genes subject to coregulation across Louisiana strains for enrichment of gene functions as follows. We downloaded N. crassa gene annotations from the FunCat database maintained by the Munich Information Center for Protein Sequences (20). We eliminated from analysis all clusters that contained <9 genes and those with <2 genes with assigned FunCat annotations; the final set comprised 143 clusters. We used a custom Perl script and the R function phyper to assess enrichment of each FunCat annotation term in each cluster. We used the Benjamini-Hochberg (66) correction for multiple hypothesis testing within each of the five FunCat levels; see Data Set S1 in the supplemental material for lists of all FunCat terms and all clusters exhibiting enrichment at a corrected P value of <0.05.
NCU04628 conservation.
Homologs of NCU04628 were identified using the Joint Genome Institute MycoCosm fungal genomics web resource (67). Multiple sequence alignment was performed using the software program MUSCLE (68).
Hyphal morphology assays.
The deletion strain for NCU04826 (FGSC 16805) (6) (mating type a) was crossed to an isogenic wild-type control (mating type A; FGSC 2489) on Westergaard synthetic crossing medium (69), and progeny were screened for hygromycin resistance on sorbose medium (70) with and without 200 µg/ml hygromycin (Sigma). Five hygromycin-resistant and five hygromycin-sensitive progeny were inoculated onto petri dishes containing Vogel’s minimal medium (VMM) (71) and allowed to grow overnight. Strains were observed under a dissecting microscope, and hyphal branching was scored as follows. For each hypha, the number of nodes nn was counted, starting from the tip and moving toward the center of the colony, until the first subtending branch that was also branching was reached. Values of nn were averaged across 20 randomly selected hyphal tips for each individual.
For imaging of colonies of strains harboring deletions in genes coregulated with NCU04826 in Fig. S1 in the supplemental material, each deletion strain in the FGSC 2489 background (6) was obtained from the Fungal Genetics Stock Center and grown on VMM overnight.
Generation of Nc_nmr6 deletion strain.
The gene knockout procedure for NCU00789 was modified from a previously published method (6) as follows. The strain FGSC 9717 (mus-51::bar a) was grown for 3 days at 30°C in the dark, followed by another 7 days at room temperature, in 100 ml VMM containing 2% (wt/vol) sucrose and 1.5% (wt/vol) agar. Conidia were collected by filtration, washed three times with ice-cold 1 M sorbitol, and resuspended in a final volume of 5 ml. An aliquot of 90 µl (~109 conidia) was mixed with 1.0 µg of DNA encoding an NCU00789 deletion cassette (the sequence of the hph hygromycin resistance gene [72] flanked by ~1 kb of the genomic region upstream of NCU00789 in the N. crassa reference genome and ~1 kb of the region downstream of the gene, kindly provided by Carol Ringelberg, Department of Genetics, Dartmouth Medical School). DNA entry into conidia was via electroporation on a Bio-Rad Gene Pulser II with the following settings: 1.5 kV, 600 Ω, and 25 µF. Treated conidia were then mixed with 900 µl ice-cold 1 M sorbitol and 30 ml of a top agar solution containing 20% sucrose, 0.5% fructose, and 0.5% glucose (FGS) warmed to 50°C. This mixture was overlaid on each of three agar plates containing FGS medium and 200 µg/ml hygromycin, and subsequently incubated at 30°C for 3 days. Individual colonies were used to inoculate agar slants (3 ml VMM containing 2% sucrose and 200 µg/ml hygromycin), from which DNA was isolated and screened by PCR with the primers 5′ TGCAATAGGTCAGGCTCT 3′ (hyg) and 5′ GCGGATAACAATTTCACACAG 3′ (NCU00789) using Phire Hot Start polymerase according to the manufacturer’s recommendations (Thermo Scientific; catalogue no. F-130).
To reduce the likelihood of background mutations in NCU00789 deletion strains, each PCR-confirmed mutant was backcrossed to a wild type as follows. FGSC 2489 was grown as a female strain on Westergaard medium (69) for 7 days to allow formation of protoperithecia and crossed with conidia from each deletion strain of the opposite mating type. Ascospores for each cross were collected, placed in sterile water, and induced to germinate via incubation at 60°C for 30 min. Treated ascospores were spread on VMM–2% sucrose plates containing 400 µg/ml hygromycin and grown at 30°C for 14 h. Germinated spores were isolated and used to inoculate 3-ml VMM–2% sucrose slants. Cultures were checked for loss of the bar gene by lack of growth on VMM without NH4NO3, 0.5% l-proline, 2% sucrose, and 400 µg/ml ignite. Progeny from each cross were also checked for integration of the hygromycin cassette by PCR as described above.
Transcriptional profiling of N. crassa deletion strains.
Following methods described elsewhere (73), we cultured, harvested, and isolated RNA from one replicate of the pp4 deletion strain (FGSC 12454) (6), one of the NCU05257 deletion strain (FGSC 16020) (6), and one of OR74A (FGSC 4200) as a wild-type control, and separately, we isolated RNA from one replicate of the NCU00789 deletion strain and an OR74A wild-type control. Multiplex library construction from each isolate’s cDNA was done according to the manufacturer’s protocol (Truseq v2 LT sample prep kit; Illumina) except that adaptors were diluted 5× before ligation to blunt end cDNA. The RNA-seq libraries were sequenced on an HiSeq2500 instrument (Illumina) as single-end 50-bp reads.
In each RNA-seq data set, we eliminated from analysis all genes with <5 mapped reads. We then assessed the significance of differential expression of each gene between each deletion strain and its respective control using the R software package DEseq (74), normalizing read counts between lanes in each comparison using the estimateSizeFactors function and using expression variance across the pooled samples as described previously (74). Genome-wide expression measurements for each deletion strain are given in Data Set S2 in the supplemental material.
We used a resampling approach to evaluate the expression response of a regulon of interest in the NCU05257 (asi-1), NCU08301 (pp4), or NCU00789 (Nc_nmr6) deletion strain transcriptional profile. For this purpose, we first tabulated the average of the log10 fold change in expression between the wild type and the deletion strain across the genes of the regulon. We then applied the same procedure to a gene set of the same size as the true regulon, randomly drawn from the pool of expressed genes (excluding those present in the regulon). The latter resampling procedure was repeated 10,000 times. Significance, in a one-tailed test of the hypothesis that the true regulon was expressed at lower levels than the null expectation, was assessed as the proportion of resampled gene sets whose average expression ratios were less than or equal to that of the true regulon.
Amino acid starvation assays.
The deletion strain for NCU05257 (FGSC 16020) was crossed to an isogenic wild-type control (FGSC 4200), and five progeny strains bearing the deletion and five wild-type progeny were recovered, as described above. Agar plugs were taken from each progeny culture and used to inoculate race tubes (75) containing VMM (71) with and without 6 mM 3-AT (Sigma). Each tube was incubated in constant light at 25°C, and the location of the hyphal front was recorded daily until it reached the opposite end of the tube. For comparative purposes, we also measured the growth rate for three replicates of a cpc-1 mutant strain (FGSC 4264) (76) on race tubes containing VMM with and without 6 mM 3-AT.
Differential expression between N. crassa populations.
We calculated normalized expression measurements for each of 19 Caribbean isolates and 3 Panamanian isolates of N. crassa as described above, using previously published RNA-seq data (11). Given these measures and the expression profiles of Louisiana isolates as described above, we tested each gene in turn for differential expression between the Caribbean and Louisiana populations using the Wilcoxon test; we then corrected these nominal empirical P values using the Benjamini-Hochberg method (66) for multiple hypothesis testing. Using the set of 1,539 genes with differential expression significant at a Benjamini-Hochberg-corrected P value of <0.05, we evaluated enrichment of FunCat annotations as above.
Curation of nitrogen metabolism gene sets in N. crassa.
To define the nitrogen metabolite repression program in N. crassa in Data Set S4 in the supplemental material, we used the NMR (nitrogen metabolite repression) signature from Saccharomyces cerevisiae (77) to identify 59 homologs in the reference sequence of N. crassa by a best-reciprocal-BLAST search (protein BLAST with E value cutoff of 1e−04). We also included NMR-controlled genes characterized for Neurospora (78–81) for a total of 66 genes.
To define a broader set of nitrogen metabolism genes in N. crassa, we downloaded expression measurements of Magnaporthe grisea under nitrogen-starved and nitrogen-replete conditions from reference 33 and retained for analysis all genes for which the absolute value of the log2 signal intensity ratio between the conditions was 2-fold or greater. We then used the FungiDB database (82) to identify N. crassa orthologs of these genes (see Data Set S4 in the supplemental material). We also added genes annotated as involved in nitrogen metabolism in N. crassa using GO (gene ontology) terms, FunCat terms, and the Broad Institute annotation (http://www.broadinstitute.org/annotation/genome/neurospora/), for a total of 1,203 genes (see Data Set S4).
As our set of candidate regulators of NMR in Data Set S4, we compiled a list of S. cerevisiae regulators from reference 29 and examined all Neurospora homologs of these genes using homology search criteria so as to include both orthologs and paralogs (protein BLAST without best-reciprocal-BLAST requirement; E value cutoff of 1e−10).
Carbon catabolite repressed genes in N. crassa.
Our list of carbon metabolite repressed genes was derived from transcriptional profiling of the cre-1 deletion strain performed previously (32). Specifically, we used the 75 genes showing expression levels that were significantly elevated in the cre-1 deletion strain compared to wild-type levels when grown on minimal medium (see Table S2 in reference 32).
Association of gene expression with genotype.
To test expression of a given nitrogen metabolism gene for association with inheritance at the DNA level across Louisiana strains, given the normalized expression measurements for a transcript and genotypes at a single-nucleotide variant, we first split the strains into two groups on the basis of inheritance at the variant and then used the Wilcoxon test with Benjamini-Hochberg correction as described above.
SUPPLEMENTAL MATERIAL
Cluster membership and functional enrichment of coregulated genes across Louisiana isolates of N. crassa. Regulon clusters: each row lists gene identifiers (IDs) for a gene cluster defined based on coregulation across wild Louisiana strains of N. crassa, with pairwise correlation among genes of a cluster at Spearman’s correlation coefficient ≥ 0.4. Red text indicates transcription factors of known function for which FunCat category annotations (20) associated with the factor were also enriched among genes of the indicated cluster other than the factor itself. Cluster enrichment, each row reports a FunCat annotation term (20) enriched among the genes of the indicated cluster inferred from expression profiles across Louisiana strains of N. crassa. Corrected P value, results of a hypergeometric test for overlap between the indicated FunCat category and the indicated cluster, corrected for multiple testing by the Benjamini-Hochberg method. Only enrichments at corrected P < 0.05 are shown. Download
Gene expression profiles of engineered N. crassa mutants. Genome-wide expression, each row reports expression measurements for one gene, and each column reports expression in one strain. For details of RNA-seq normalization, see Materials and Methods in the main text. DE genes, each tab reports all genes significantly differentially expressed, at corrected P values < 0.1, in a Wilcoxon test for differential expression between the indicated deletion strain and a wild-type control, corrected for multiple testing by the Benjamini-Hochberg method. KO/WT expression, log10 of the ratio of expression of the indicated gene between the deletion strain and an isogenic wild-type strain. Download
Genes differentially expressed between the Caribbean and Louisiana populations of N. crassa. Corrected P value, results of a Wilcoxon test for differential expression of the indicated gene between Louisiana strains and Caribbean strains of N. crassa, corrected for multiple testing by the Benjamini-Hochberg method. Columns labeled with “MEDIAN_EXP” report median expression of the indicated gene across strains of the indicated population: LA, Louisiana; CA, Caribbean; PAN, Panama. The last column reports the ratio between the median expression of the indicated gene among Louisiana strains and the median expression among Caribbean strains. Only genes with corrected P values < 0.05 are shown. Download
N. crassa genes with putative functions related to nitrogen metabolism. NMR genes, known and inferred targets of the nitrogen metabolite repression (NMR) program in N. crassa. The first nine lines report genes previously shown to be targets of the NMR program in N. crassa. All other lines list inferred N. crassa homologs of S. cerevisiae NMR targets from reference 77. NMR regulators: inferred N. crassa homologs of master regulators of the nitrogen metabolite repression program. Shown are the results of sequence searches of the N. crassa proteome for homologs of S. cerevisiae genes annotated as master regulators of the nitrogen metabolite repression program (29). For details of sequence search methods, see Materials and Methods in the main text. M. grisea N starvation genes, inferred nitrogen acquisition and metabolism genes in N. crassa. Shown are the results of sequence searches of the N. crassa proteome for homologs of Magnaporthe grisea genes responsive to nitrogen starvation according to reference 33. For details of sequence search methods, see Materials and Methods. For N. crassa nitrogen genes, shown is the union of the sets of genes whose annotation (http://www.broadinstitute.org/annotation/genome/neurospora/), Gene Ontology term, and/or FunCat terms were related to nitrogen acquisition or metabolism. Download
Deletion of genes coregulated with hbc-1 (NCU04826) confers morphological defects. Each micrograph shows hyphal architecture of a colony of a strain harboring a deletion in a gene coregulated with hbc-1 across Louisiana isolates of N. crassa (see Table S2). Download
The histidine analog 3-AT compromises growth of a cpc-1 mutant. Each column reports the distribution, across three biological replicates under one environmental condition, of the growth rate of an engineered cpc-1 deletion strain. VMM, Vogel’s minimal medium; VMM + 3AT, Vogel’s minimal medium containing 6 mM 3-AT. Symbols are as in Fig. 1C of the main text. *, the cpc-1 mutant grows significantly slower in the presence of 3-AT (one-sided Wilcoxon test P value = 0.05). Download
Numbers of clusters of coregulated genes inferred from expression profiles of Louisiana strains exceed the chance expectation. Each row reports the results of inference of clusters of coregulated genes from genome-wide transcriptional profiles of wild Louisiana isolates of N. crassa at a different stringency. In a given row, the first column reports a value of the Spearman correlation coefficient measuring the coexpression between two genes across strains. The second column reports the number of gene clusters emerging from clustering analysis of expression data using the indicated cutoff. In the third column, the first value reports the median number of genes in the inferred clusters, and the values in parentheses report the maximum and minimum cluster sizes. The fourth column reports the total number of genes falling into clusters. The last column reports the number of gene clusters emerging from clustering analysis of permuted (null) expression data. Red type indicates results for the correlation coefficient used in this study.
Genes coregulated with hbc-1 (NCU04826) and asi-1 (NCU05257) across Louisiana strains. Shown are annotations (http://www.broadinstitute.org/annotation/genome/neurospora/) of the sets of genes coregulated with hbc-1 and asi-1 (red type) across wild Louisiana isolates of N. crassa (cluster 48 and 15, respectively; see Data Set S1).
Functional categories enriched in the transcriptional signatures of asi-1 (NCU05257) and Nc_nmr6 (NCU00789). Shown are the results of FunCat enrichment analysis (20) of the genes differentially expressed between a strain bearing an engineered mutation in asi-1 (top) or Nc_nmr6 (bottom) and an isogenic wild-type strain (see Data Set S2). Corrected (P) results of a hypergeometric test for overlap between the indicated FunCat category and the set of differentially expressed genes, corrected for multiple testing by the Benjamini-Hochberg method, are shown. Only groups enriched in each analysis at corrected P values < 0.05 are shown.
Strains used in this study. Mat, mating type. FGSC, strain identification number from the Fungal Genetics Stock Center (http://www.fgsc.net/Neurospora/). Perkins, original strain identification number assigned by David Perkins (13–15).
ACKNOWLEDGMENTS
We thank J. Welch for strain handling and microscopy and C. Hall for expression data processing.
This work was supported by NIH R24 GM081597 to J.W.T., N.L.G., and R.B.B. We are pleased to acknowledge use of materials generated by NIH Program Project Grant P01GM068087 (functional analysis of a model filamentous fungus).
Footnotes
Citation Ellison CE, Kowbel D, Glass NL, Taylor JW, Brem RB. 2014. Discovering functions of unannotated genes from a transcriptome survey of wild fungal isolates. mBio 5(2):e01046-13. doi:10.1128/mBio.01046-13.
REFERENCES
- 1. Gow NAR, Gadd GM. 1995. The growing fungus, 1st ed. Chapman and Hall, London, United Kingdom [Google Scholar]
- 2. Blackwell M. 2011. The fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 98:426–438. 10.3732/ajb.1000298 [DOI] [PubMed] [Google Scholar]
- 3. Christie KR, Hong EL, Cherry JM. 2009. Functional annotations for the Saccharomyces cerevisiae genome: the knowns and the known unknowns. Trends Microbiol. 17:286–294. 10.1016/j.tim.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kyrpides NC. 1999. Genomes OnLine Database (GOLD 1.0): a monitor of complete and ongoing genome projects world-wide. Bioinformatics 15:773–774. 10.1093/bioinformatics/15.9.773 [DOI] [PubMed] [Google Scholar]
- 5. Arvas M, Kivioja T, Mitchell A, Saloheimo M, Ussery D, Penttila M, Oliver S. 2007. Comparison of protein coding gene contents of the fungal phyla Pezizomycotina and Saccharomycotina. BMC Genomics 8:325. 10.1186/1471-2164-8-325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC. 2006. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. U. S. A. 103:10352–10357. 10.1073/pnas.0601456103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kasuga T, Mannhaupt G, Glass NL. 2009. Relationship between phylogenetic distribution and genomic features in Neurospora crassa. PLoS One 4:e5286. 10.1371/journal.pone.0005286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rockman MV, Kruglyak L. 2006. Genetics of global gene expression. Nat. Rev. Genet. 7:862–872. 10.1038/nrg1964 [DOI] [PubMed] [Google Scholar]
- 9. Nayak RR, Kearns M, Spielman RS, Cheung VG. 2009. Coexpression network based on natural variation in human gene expression reveals gene interactions and functions. Genome Res. 19:1953–1962. 10.1101/gr.097600.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Connelly CF, Akey JM. 2012. On the prospects of whole-genome association mapping in Saccharomyces cerevisiae. Genetics 191:1345–1353. 10.1534/genetics.112.141168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellison CE, Hall C, Kowbel D, Welch J, Brem RB, Glass NL, Taylor JW. 2011. Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc. Natl. Acad. Sci. U. S. A. 108:2831–2836. 10.1073/pnas.1014971108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dettman JR, Jacobson DJ, Taylor JW. 2003. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 57:2703–2720. 10.1111/j.0014-3820.2003.tb01514.x [DOI] [PubMed] [Google Scholar]
- 13. Turner BC, Perkins DD, Fairfield A. 2001. Neurospora from natural populations: a global study. Fungal Genet. Biol. 32:67–92. 10.1006/fgbi.2001.1247 [DOI] [PubMed] [Google Scholar]
- 14. Perkins DD, Turner BC. 1988. Neurospora from natural populations toward the population biology of a haploid eukaryote. Exp. Mycol. 12:91–131. 10.1016/0147-5975(88)90001-1 [DOI] [Google Scholar]
- 15. Perkins DD, Turner BC, Barry EG. 1976. Strains of Neurospora collected from nature. Evolution 30:281–313. 10.2307/2407702 [DOI] [PubMed] [Google Scholar]
- 16. Jacobson DJ, Dettman JR, Adams RI, Boesl C, Sultana S, Roenneberg T, Merrow M, Duarte M, Marques I, Ushakova A, Carneiro P, Videira A, Navarro-Sampedro L, Olmedo M, Corrochano LM, Taylor JW. 2006. New findings of Neurospora in Europe and comparisons of diversity in temperate climates on continental scales. Mycologia 98:550–559. 10.3852/mycologia.98.4.550 [DOI] [PubMed] [Google Scholar]
- 17. Jacobson DJ, Powell AJ, Dettman JR, Saenz GS, Barton MM, Hiltz MD, Dvorachek WH, Jr, Glass NL, Taylor JW, Natvig DO. 2004. Neurospora in temperate forests of western North America. Mycologia 96:66–74. 10.2307/3761989 [DOI] [PubMed] [Google Scholar]
- 18. Mir-Rashed N, Jacobson DJ, Dehghany MR, Micali OC, Smith ML. 2000. Molecular and functional analyses of incompatibility genes at het-6 in a population of Neurospora crassa. Fungal Genet. Biol. 30:197–205. 10.1006/fgbi.2000.1218 [DOI] [PubMed] [Google Scholar]
- 19. Palma-Guerrero J, Hall CR, Kowbel D, Welch J, Taylor JW, Brem RB, Glass NL. 2013. Genome wide association identifies novel loci involved in fungal communication. PLoS Genet. 9:e1003669. 10.1371/journal.pgen.1003669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, Mewes HW. 2004. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32:5539–5545. 10.1093/nar/gkh894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunlap JC, Borkovich KA, Henn MR, Turner GE, Sachs MS, Glass NL, McCluskey K, Plamann M, Galagan JE, Birren BW, Weiss RL, Townsend JP, Loros JJ, Nelson MA, Lambreghts R, Colot HV, Park G, Collopy P, Ringelberg C, Crew C, Litvinkova L, DeCaprio D, Hood HM, Curilla S, Shi M, Crawford M, Koerhsen M, Montgomery P, Larson L, Pearson M, Kasuga T, Tian C, Baştürkmen M, Altamirano L, Xu J. 2007. Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Adv. Genet. 57:49–96. 10.1016/S0065-2660(06)57002-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Borkovich KA, Alex LA, Yarden O, Freitag M, Turner GE, Read ND, Seiler S, Bell-Pedersen D, Paietta J, Plesofsky N, Plamann M, Goodrich-Tanrikulu M, Schulte U, Mannhaupt G, Nargang FE, Radford A, Selitrennikoff C, Galagan JE, Dunlap JC, Loros JJ, Catcheside D, Inoue H, Aramayo R, Polymenis M, Selker EU, Sachs MS, Marzluf GA, Paulsen I, Davis R, Ebbole DJ, Zelter A, Kalkman ER, O’Rourke R, Bowring F, Yeadon J, Ishii C, Suzuki K, Sakai W, Pratt R. 2004. Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol. Mol. Biol. Rev. 68:1–108. 10.1128/MMBR.68.1.1-108.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tian C, Kasuga T, Sachs MS, Glass NL. 2007. Transcriptional profiling of cross pathway control in Neurospora crassa and comparative analysis of the Gcn4 and CPC1 regulons. Eukaryot. Cell 6:1018–1029. 10.1128/EC.00078-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roche CM, Blanch HW, Clark DS, Glass NL. 2013. Physiological role of acyl coenzyme A synthetase homologs in lipid metabolism in Neurospora crassa. Eukaryot. Cell 12:1244–1257. 10.1128/EC.00079-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ebbole DJ, Paluh JL, Plamann M, Sachs MS, Yanofsky C. 1991. cpc-1, the general regulatory gene for genes of amino acid biosynthesis in Neurospora crassa, is differentially expressed during the asexual life cycle. Mol. Cell. Biol. 11:928–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sachs MS, Yanofsky C. 1991. Developmental expression of genes involved in conidiation and amino acid biosynthesis in Neurospora crassa. Dev. Biol. 148:117–128. 10.1016/0012-1606(91)90322-T [DOI] [PubMed] [Google Scholar]
- 27. Marzluf GA. 1997. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reinert WR, Marzluf GA. 1975. Genetic and metabolic control of the purine catabolic enzymes of Neurospora crasse. Mol. Gen. Genet. 139:39–55. 10.1007/BF00267994 [DOI] [PubMed] [Google Scholar]
- 29. Cooper TG. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol. Rev. 26:223–238. 10.1111/j.1574-6976.2002.tb00612.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cha J, Chang SS, Huang G, Cheng P, Liu Y. 2008. Control of WHITE COLLAR localization by phosphorylation is a critical step in the circadian negative feedback process. EMBO J. 27:3246–3255. 10.1038/emboj.2008.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen PT, Philp A, Vázquez-Martin C. 2005. Protein phosphatase 4—from obscurity to vital functions. FEBS Lett. 579:3278–3286. 10.1016/j.febslet.2005.04.070 [DOI] [PubMed] [Google Scholar]
- 32. Sun J, Glass NL. 2011. Identification of the CRE-1 cellulolytic regulon in Neurospora crassa. PLoS One 6:e25654. 10.1371/journal.pone.0025654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donofrio NM, Oh Y, Lundy R, Pan H, Brown DE, Jeong JS, Coughlan S, Mitchell TK, Dean RA. 2006. Global gene expression during nitrogen starvation in the rice blast fungus, Magnaporthe grisea. Fungal Genet. Biol. 43:605–617. 10.1016/j.fgb.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 34. De Smet R, Marchal K. 2010. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8:717–729 10.1038/nrmicro2419 [DOI] [PubMed] [Google Scholar]
- 35. Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, The DREAM5 Consortium. Kellis M, Collins JJ, Stolovitzky G. 2012. Wisdom of crowds for robust gene network inference. Nat. Methods 9:796–804. 10.1038/nmeth.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guan Y, Dunham M, Caudy A, Troyanskaya O. 2010. Systematic planning of genome-scale experiments in poorly studied species. PLoS Comput. Biol. 6:e1000698. 10.1371/journal.pcbi.1000698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes TR, de Boer CG. 2013. Mapping yeast transcriptional networks. Genetics 195:9–36. 10.1534/genetics.113.153262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harris SD. 2008. Branching of fungal hyphae: regulation, mechanisms and comparison with other branching systems. Mycologia 100:823–832. 10.3852/08-177 [DOI] [PubMed] [Google Scholar]
- 39. Seiler S, Vogt N, Ziv C, Gorovits R, Yarden O. 2006. The STE20/germinal center kinase POD6 interacts with the NDR kinase COT1 and is involved in polar tip extension in Neurospora crassa. Mol. Biol. Cell 17:4080–4092. 10.1091/mbc.E06-01-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yarden O, Plamann M, Ebbole DJ, Yanofsky C. 1992. cot-1, a gene required for hyphal elongation in Neurospora crassa, encodes a protein kinase. EMBO J. 11:2159–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vogt N, Seiler S. 2008. The RHO1-specific GTPase-activating protein LRG1 regulates polar tip growth in parallel to Ndr kinase signaling in Neurospora. Mol. Biol. Cell 19:4554–4569. 10.1091/mbc.E07-12-1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Winkelmann G. 2002. Microbial siderophore-mediated transport. Biochem. Soc. Trans. 30:691–696. 10.1042/bst0300691 [DOI] [PubMed] [Google Scholar]
- 43. Mercier A, Labbé S. 2010. Iron-dependent remodeling of fungal metabolic pathways associated with ferrichrome biosynthesis. Appl. Environ. Microbiol. 76:3806–3817. 10.1128/AEM.00659-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Broach JR. 2012. Nutritional control of growth and development in yeast. Genetics 192:73–105. 10.1534/genetics.111.135731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sancar G, Sancar C, Brügger B, Ha N, Sachsenheimer T, Gin E, Wdowik S, Lohmann I, Wieland F, Höfer T, Diernfellner A, Brunner M. 2011. A global circadian repressor controls antiphasic expression of metabolic genes in Neurospora. Mol. Cell 44:687–697. 10.1016/j.molcel.2011.10.019 [DOI] [PubMed] [Google Scholar]
- 46. Johnston M. 1999. Feasting, fasting and fermenting. Glucose sensing in yeast and other cells. Trends Genet. 15:29–33. 10.1016/S0168-9525(98)01637-0 [DOI] [PubMed] [Google Scholar]
- 47. Thevelein JM, Geladé R, Holsbeeks I, Lagatie O, Popova Y, Rolland F, Stolz F, van de Velde S, Van Dijck P, Vandormael P, Van Nuland A, Van Roey K, Van Zeebroeck G, Yan B. 2005. Nutrient sensing systems for rapid activation of the protein kinase A pathway in yeast. Biochem. Soc. Trans. 33:253–256. 10.1042/BST0330253 [DOI] [PubMed] [Google Scholar]
- 48. Andrade SL, Einsle O. 2007. The Amt/Mep/Rh family of ammonium transport proteins. Mol. Membr. Biol. 24:357–365. 10.1080/09687680701388423 [DOI] [PubMed] [Google Scholar]
- 49. Ebbole DJ. 1998. Carbon catabolite repression of gene expression and conidiation in Neurospora crassa. Fungal Genet. Biol. 25:15–21. 10.1006/fgbi.1998.1088 [DOI] [PubMed] [Google Scholar]
- 50. Dunn-Coleman NS, Tomsett AB, Garrett RH. 1981. The regulation of nitrate assimilation in Neurospora crassa: biochemical analysis of the nmr-1 mutants. Mol. Gen. Genet. 182:234–239. 10.1007/BF00269663 [DOI] [PubMed] [Google Scholar]
- 51. Wilson RA, Gibson RP, Quispe CF, Littlechild JA, Talbot NJ. 2010. An NADPH-dependent genetic switch regulates plant infection by the rice blast fungus. Proc. Natl. Acad. Sci. U. S. A. 107:21902–21907. 10.1073/pnas.1006839107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson RA, Jenkinson JM, Gibson RP, Littlechild JA, Wang ZY, Talbot NJ. 2007. Tps1 regulates the pentose phosphate pathway, nitrogen metabolism and fungal virulence. EMBO J. 26:3673–3685. 10.1038/sj.emboj.7601795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fernandez J, Wright JD, Hartline D, Quispe CF, Madayiputhiya N, Wilson RA. 2012. Principles of carbon catabolite repression in the rice blast fungus: Tps1, Nmr1-3, and a MATE-family pump regulate glucose metabolism during infection. PLoS Genet. 8:e1002673. 10.1371/journal.pgen.1002673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bhosale R, Jewell JB, Hollunder J, Koo AJ, Vuylsteke M, Michoel T, Hilson P, Goossens A, Howe GA, Browse J, Maere S. 2013. Predicting gene function from uncontrolled expression variation among individual wild-type Arabidopsis plants. Plant Cell 25:2865–2877. 10.1105/tpc.113.112268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hess DC, Myers CL, Huttenhower C, Hibbs MA, Hayes AP, Paw J, Clore JJ, Mendoza RM, Luis BS, Nislow C, Giaever G, Costanzo M, Troyanskaya OG, Caudy AA. 2009. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 5:e1000407. 10.1371/journal.pgen.1000407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo F, Yang Y, Zhong J, Gao H, Khan L, Thompson DK, Zhou J. 2007. Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinformatics 8:299. 10.1186/1471-2105-8-299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang W, Morris QD, Chang R, Shai O, Bakowski MA, Mitsakakis N, Mohammad N, Robinson MD, Zirngibl R, Somogyi E, Laurin N, Eftekharpour E, Sat E, Grigull J, Pan Q, Peng WT, Krogan N, Greenblatt J, Fehlings M, van der Kooy D, Aubin J, Bruneau BG, Rossant J, Blencowe BJ, Frey BJ, Hughes TR. 2004. The functional landscape of mouse gene expression. J. Biol. 3:21. 10.1186/jbiol16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lan H, Carson R, Provart NJ, Bonner AJ. 2007. Combining classifiers to predict gene function in Arabidopsis thaliana using large-scale gene expression measurements. BMC Bioinformatics 8:358. 10.1186/1471-2105-8-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang J, Lu K, Xiang Y, Islam M, Kotian S, Kais Z, Lee C, Arora M, Liu HW, Parvin JD, Huang K. 2012. Weighted frequent gene co-expression network mining to identify genes involved in genome stability. PLoS Comput. Biol. 8:e1002656. 10.1371/journal.pcbi.1002656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCluskey K. 2003. The Fungal Genetics Stock Center: from molds to molecules. Adv. Appl. Microbiol. 52:245–262. 10.1016/S0065-2164(03)01010-4 [DOI] [PubMed] [Google Scholar]
- 61. Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen CB, Butler J, Endrizzi M, Qui D, Ianakiev P, Bell-Pedersen D, Nelson MA, Werner-Washburne M, Selitrennikoff CP, Kinsey JA, Braun EL, Zelter A, Schulte U, Kothe GO, Jedd G, Mewes W, Staben C, Marcotte E, Greenberg D, Roy A, Foley K, Naylor J, Stange-Thomann N, Barrett R, Gnerre S, Kamal M, Kamvysselis M, Mauceli E, Bielke C, Rudd S, Frishman D, Krystofova S, Rasmussen C, Metzenberg RL, Perkins DD, Kroken S, Cogoni C, Macino G, Catcheside D, Li WX, Pratt RJ, Osmani SA, DeSouza CPC, Glass L, Orbach MJ, Berglund JA, Voelker R, Yarden O, Plamann M, Seller S, Dunlap J, Radford A, Aramayo R, Natvig DO, Alex LA, Mannhaupt G, Ebbole DJ, Freitag M, Paulsen I, Sachs MS, Lander ES, Nusbaum C, Birren B. 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868. 10.1038/nature01554 [DOI] [PubMed] [Google Scholar]
- 62. Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11:94. 10.1186/1471-2105-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Team RDC 2009. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 65. Horan K, Jang C, Bailey-Serres J, Mittler R, Shelton C, Harper JF, Zhu JK, Cushman JC, Gollery M, Girke T. 2008. Annotating genes of known and unknown function by large-scale coexpression analysis. Plant Physiol. 147:41–57. 10.1104/pp.108.117366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Stat. Methodol. 57:289–300 [Google Scholar]
- 67. Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, Otillar R, Poliakov A, Ratnere I, Riley R, Smirnova T, Rokhsar D, Dubchak I. 2012. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 40:D26–D32. 10.1093/nar/gkr947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Westergaard M, Mitchell HK. 1947. Neurospora. V. A synthetic medium favoring sexual reproduction. Am. J. Bot. 34:573–577. 10.2307/2437339 [DOI] [Google Scholar]
- 70. Tatum EL, Barratt RW, Cutter VM., Jr. 1949. Chemical induction of colonial paramorphs in Neurospora and Syncephalastrum. Science 109:509–511. 10.1126/science.109.2838.509 [DOI] [PubMed] [Google Scholar]
- 71. Vogel HJ. 1956. A convenient growth medium for Neurospora (Medium N). Microb. Genet. Bull. 13:42–43
- 72. Gritz L, Davies J. 1983. Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 25:179–188. 10.1016/0378-1119(83)90223-8 [DOI] [PubMed] [Google Scholar]
- 73. Hall C, Welch J, Kowbel DJ, Glass NL. 2010. Evolution and diversity of a fungal self/nonself recognition locus. PLoS One 5:e14055. 10.1371/journal.pone.0014055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ryan FJ, Beadle GW, Tatum EL. 1943. The tube method of measuring the growth rate of Neurospora. Am. J. Bot. 30:784–799. 10.2307/2437554 [DOI] [Google Scholar]
- 76. Paluh JL, Plamann M, Krüger D, Barthelmess IB, Yanofsky C, Perkins DD. 1990. Determination of the inactivating alterations in two mutant alleles of the Neurospora crassa cross-pathway control gene cpc-1. Genetics 124:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, van Helden J, André B. 2007. Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 27:3065–3086. 10.1128/MCB.01084-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang LW, Marzluf GA. 1980. Purification and characterization of uricase, a nitrogen-regulated enzyme from Neurospora crassa. Arch. Biochem. Biophys. 201:185–193. 10.1016/0003-9861(80)90501-9 [DOI] [PubMed] [Google Scholar]
- 79. DeBusk RM, Ogilvie S. 1984. Regulation of amino acid utilization in Neurospora crassa: effect of nmr-1 and ms-5 mutations. J. Bacteriol. 160:656–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schluttig A, Fritsche W. 1975. Repression of urease biosynthesis in Neurospora crassa by ammonium ions. Z. Allg. Mikrobiol. 15:371–376. 10.1002/jobm.3630150509 [DOI] [PubMed] [Google Scholar]
- 81. Griffith AB, Garrett RH. 1988. Xanthine dehydrogenase expression in Neurospora crassa does not require a functional nit-2 regulatory gene. Biochem. Genet. 26:37–52. 10.1007/BF00555487 [DOI] [PubMed] [Google Scholar]
- 82. Stajich JE, Harris T, Brunk BP, Brestelli J, Fischer S, Harb OS, Kissinger JC, Li W, Nayak V, Pinney DF, Stoeckert CJ, Jr, Roos DS. 2012. FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 40:D675–D681. 10.1093/nar/gkr918 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cluster membership and functional enrichment of coregulated genes across Louisiana isolates of N. crassa. Regulon clusters: each row lists gene identifiers (IDs) for a gene cluster defined based on coregulation across wild Louisiana strains of N. crassa, with pairwise correlation among genes of a cluster at Spearman’s correlation coefficient ≥ 0.4. Red text indicates transcription factors of known function for which FunCat category annotations (20) associated with the factor were also enriched among genes of the indicated cluster other than the factor itself. Cluster enrichment, each row reports a FunCat annotation term (20) enriched among the genes of the indicated cluster inferred from expression profiles across Louisiana strains of N. crassa. Corrected P value, results of a hypergeometric test for overlap between the indicated FunCat category and the indicated cluster, corrected for multiple testing by the Benjamini-Hochberg method. Only enrichments at corrected P < 0.05 are shown. Download
Gene expression profiles of engineered N. crassa mutants. Genome-wide expression, each row reports expression measurements for one gene, and each column reports expression in one strain. For details of RNA-seq normalization, see Materials and Methods in the main text. DE genes, each tab reports all genes significantly differentially expressed, at corrected P values < 0.1, in a Wilcoxon test for differential expression between the indicated deletion strain and a wild-type control, corrected for multiple testing by the Benjamini-Hochberg method. KO/WT expression, log10 of the ratio of expression of the indicated gene between the deletion strain and an isogenic wild-type strain. Download
Genes differentially expressed between the Caribbean and Louisiana populations of N. crassa. Corrected P value, results of a Wilcoxon test for differential expression of the indicated gene between Louisiana strains and Caribbean strains of N. crassa, corrected for multiple testing by the Benjamini-Hochberg method. Columns labeled with “MEDIAN_EXP” report median expression of the indicated gene across strains of the indicated population: LA, Louisiana; CA, Caribbean; PAN, Panama. The last column reports the ratio between the median expression of the indicated gene among Louisiana strains and the median expression among Caribbean strains. Only genes with corrected P values < 0.05 are shown. Download
N. crassa genes with putative functions related to nitrogen metabolism. NMR genes, known and inferred targets of the nitrogen metabolite repression (NMR) program in N. crassa. The first nine lines report genes previously shown to be targets of the NMR program in N. crassa. All other lines list inferred N. crassa homologs of S. cerevisiae NMR targets from reference 77. NMR regulators: inferred N. crassa homologs of master regulators of the nitrogen metabolite repression program. Shown are the results of sequence searches of the N. crassa proteome for homologs of S. cerevisiae genes annotated as master regulators of the nitrogen metabolite repression program (29). For details of sequence search methods, see Materials and Methods in the main text. M. grisea N starvation genes, inferred nitrogen acquisition and metabolism genes in N. crassa. Shown are the results of sequence searches of the N. crassa proteome for homologs of Magnaporthe grisea genes responsive to nitrogen starvation according to reference 33. For details of sequence search methods, see Materials and Methods. For N. crassa nitrogen genes, shown is the union of the sets of genes whose annotation (http://www.broadinstitute.org/annotation/genome/neurospora/), Gene Ontology term, and/or FunCat terms were related to nitrogen acquisition or metabolism. Download
Deletion of genes coregulated with hbc-1 (NCU04826) confers morphological defects. Each micrograph shows hyphal architecture of a colony of a strain harboring a deletion in a gene coregulated with hbc-1 across Louisiana isolates of N. crassa (see Table S2). Download
The histidine analog 3-AT compromises growth of a cpc-1 mutant. Each column reports the distribution, across three biological replicates under one environmental condition, of the growth rate of an engineered cpc-1 deletion strain. VMM, Vogel’s minimal medium; VMM + 3AT, Vogel’s minimal medium containing 6 mM 3-AT. Symbols are as in Fig. 1C of the main text. *, the cpc-1 mutant grows significantly slower in the presence of 3-AT (one-sided Wilcoxon test P value = 0.05). Download
Numbers of clusters of coregulated genes inferred from expression profiles of Louisiana strains exceed the chance expectation. Each row reports the results of inference of clusters of coregulated genes from genome-wide transcriptional profiles of wild Louisiana isolates of N. crassa at a different stringency. In a given row, the first column reports a value of the Spearman correlation coefficient measuring the coexpression between two genes across strains. The second column reports the number of gene clusters emerging from clustering analysis of expression data using the indicated cutoff. In the third column, the first value reports the median number of genes in the inferred clusters, and the values in parentheses report the maximum and minimum cluster sizes. The fourth column reports the total number of genes falling into clusters. The last column reports the number of gene clusters emerging from clustering analysis of permuted (null) expression data. Red type indicates results for the correlation coefficient used in this study.
Genes coregulated with hbc-1 (NCU04826) and asi-1 (NCU05257) across Louisiana strains. Shown are annotations (http://www.broadinstitute.org/annotation/genome/neurospora/) of the sets of genes coregulated with hbc-1 and asi-1 (red type) across wild Louisiana isolates of N. crassa (cluster 48 and 15, respectively; see Data Set S1).
Functional categories enriched in the transcriptional signatures of asi-1 (NCU05257) and Nc_nmr6 (NCU00789). Shown are the results of FunCat enrichment analysis (20) of the genes differentially expressed between a strain bearing an engineered mutation in asi-1 (top) or Nc_nmr6 (bottom) and an isogenic wild-type strain (see Data Set S2). Corrected (P) results of a hypergeometric test for overlap between the indicated FunCat category and the set of differentially expressed genes, corrected for multiple testing by the Benjamini-Hochberg method, are shown. Only groups enriched in each analysis at corrected P values < 0.05 are shown.
Strains used in this study. Mat, mating type. FGSC, strain identification number from the Fungal Genetics Stock Center (http://www.fgsc.net/Neurospora/). Perkins, original strain identification number assigned by David Perkins (13–15).