

ABSTRACT
Positive-stranded viruses synthesize their RNA in membrane-bound organelles, but it is not clear how this benefits the virus or the host. For coronaviruses, these organelles take the form of double-membrane vesicles (DMVs) interconnected by a convoluted membrane network. We used electron microscopy to identify murine coronaviruses with mutations in nsp3 and nsp14 that replicated normally while producing only half the normal amount of DMVs under low-temperature growth conditions. Viruses with mutations in nsp5 and nsp16 produced small DMVs but also replicated normally. Quantitative reverse transcriptase PCR (RT-PCR) confirmed that the most strongly affected of these, the nsp3 mutant, produced more viral RNA than wild-type virus. Competitive growth assays were carried out in both continuous and primary cells to better understand the contribution of DMVs to viral fitness. Surprisingly, several viruses that produced fewer or smaller DMVs showed a higher fitness than wild-type virus at the reduced temperature, suggesting that larger and more numerous DMVs do not necessarily confer a competitive advantage in primary or continuous cell culture. For the first time, this directly demonstrates that replication and organelle formation may be, at least in part, studied separately during infection with positive-stranded RNA virus.
IMPORTANCE
The viruses that cause severe acute respiratory syndrome (SARS), poliomyelitis, and hepatitis C all replicate in double-membrane vesicles (DMVs). The big question about DMVs is why they exist in the first place. In this study, we looked at thousands of infected cells and identified two coronavirus mutants that made half as many organelles as normal and two others that made typical numbers but smaller organelles. Despite differences in DMV size and number, all four mutants replicated as efficiently as wild-type virus. To better understand the relative importance of replicative organelles, we carried out competitive fitness experiments. None of these viruses was found to be significantly less fit than wild-type, and two were actually fitter in tests in two kinds of cells. This suggests that viruses have evolved to have tremendous plasticity in the ability to form membrane-associated replication complexes and that large and numerous DMVs are not exclusively associated with efficient coronavirus replication.
INTRODUCTION
Positive-stranded RNA viruses form intracellular membrane-bound organelles after infection, and these organelles are believed to facilitate intracellular RNA replication (1, 2). In the coronaviruses, which cause diseases in humans, including severe acute respiratory syndrome (SARS) and Middle Eastern respiratory syndrome (MERS), these replicative structures take the form of double-membrane vesicles (DMV), which later develop into clusters where the outer DMV membranes are connected by a network of convoluted membrane (CM) tubules (3–5). DMV clusters are derived from the endoplasmic reticulum (ER) or ER-Golgi intermediate compartment (4, 6) and contain newly synthesized viral RNA and viral replicase proteins (4, 7–13). Previous reports have linked organelle formation to ongoing RNA replication (4, 14, 15). This has been interpreted as evidence that DMVs either are required for coronavirus RNA synthesis or are generated as a result of viral RNA synthesis (4, 14).
The SARS coronavirus (SARS-CoV) replicative machinery consists of up to sixteen nonstructural proteins (nsp1 to nsp16), which are processed from large polyproteins (reviewed in reference 16). The conserved gene cluster from the nsp3 gene to the nsp6 gene contains all the factors necessary for DMV formation: nsp3, nsp4, and nsp6 are needed to form DMV clusters (17) after essential processing by proteinases located in nsp3 (18) and nsp5 (19). Electron microscopy suggests that nsp3 induces membrane proliferation (17). nsp3 and nsp4 localize predominantly to the CM in DMV clusters (4, 9, 20) and are sufficient to induce structures resembling CMs in the absence of other viral proteins (17). Mutations in nsp4 have been linked to formation of aberrant DMVs with apparent defects in membrane pairing (21). nsp6 from several coronaviruses causes single-membrane LC3-containing vesicles (22) to accumulate around microtubule organization centers (17), although it is not clear how this is connected to the formation of DMV clusters.
Some replicase subunits work together in units known as cistrons. Five cistrons (0, I, II, IV, and VI) have been defined, with gaps left in the naming scheme in case new cistrons are discovered that map to nsp13 and nsp15, respectively (14, 23). Complementation studies using temperature-sensitive (ts) mutants of mouse hepatitis virus (MHV) have showed that mutations in nsp3, nsp12, nsp14, and nsp16 protein define distinct cistrons (cistrons 0, II, IV, and VI, respectively), while mutations in nsp5 and nsp10 proteins define a single cistron (cistron I) (14, 23–25). Temperature-sensitive mutations in coronavirus nsp3, nsp5, nsp10, nsp12, nsp14, and nsp16 have been reported, all of which permit RNA synthesis at 33°C but cause RNA synthesis defects at 40°C (14, 24–28).
In this study, we examined the intracellular organelle phenotype of MHV strains with temperature-sensitive (ts) mutations in each of the known cistrons. Both mutant and control viruses were grown at 33°C, a temperature selected to allow full growth of mutant viruses. We chose to work with ts viruses as a pool of ready-made replicase point mutants that have been preselected for both robust growth at 33°C and functional importance, since each of the ts mutations is lethal at 40°C. We carried out these tests in order both to better understand the connection between organelles and viral RNA synthesis and to examine whether mutation of critical residues produces a detectable effect on the replicative organelle. To do this, we quantified virus release, DMV formation, RNA synthesis, and competitive fitness in a variety of cell types.
RESULTS
Genetics and growth of mutant viruses.
Wild-type MHV-inf-1 and five mutant MHV strains selected on the basis of defective RNA synthesis at 40°C were used in this study (Fig. 1). Brts31 contains a mutation in a region that likely forms an interdomain flexible linker in nsp3 (29, 30), which paradoxically interferes with the function of the nsp5 main proteinase (Mpro) at 40°C (14). Albts16 has a mutation in a loop within the carboxyl-terminal domain of Mpro (23, 31). Albts22 has a mutation in the predicted thumb domain of the polymerase fold near the carboxyl terminus of nsp12 (23, 32). Wüts18 has a mutation on the surface of the viral 2′-O-methyltransferase (23, 33). The last mutant chosen was the previously undescribed Brts105, which contains a single proline to serine substitution (CCA to UCA) at position 19320 in sequence AY700211 (Fig. 1). The Brts105 mutation falls in the viral N7-methyltransferase domain of nsp14 (34), between the previously characterized Wüts38 and Albts17 mutations, both of which map to cistron IV (23).
FIG 1 .
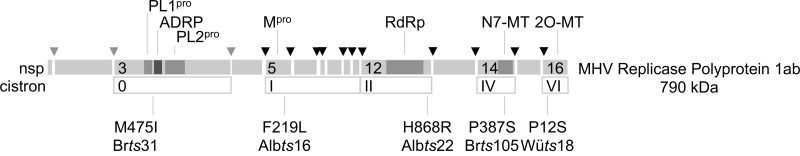
Genetic characteristics of MHV mutants. The schematic shows amino acid changes responsible for temperature-sensitive phenotypes of viruses used in this study. Amino acid positions are numbered from the start of each nonstructural protein. Functions of potentially affected domains that are marked include papain-like proteinases (PL1pro and PL2pro), ADP-ribose 1ʺ phosphatase (ADRP), main proteinase (Mpro), RNA-dependent RNA polymerase (RdRp), and methyltransferases (N7-MT and 2O-MT).
Brts105 was subjected to several tests to characterize its growth at 33°C and 40°C. The replication of Brts105 was compared to that of both the wild type and the better-characterized Brts31 mutant in primary mouse embryo fibroblast cells (Fig. 2A). Brts105 growth was slightly delayed compared to that of the wild type and Brts31 but reached a similar peak titer. Neither Brts105 nor Brts31 virus grew at 40°C (Fig. 2B), and Brts105 did not produce RNA at 40°C (Fig. 2C), demonstrating that growth of both viruses was temperature sensitive.
FIG 2 .

Characterization of Brts105 growth. Viruses were inoculated at a multiplicity of 10 PFU/cell on primary MEFs (mouse embryo fibroblasts) at 33°C for 1 h and then incubated at either 33°C (A) or 40°C (B) for the duration of the experiment. Viral growth was measured by plaque assay at 33°C. The average of three replicates is shown. Dotted lines indicate the lower limit of detection for the plaque assay. (C) Northern blots show the effect of temperature on viral RNA production by the wild type and Brts105. Bands corresponding to viral subgenomic RNAs 4 to 7 are indicated. (D to F) The relative severity of ts phenotypes was characterized by a growth stop/resume assay. Growth was stopped 4.5 h after inoculation by incubating ts virus at 40°C or by treating wild-type virus with 10 µg/ml cycloheximide (CHX). After 3 h of arrested growth, the inhibition was either maintained to the end of the experiment (Stopped) or released (Resumed). Virus growth was measured by plaque assay, and the results for Brts105 are shown in panel E. The averages for 6 replicates are shown. The titer ratio (resumed to stopped) from panel E is shown in comparison to other mutants and CHX treatment in panel F. Statistically significant increases in titer after resumption are indicated (*, P ≤ 0.05; ns, not significant).
Functional characterization of ts viruses.
To better understand the functional consequences of activating the ts phenotype of Brts105, we performed a stop/resume assay, which was designed as a simple tool to compare the severity of ts phenotypes. The assay takes advantage of the eclipse phase, during which RNA replication takes place but no new virus emerges, which lasts for 6 h after inoculation (5). The stop/resume assay works by stopping RNA synthesis 4.5 h after inoculation, during the eclipse phase. After 3 h under stopped conditions, RNA synthesis was then either resumed for 4 h, in order to allow the release of new virus with the participation of previously synthesized components, or kept under stopped conditions (Fig. 2D). If the resumed titer was significantly higher than the stopped titer (typically about a 10-fold increase), then the ts phenotype was classified as mild, meaning that some residual function or functional components were available after the stoppage period, but if the titer did not increase significantly after resumption, then the ts phenotype was classified as severe.
The results of a stop/resume assay for Brts105 are shown in Fig. 2E and were typical of the mild phenotype. Stop/resume assays were performed at least twice, with three replicates per time point. Results for other viruses are summarized in Fig. 2F. Brts105, Brts31, and Albts16 displayed mild ts phenotypes. This was consistent with the previously described mild ts phenotype of Albts16 (23). The ts phenotype of Albts22 was found to be severe, as previously reported (23), and was equivalent in severity to treatment with cycloheximide, which blocks translation and has been shown to result in inactivation of replicase components in about 1 h (23, 35, 36).
We next compared the growth of the panel of viruses at 33°C on continuous 17Clone-1 fibroblasts (17Cl-1), primary mouse embryo fibroblasts (MEFs), adipose-derived mesenchymal stem cells (ADMSC), and bone marrow-derived stem cells (BMSC) (Table 1). All of the mutants except Albts22 grew to approximately wild-type levels at 33°C in every cell type tested. Albts16 grew slightly slower than the wild type in 17Cl-1 cells but grew to wild-type levels in MEFs. Albts22 consistently produced about one-tenth as many infectious progeny as wild-type virus. These results showed that, except for Albts22, the panel of mutants displayed growth characteristics similar to those of wild-type virus at 33°C. These results were consistent with previous results, which showed that Wüts18 (37) and Brts31 (14) grow to wild-type levels at 33°C, while growth of Albts22 is attenuated even at 30°C (23).
TABLE 1 .
Comparative growth characteristics of MHV mutantsa
| Cell line | Conditions |
Titer (log) of wild typeb |
Titer difference relative to wild type (log) |
|||||
|---|---|---|---|---|---|---|---|---|
| Time (h) | Temp (°C) | Brts31 | Albts16 | Brts105 | Wüts18 | Albts22 | ||
| 17Cl-1 | 14 | 33 | 6.9 ± 0.3 | 0 | −0.6 | −0.2 | 0.4 | −0.8 |
| MEF | 24 | 33 | 5.3 ± 0.4 | 0.1 | −0.4 | −0.3 | 0.1 | −1 |
| MEF | 48 | 33 | 7.1 ± 0.2 | −0.2 | NDc | 0 | ND | ND |
| MEF | 72 | 33 | 4.6 ± 0.4 | ND | 0 | ND | ND | ND |
| ADMSC | 48 | 33 | 6.2 ± 0.1 | 0.1 | ND | ND | ND | ND |
| BMSC | 18 | 33 | 3.8 ± 0.2 | 0 | ND | ND | ND | ND |
| 17Cl-1 | 14 | 40 | 5.1 ± 0.2 | ≤−3.8 | −3.4 | ≤−3.8 | −2.7 | ≤−3.8 |
| MEF | 24 | 40 | 5.6 ± 0.2 | ≤−4.3 | ≤−4.3 | ≤−4.3 | ≤−4.3 | −3.5 |
Effects of mutations on viral growth were assessed at permissive (33°C) and nonpermissive (40°C) temperatures in continuous (17Cl-1) and primary (MEF, ADMSC, and BMSC) cell cultures. Titer differences were calculated from a minimum of three replicates.
The wild-type titer is shown as the mean ± standard deviation.
ND, not determined.
Aberrant DMV formation at 33°C.
Cells were infected at high multiplicity with wild-type or mutant viruses and then examined by electron microscopy to investigate the effects of mutations on replicative organelle formation at 33°C. One randomly oriented ultrathin section per cell was examined in order to compare the sizes and numbers of intracellular organelles virions, as described previously (17).
Clusters of DMVs were observed next to regions of CM in 17Cl-1 cells infected with wild-type and mutant viruses (Fig. 3A). However, all of the mutants produced smaller DMVs than wild-type virus, varying from nearly wild-type size (Brts105) to approximately 10% smaller (Brts31, Wüts18, and Albts22) to 17% smaller (Albts16). Many DMV-containing cells also contained intracellular virus particles (Fig. 3B), which did not differ significantly in size from wild-type controls.
FIG 3 .

Replicative organelles formed at 33°C by MHV mutants in 17Cl-1 cells. (A) Positions of double-membrane vesicles (stars), intracellular virus (arrowhead), and convoluted membrane (c) in infected cells are marked. (B) Magnified images of intracellular virus particles from each virus.
DMV size was measured in order to better understand the effects of the mutations on DMV synthesis. Altogether, 4,646 DMVs were measured at various times after infection in three continuous cell lines. Under every condition that was tested, mutant viruses formed significantly smaller DMVs than wild-type viruses (Table 2). The only revertant virus tested produced DMVs of wild-type size (Wüts18 revertant in L929 cells in Table 2). To control for the possibility that DMV size differences were due to variation in preparation for electron microscopy, 1,352 intracellular virions were measured from the same micrographs. No significant differences in intracellular virion size were observed (Table 2). Together, this demonstrated that Brts31, Albts16, Albts22, Brts105, and Wüts18 have small DMV phenotypes at 33°C.
TABLE 2 .
DMV and virion size in infected cells
| Conditions | Virus | ts | Double-membrane vesicles |
Intracellular virions |
||||
|---|---|---|---|---|---|---|---|---|
| n | Sizea (nm) | Pb | n | Sizea (nm) | P | |||
| L929, 37°C, 5 hpic | Wild-type | 302 | 179 ± 36 | 63 | 75 ± 9 | |||
| Wüts18 | nsp16 | 196 | 157 ± 61 | 2 × 10−6 | 77 | 75 ± 8 | NSd | |
| Revertant | nsp16e | 165 | 178 ± 31 | NS | 42 | 77 ± 13 | NS | |
| DBT, 33°C, 5.5 hpi | Wild type | 468 | 228 ± 45 | 74 | 69 ± 8 | |||
| Brts31 | nsp3 | 58 | 195 ± 38 | 2 × 10−6 | 70 | 69 ± 9 | NS | |
| DBT, 40°C, 5.5 hpif | Wild type | 841 | 218 ± 50 | 9 × 10−5 | 141 | 71 ± 10 | NS | |
| 17cl1, 33°C,10 hpi | Wild type | 604 | 228 ± 36 | 169 | 68 ± 10 | |||
| Brts31 | nsp3 | 403 | 208 ± 34 | 5 × 10−19 | 263 | 68 ± 10 | NS | |
| Albts16 | nsp5 | 459 | 189 ± 33 | 8 × 10−66 | 102 | 70 ± 8 | NS | |
| Brts105 | nsp14 | 463 | 220 ± 36 | 2 × 10−4 | 278 | 69 ± 10 | NS | |
| Wüts18 | nsp16 | 456 | 211 ± 35 | 2 × 10−15 | 39 | 67 ± 12 | NS | |
| Albts22g | nsp12 | 231 | 204 ± 43 | 2 × 10−13 | 34 | 68 ± 11 | NS | |
Size was calculated as perimeter length divided by π; data are means ± standard deviations.
Compared to the wild-type (Wüts18) or revertant (Brts31) control by Mann-Whitney test.
Wüts18 and the revertant grow to wild-type levels at 37°C (41).
NS, not significantly different from the corresponding wild-type control.
Containing the original ts mutation in nsp16 plus a compensating mutation in nsp16 N43S.
Incubated at 33°C for 3.5 h and then shifted to 40°C.
Attenuated growth at 33°C compared to the wild type.
Fewer DMVs formed at 33°C.
Ultrathin sections of infected 17Cl-1 and DBT cells were next examined for the prevalence of visible signs of infection in order to determine whether any of the mutations affected the number of DMV clusters present. For the combined viruses, 2,285 ultrathin cell sections were examined for the presence of intracellular signs of infection. Sections containing intracellular virions or DMV clusters were counted and photographed, and the number of each was recorded. Cells inoculated with Brts31 and Brts105 each contained about half the expected number of DMV-positive cells at 33°C compared to the wild type, despite a similar prevalence of cells with intracellular virions visible (Table 3). The prevalence of DMV and virion-containing cells did not differ from that in the wild-type controls for Albts16 and Wüts18, but Albts22 produced significantly fewer signs of infection (Table 3). These results demonstrated that Albts22, Brts31, and Brts105 produced fewer DMV clusters than wild-type virus.
TABLE 3 .
DMV and intracellular virion (IV) prevalence in infected cells
| Conditions | Virus | ts | No. of cells |
Prevalence of intracellular signs of infection |
No. per cell section when present |
Rel. DMV capacity (%)b |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DMV (%) | Pa | IV (%) | P | DMV | P | IV | P | |||||
| DBT, 33°C, 5.5 hpi | Wild type | 323 | 6 | 7 | − | 18 ± 20 | 3 ± 4 | − | 100 ± 60 | |||
| Brts31 | nsp3 | 753 | 2 | 8 × 10−4 | 7 | NSc | 6 ± 6 | 1 × 10−2 | 3 ± 4 | NS | 20 ± 12 | |
| 17Cl-1, 33°C, 10 hpi | Wild type | 161 | 40 | 29 | 9 ± 9 | 4 ± 3 | 100 ± 48 | |||||
| Brts31 | nsp3 | 238 | 24 | 4 × 10−4 | 25 | NS | 7 ± 8 | 4 × 10−4 | 4 ± 6 | NS | 35 ± 18 | |
| Albts16 | nsp5 | 120 | 37 | NS | 19 | NS | 9 ± 8 | NS | 4 ± 4 | NS | 53 ± 28 | |
| Brts105 | nsp14 | 230 | 22 | 1 × 10−4 | 32 | NS | 9 ± 13 | NS | 4 ± 4 | NS | 49 ± 25 | |
| Wüts18 | nsp16 | 140 | 36 | NS | 20 | NS | 9 ± 10 | NS | 2 ± 1 | 7 × 10−3 | 71 ± 36 | |
| Albts22d | nsp12 | 320 | 13 | 1 × 10−5 | 9 | 1 × 10−5 | 6 ± 5 | NS | 1 ± 0.4 | 1 × 10−5 | 16 ± 10 | |
Calculated by Fisher’s exact test.
Relative DMV capacity was calculated as follows: average DMV size (± standard deviation) × percentage of cell sections with DMVs × DMVs per section, relative to wild-type.
Not significantly different from the corresponding wild-type control.
Attenuated growth at 33°C compared to the wild type.
The number of DMVs per cell was measured in order to better quantify differences in organelle prevalence. Brts31 DMV clusters contained about a three-quarters as many DMVs as wild-type clusters both early and late in infection (Table 3). Albts22 clusters contained about two-thirds as many DMVs as wild-type clusters, consistent with the severely attenuated growth. For all viruses examined, there was one CM present per approximately 7 DMVs and 1 to 5 DMV clusters per cell section, when present (data not shown). Collectively these data distinguish two low-DMV phenotypes that occurred without attenuation of the virus: Brts105 formed fewer DMVs of near-normal size, while Brts31 formed fewer DMVs of smaller-than-normal size.
DMV capacity and RNA synthesis.
Viral RNA accumulates in DMVs, leading to the interpretation that DMV clusters are the primary site of RNA replication (4), although a recent study has called into question whether all DMVs are active in RNA synthesis (38). In this case, the total DMV capacity in an infected cell may limit the amount of viral RNA synthesis that can take place inside a cell. To estimate total DMV capacity, DMV size (Table 2) was multiplied by the relative percentage of DMV-containing cells (Table 3) and multiplied by the number of DMVs per cluster (Table 3) to estimate the relative DMV capacity of cells infected with wild-type and mutant viruses. Results were normalized to those for wild-type controls grown at 33°C and are shown in Table 3.
It has been previously shown that wild-type MHV produces about twice as much viral RNA at 40°C as at 33°C (23) (Fig. 2C). To test whether this increased RNA synthesis was reflected in the number or size of replicative organelles, cells infected with wild-type virus and incubated at 40°C and 33°C were examined. Organelles formed at 40°C were smaller (Table 2) but more numerous (31 per cell compared to 18 per cell at 33°C), effectively doubling DMV capacity (Fig. 4A). These results demonstrated that conditions reported to increase RNA yield correlated with increased DMV capacity for wild-type MHV.
FIG 4 .

RNA synthesis and relative DMV capacity. (A) Relative DMV capacity was estimated as DMV size (± standard deviation) × percentage of cell sections with visible DMVs × average number of DMVs per cell and expressed as a percentage of wild-type DMV capacity (indicated by the dotted line). (B) Viral genomic and subgenomic RNA synthesis at 10 h postinoculation in 17Cl-1 cells was measured by quantitative RT-PCR and is expressed as an amount relative to wild-type RNA synthesis. Statistically significant differences in RNA load are indicated (***, P ≤ 0.001; ns, not significant).
Albts22 displayed the lowest relative DMV capacity among the viruses we examined (Fig. 4A). The nearly tenfold reduction in Albts22 DMV capacity compared to wild-type corresponded well with nearly tenfold-lower production of infectious progeny (Table 1). However, DMV capacity correlated poorly with yield for Brts31, which produced infectious progeny at wild-type levels despite having only 35% of the DMV capacity of wild-type virus (Table 3). This suggested two possibilities: that Brts31 packages a reduced amount of RNA more efficiently than wild-type virus, or that Brts31 synthesizes normal amounts of RNA from a smaller-than-normal number of organelles.
To discriminate between these possibilities, wild-type and Brts31 RNA synthesis was compared by quantitative RT-PCR. Genomic and subgenomic RNA 7 synthesis was quantified using primers specific to the nsp3-encoding region and across the leader-body junction of subgenomic RNA 7, respectively. Brts31 synthesized somewhat larger amounts of genomic and subgenomic RNA than the wild type at 33°C, as shown in Fig. 4B. Together, these data demonstrate that efficient RNA production is not necessarily correlated with large or numerous DMVs.
DMVs and fitness.
To better understand the role of DMV formation in the virus replication cycle, we carried out competitive fitness assays on all the viruses that grew to approximately wild-type titers. In these assays, an equal number of plaque-forming units of wild-type virus and one ts virus were added at low multiplicity to either continuous or primary cells, which were then incubated at 33°C. Fitness was calculated as the ratio of mutant virus to wild-type virus present in the supernatant at the end of the competition. In this assay, a ratio of 1 would indicate that the viruses were equally fit, higher ratios indicate that the mutant virus is fitter than the wild type, and lower ratios indicate that the mutant virus is less fit than the wild type. Viruses were differentiated by means of plaque assays initiated at 33°C for 1 h and then continued by incubation at either 40°C (where only the wild type forms plaques) or 33°C (where both viruses form plaques). The assay was validated by performing differential plaque assays on pure samples of the wild type and Brts105 (Fig. 5A). The number of plaques formed by the wild type did not differ significantly at the two temperatures. Brts105 formed plaques in 33°C plaque assays but not in plaque assays initiated at 33°C and then continued by incubation at 40°C (Fig. 5A). This demonstrated that titers of wild-type and ts viruses can be distinguished by differential plaque assays and that the reversion rate of Brts105 was less than 10−5 and therefore would not detectably complicate the interpretation of competition experiments, which typically produced more than 106 PFU/ml.
FIG 5 .

Viral fitness assay validation and results. (A) Numbers of plaques generated by samples of the wild type and Brts105. Plaque assays were inoculated at 33°C for 1 h and then incubated at 33°C and 40°C. Results were normalized to the average titer obtained from the 33°C plaque assays and are averages for 14 replicates for the wild type or 8 replicates for Brts105. A titer below the 25-PFU/ml lower limit of detection is indicated (<). (B) Relative fitness of MHV mutants in continuous 17Cl-1 and primary MEFs, as determined by plaque assays at both 33°C and 40°C for each sample. Fitness is expressed as (33°C titer − 40°C titer)/40°C titer. Error bars show standard deviation. Statistically significant differences between the titer of ts virus (33°C titer − 40°C titer) and wild-type virus (40°C titer) are indicated (*, P ≤ 0.05; ns, not significant; nt, conditions not tested). (C) DNA sequencing results for the wild type, Brts105, and a 1:1 mixture of wild-type and Brts105 viruses. (D) Results of competitive fitness assays performed in triplicate with 1:1, 3:1, and 5:1 ratios of Brts105 to wild-type virus as determined by RT-PCR and DNA sequencing. Results are ratios of the two viruses based on peak height from direct cDNA sequencing and are from three replicate sequences.
Brts105 and Wüts18 showed a small but statistically significant increase in fitness compared to the wild type, Brts31 and Albts16 did not differ significantly in fitness from the wild type, and Albts22 appeared somewhat but not significantly less fit than the wild type in continuous cells (Fig. 5B). Results from primary MEFs agreed with continuous-cell results. This also showed that Brts105 and Wüts18 are cold adapted compared to wild-type virus, in addition to being ts. This demonstrated that viruses that produce larger replicative organelles (Fig. 5) (Albts16, Wüts18, and Brts31 versus the wild type) or more numerous replicative organelles (Fig. 5) (Brts31 and Brts105 versus the wild type) do not have a competitive disadvantage per se under these assay conditions.
In order to further test whether the differential plaque assay results reflected the proportion of wild-type and ts RNA released into the supernatant at the end of a competition assay in 17Cl-1 cells, RT-PCR and DNA sequencing (39) were performed for competition assays containing Brts105 and wild-type virus (Fig. 5C and D). In this assay, three replicates gave a fitness of 0.8 ± 0.1 for Brts105, which fell within the range of results recorded by plaque assay (0.4 to 6.4). The parity between Brts105 and the wild type was maintained even when the relative abundance of each virus at the beginning of the competition was altered (Fig. 5D). This confirmed that Brts105 was not significantly less fit than wild-type virus under these test conditions.
DISCUSSION
The formation of replicative organelles is a conserved strategy among positive-stranded viruses and has led to the assumption that viral organelles play an important and highly conserved role in intracellular RNA replication (1, 2). Previous studies had shown that newly synthesized viral RNA can be detected as early as 1 h after inoculation (40), but the earliest time at which DMVs have been detected is 2 h after inoculation (4, 5), and complete organelles with CM and DMVs were first reported 3 h after inoculation with MHV (5) or 4 h after inoculation with SARS-CoV (4), suggesting that the replicative organelle may be a lagging indicator of viral RNA synthesis. However, other studies have shown that replicative organelles disappear within about 2 h of treatments that strongly inhibit viral RNA synthesis (14, 15). In this study, we documented viruses with a range of organelle sizes and prevalence that were sufficient to support wild-type levels of viral growth. This included two viruses of the six tested (Brts31 and Brts105) that replicated normally with half the normal complement of organelles or less. These results extend findings that attenuated nsp4 mutant viruses produce fewer organelles than normal (21). Together, these results suggest that there is a considerable plasticity in the replication complex, since a variety of sizes and numbers of DMVs can support RNA synthesis.
The data presented here do not necessarily disprove that DMV clusters are needed for intracellular coronavirus RNA synthesis but demonstrate that the link may be looser than anticipated. This finding was anticipated by a recent study that demonstrated that subcellular compartments that contain double-stranded RNA (dsRNA) (i.e., probable DMVs) were not necessarily sites of active RNA replication (38). We unexpectedly found that viruses that produced larger and more numerous DMVs did not have a consistent fitness advantage in primary or continuous cells under test conditions. However, it is quite possible that further testing in primary macrophages or live animals would reveal a competitive advantage for increased organelle numbers or increased DMV capacity. A previous analysis of coronavirus RNA replication had shown that viral dsRNA colocalizes well with nonstructural proteins early in infection but that the correlation becomes less clear over time (38). Nevertheless, these results are the first direct evidence to our knowledge that intracellular RNA synthesis can be at least partially decoupled from formation of replicative organelles. We also report here for the first time that viruses with mutations in all 5 known cistrons of the MHV replicase form either slightly or substantially smaller DMVs, suggesting that while DMVs are formed primarily by nsp3, nsp4, and nsp6 mutants (17), mutations in any part of the replicase locus can potentially restrict DMV formation. This suggests that the size and potentially the form of the replicative organelle may be dependent on factors other than the amount of RNA synthesis, such as the efficiency of polyprotein processing.
DMVs are complex, containing viral replicase subunits, RNA, host membranes, and host proteins (4, 5, 7–13, 41, 42), so some changes in DMV size and number may be related to the efficiency of the many poorly understood processes that contribute to DMV formation. The results presented here should be taken with two caveats: that the effects of the mutations may be indirect, and that comparisons of growth and competition could be performed only at reduced temperature. Although all ts viruses except for Albts22 matched the output of the wild type at reduced temperature, reduced temperature may affect replication and the processes that lead to DMV formation differently.
The effect of the Brts31 mutation in reducing DMV formation may be explained by the essential role of nsp3 in organelle formation (17). However, we could not find a simple explanation for finding that the Brts105 mutation in nsp14, located over 15,000 nucleotides (nt) downstream of the Brts31 mutation, in a domain which had not previously been shown to be essential for DMV synthesis, also reduced organelle formation. While it is possible that the N7 methyltransferase activity of nsp14 is involved in an early step of DMV formation, a more likely explanation is that the mutation alters long-range protein folding or protein processing that occurs upstream of DMV formation, as observed previously for mutations in nsp10 and nsp3 (14, 27). However, further work will be needed to study the effects of the Brts105 mutation on the function of other essential replicase proteins such as nsp5 (14, 27) or protein complexes such as the nsp10-nsp14-nsp16 RNA cap methylation complex (43). Notwithstanding, the results presented here constitute the first demonstration of a functional linkage between nsp14 and the DMV scaffold proteins.
MATERIALS AND METHODS
Viruses and cells.
Wild-type control virus MHV-A59 and Brts31 were propagated from recombinant vaccinia virus-based infectious clones as described previously (14). MHV-Brts105 was isolated following selection with 150 µg/ml of 5-fluorouracil at 33°C for 16 h using the procedure described for Brts31 (14). MHV-Brts105, Albts16, Wüts18, and Albts22 mutants were plaque purified twice on 17Cl-1 cells and completely sequenced to confirm the presence of the appropriate ts mutation and the absence of other changes. 17Cl-1, DBT, and L929 cells were grown in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 5% tryptose phosphate broth (TPB; Sigma), 10% heat-inactivated fetal bovine serum (FBS; Biosera), antibiotics, and nonessential amino acids (NEAA; Invitrogen). Primary mouse embryo fibroblasts (Lonza), adipose-derived mesenchymal stem cells (Cyagen), and bone marrow-derived mesenchymal stem cells (obtained from Darwin Prockop at the Health Science Center of Texas A&M University, Bryan, TX) were cultured in DMEM supplemented with 10% FBS, NEAA, and antibiotics. Virus titers were determined by plaque assay on 17Cl-1 cells.
Northern blotting.
RNA for Northern blotting was obtained by infecting ~107 17Cl-1 cells with virus at a multiplicity of 10 PFU per cell for 8 h at 33°C or 6 h at 39.5°C and isolating total RNA using the Trizol reagent (Invitrogen, Paisley, United Kingdom) as described by the manufacturer. The poly(A)-containing RNA was isolated using oligo(dT)25 Dynabeads (Dynal, Oslo, Norway) as previously described (44). The RNA was then incubated with formamide (50%) and formaldehyde (2.2 M) at 70°C for 10 min and electrophoresed in a 1% agarose gel containing 20 mM MOPS (3-N-morpholinopropanesulfonic acid) and 600 mM formaldehyde. After electrophoresis, the gel was soaked in 0.05 N NaOH, neutralized, and equilibrated with 20× SSC (3 M NaCl, 0.3 M sodium citrate, 1 mM EDTA) before being vacuum blotted onto a nylon membrane (Optitran BA-S 83; Schleicher and Schuell, Sigma-Aldrich, Poole, United Kingdom). The RNA was cross-linked to the membrane using UV light, and MHV RNAs were detected by hybridization with a 466-bp, α-32P-, random-prime-labeled PCR product corresponding to sequences in the nucleocapsid protein open reading frame of the MHV-A59 genome (45).
Quantitative reverse transcriptase PCR (qRT-PCR).
Duplicate flasks were prepared so that RNA extraction and DMV analysis could be performed on samples from the same experiment. Total cellular RNA was extracted from infected cells 10 h after inoculation using an RNeasy minikit (Qiagen). The RNA integrity was analyzed with the 2100 Bioanalyzer using the RNA 6000 Nano kit (Agilent). Both cDNA synthesis and PCR were performed in a single tube from total RNA using the SensiMix SYBR one-step kit (Bioline). Genomic RNA was quantified by amplifying a 124-bp region from the nsp3 gene with the forward primer 5′ AATAAGCAGGAGGCAAATGTTC 3′ and the reverse primer 5′ CCCACAACACAATGAAACAAATC 3′. A 110-nt amplicon spanning the leader-body junction of sgRNA7 was amplified using forward primer 5′ GTACCCTCTCAACTCTAAAAC 3′ and reverse primer 5′ GCGGTTTACAGAGGAGCTT 3′. Amplifications were carried out in triplicate, with a 250 nM primer concentration in 25-µl reaction volumes. Melt curve analysis was performed to confirm PCR product specificity. Absolute quantitation was performed by comparison to a standard curve of cloned amplicons.
Electron microscopy.
Cells were inoculated with at least 20 PFU of virus per cell and maintained at 33°C. Infected cells were fixed in electron microscopy-grade 4% glutaraldehyde (Sigma) in 0.1 M cacodylate buffer, scraped from the plate, and then postfixed in 0.1 M cacodylate-buffered 1% osmium tetroxide for 1 h. The cells were dehydrated in increasing concentrations of acetone and then embedded in Agar 100 resin (Agar Scientific). Ultrathin sections that produced a silver interference pattern (50 to 60 nm in thickness) were taken from embedded samples and poststained with 0.5% uranyl acetate. Electron microscopy was performed at 80 kV on a Philips CM20 equipped with a 2K charge-coupled devide (CCD) camera or a Hitachi H600 equipped with film. Film negatives were scanned into digital files using a Microtek i800 scanner. Cell sections used here each contained a single visible nucleus, with intact nuclear and plasma membranes. Images of DMV- and virion-containing cells meeting these criteria were captured for later analysis.
Measurement of intracellular features was done using the boxer module of EMAN (46). The longest and shortest diameters of each DMV and intracellular virus particle were measured twice each by two observers. The standard deviation between repeated measurements was about 2 pixels on average, which equated to approximately 7 nm at the level of the specimen. Since most of the DMVs and virions had an elliptical profile, size was estimated by Ramanujan’s first approximation for the perimeter of ellipse, and the result was divided by π to approximate diameter.
Competitive fitness assays.
Continuous 17Cl-1 cells or primary MEFs (mouse embryo fibroblasts) were inoculated with a mixed virus pool containing 0.005 PFU per cell each of wild-type virus and one ts mutant. Cells were incubated at 33°C for 24 h on 17Cl-1 or 48 h on MEFs to allow the viruses to complete several rounds of replication. Relative amounts of wild-type and mutant virus were quantified either by RT-PCR following the method of Hall and Little (39) or by two replicate plaque assays, of which both were inoculated at 33°C for 1 h and then one was incubated at 33°C and the other at 40°C for 3 days. For RT-PCR quantification, released virus was concentrated by precipitation with 10% polyethylene glycol 8000 and purified by pelleting through a sucrose-TNE (10 mM Tris, 100 mM NaCl, 1 mM EDTA) cushion (25% [wt/vol] sucrose in 50 mM Tris-HCl [pH 7.4], 100 mM NaCl, and 0.1 mM EDTA). RNA was extracted from purified virus using an RNeasy minikit (Qiagen). Purified wild-type and Brts105 RNAs were mixed in molar ratios from 1:5 to 5:1 for use as standards at this stage. Virion RNA and standards were converted to cDNA by reverse transcription with random hexamer primers and amplified with primers bracketing the mutation in Brts105, forward primer 5′ GCAATAGGTATGATGTGTG 3′ and reverse primer 5′ GCTGTGGTTGCCGTATTGTAAG 3′. Amplicons were then sequenced commercially (Source Bioscience) using the same MHV-specific primers. Results were regressed to the proportion of sequencing peak height, normalized to standards.
ACKNOWLEDGMENTS
This work was supported by a studentship from the Iraqi Ministry of Higher Education and Scientific Research (H.M.N.A.) and the Swiss National Science Foundation (310030_149784; V.T.). Production of bone marrow-derived stem cells was funded by NIH grant P40RR017447.
Footnotes
Citation Al-Mulla HMN, Turrell L, Smith NM, Payne L, Baliji S, Züst R, Thiel V, Baker SC, Siddell SG, Neuman BW. 2014. Competitive fitness in coronaviruses is not correlated with size or number of double-membrane vesicles under reduced-temperature growth conditions. mBio 5(2):e01107-13. doi:10.1128/mBio.01107-13.
REFERENCES
- 1. den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. 10.1016/j.chom.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363–374. 10.1038/nrmicro1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens RW, Posthuma CC, van der Meer Y, Bárcena M, Haagmans BL, Snijder EJ, van den Hoogen BG. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 94:1749–1760. 10.1099/vir.0.052910-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226. 10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ulasli M, Verheije MH, de Haan CA, Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 12:844–861. 10.1111/j.1462-5822.2010.01437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, Onderwater JJ, van der Meulen J, Koerten HK, Mommaas AM. 2006. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80:5927–5940. 10.1128/JVI.02501-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deming DJ, Graham RL, Denison MR, Baric RS. 2007. Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 81:10280–10291. 10.1128/JVI.00017-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76:3697–3708. 10.1128/JVI.76.8.3697-3708.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hagemeijer MC, Verheije MH, Ulasli M, Shaltiël IA, de Vries LA, Reggiori F, Rottier PJ, de Haan CA. 2010. Dynamics of coronavirus replication-transcription complexes. J. Virol. 84:2134–2149. 10.1128/JVI.01716-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oostra M, te Lintelo EG, Deijs M, Verheije MH, Rottier PJ, de Haan CA. 2007. Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J. Virol. 81:12323–12336. 10.1128/JVI.01506-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. 2010. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7:500–508. 10.1016/j.chom.2010.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi ST, Schiller JJ, Kanjanahaluethai A, Baker SC, Oh JW, Lai MM. 1999. Colocalization and membrane association of murine hepatitis virus gene 1 products and de novo-synthesized viral RNA in infected cells. J. Virol. 73:5957–5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJ, Locker JK. 1999. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 73:7641–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stokes HL, Baliji S, Hui CG, Sawicki SG, Baker SC, Siddell SG. 2010. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 84:10148–10158. 10.1128/JVI.00901-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verheije MH, Raaben M, Mari M, Te Lintelo EG, Reggiori F, van Kuppeveld FJ, Rottier PJ, de Haan CA. 2008. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 4:e1000088. 10.1371/journal.ppat.1000088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denison MR, Spaan WJ, van der Meer Y, Gibson CA, Sims AC, Prentice E, Lu XT. 1999. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol. 73:6862–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. 2013. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4:e00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ziebuhr J, Schelle B, Karl N, Minskaia E, Bayer S, Siddell SG, Gorbalenya AE, Thiel V. 2007. Human coronavirus 229E papain-like proteases have overlapping specificities but distinct functions in viral replication. J. Virol. 81:3922–3932. 10.1128/JVI.02091-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fang S, Shen H, Wang J, Tay FP, Liu DX. 2010. Functional and genetic studies of the substrate specificity of coronavirus infectious bronchitis virus 3C-like proteinase. J. Virol. 84:7325–7336. 10.1128/JVI.02490-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stertz S, Reichelt M, Spiegel M, Kuri T, Martínez-Sobrido L, García-Sastre A, Weber F, Kochs G. 2007. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361:304–315. 10.1016/j.virol.2006.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gadlage MJ, Sparks JS, Beachboard DC, Cox RG, Doyle JD, Stobart CC, Denison MR. 2010. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 84:280–290. 10.1128/JVI.01772-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T. 2011. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7:1335–1347. 10.4161/auto.7.11.16642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawicki SG, Sawicki DL, Younker D, Meyer Y, Thiel V, Stokes H, Siddell SG. 2005. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 1:e39. 10.1371/journal.ppat.0010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beachboard DC, Lu X, Baker SC, Denison MR. 2013. Murine hepatitis virus nsp4 N258T mutants are not temperature-sensitive. Virology 435:210–213. 10.1016/j.virol.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sturman LS, Eastwood C, Frana MF, Duchala C, Baker F, Ricard CS, Sawicki SG, Holmes KV. 1987. Temperature-sensitive mutants of MHV-A59. Adv. Exp. Med. Biol. 218:159–168. 10.1007/978-1-4684-1280-2_20 [DOI] [PubMed] [Google Scholar]
- 26. Chang GH, Luo BJ, Lu P, Lin L, Wu XY, Li J, Hu Y, Zhu QY. 2011. Construction and genetic analysis of murine hepatitis virus strain A59 Nsp16 temperature sensitive mutant and the revertant virus. Virol. Sin. 26:19–29. 10.1007/s12250-011-3145-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donaldson EF, Graham RL, Sims AC, Denison MR, Baric RS. 2007. Analysis of murine hepatitis virus strain A59 temperature-sensitive mutant TS-LA6 suggests that nsp10 plays a critical role in polyprotein processing. J. Virol. 81:7086–7098. 10.1128/JVI.00049-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stobart CC, Lee AS, Lu X, Denison MR. 2012. Temperature-sensitive mutants and revertants in the coronavirus nonstructural protein 5 protease (3CLpro) define residues involved in long-distance communication and regulation of protease activity. J. Virol. 86:4801–4810. 10.1128/JVI.06754-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson MA, Chatterjee A, Neuman BW, Wüthrich K. 2010. SARS coronavirus unique domain: three-domain molecular architecture in solution and RNA binding. J. Mol. Biol. 400:724–742. 10.1016/j.jmb.2010.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neuman BW, Joseph JS, Saikatendu KS, Serrano P, Chatterjee A, Johnson MA, Liao L, Klaus JP, Yates JR, III, Wüthrich K, Stevens RC, Buchmeier MJ, Kuhn P. 2008. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82:5279–5294. 10.1128/JVI.02631-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- 32. Xu X, Liu Y, Weiss S, Arnold E, Sarafianos SG, Ding J. 2003. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 31:7117–7130. 10.1093/nar/gkg916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Decroly E, Debarnot C, Ferron F, Bouvet M, Coutard B, Imbert I, Gluais L, Papageorgiou N, Sharff A, Bricogne G, Ortiz-Lombardia M, Lescar J, Canard B. 2011. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 7:e1002059. 10.1371/journal.ppat.1002059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Y, Cai H, Pan J, Xiang N, Tien P, Ahola T, Guo D. 2009. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 106:3484–3489. 10.1073/pnas.0808790106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sawicki SG, Sawicki DL. 1986. Coronavirus minus-strand RNA synthesis and effect of cycloheximide on coronavirus RNA synthesis. J. Virol. 57:328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang T, Sawicki SG. 2001. Mouse hepatitis virus minus-strand templates are unstable and turnover during viral replication. Adv. Exp. Med. Biol. 494:491–497. 10.1007/978-1-4615-1325-4_71 [DOI] [PubMed] [Google Scholar]
- 37. Chang GH, Oliver E, Stanton I, Wilson M, Luo BJ, Lin L, Davidson A, Siddell S. 2011. Genetic analysis of murine hepatitis virus non-structural protein 16. J. Gen. Virol. 92:122–127. 10.1099/vir.0.026781-0 [DOI] [PubMed] [Google Scholar]
- 38. Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ, de Haan CA. 2012. Visualizing coronavirus RNA synthesis in time by using click chemistry. J. Virol. 86:5808–5816. 10.1128/JVI.07207-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hall GS, Little DP. 2007. Relative quantitation of virus population size in mixed genotype infections using sequencing chromatograms. J. Virol. Methods 146:22–28. 10.1016/j.jviromet.2007.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. An S, Maeda A, Makino S. 1998. Coronavirus transcription early in infection. J. Virol. 72:8517–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Hemert MJ, de Wilde AH, Gorbalenya AE, Snijder EJ. 2008. The in vitro RNA synthesizing activity of the isolated arterivirus replication/transcription complex is dependent on a host factor. J. Biol. Chem. 283:16525–16536. 10.1074/jbc.M708136200 [DOI] [PubMed] [Google Scholar]
- 42. van Hemert MJ, van den Worm SH, Knoops K, Mommaas AM, Gorbalenya AE, Snijder EJ. 2008. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 4:e1000054. 10.1371/journal.ppat.1000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, Decroly E. 2010. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 6:e1000863. 10.1371/journal.ppat.1000863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thiel V, Rashtchian A, Herold J, Schuster DM, Guan N, Siddell SG. 1997. Effective amplification of 20-kb DNA by reverse transcription PCR. Anal. Biochem. 252:62–70. 10.1006/abio.1997.2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Skinner MA, Siddell SG. 1983. Coronavirus JHM: nucleotide sequence of the mRNA that encodes nucleocapsid protein. Nucleic Acids Res. 11:5045–5054. 10.1093/nar/11.15.5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97. 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]