

Abstract
Atypical Parkinsonism associated with white matter pathology has been described in cerebrovascular diseases, mitochondrial cytopathies, osmotic demyelinating disorders, leukoencephalopathies including leukodystrophies, and others. Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is an autosomal dominant disorder with symptomatic onset in midlife and death within a few years after symptom onset. Neuroimaging reveals cerebral white matter lesions that are pathologically characterized by non-inflammatory myelin loss, reactive astrocytosis, and axonal spheroids. Most cases are caused by mutations in the colony-stimulating factor 1 receptor (CSF1R) gene.
We studied neuropathologically verified HDLS patients with CSF1R mutations to assess Parkinsonian features. Ten families were evaluated with 16 affected individuals. During the course of the illness, all patients had at least some degree of bradykinesia. Fifteen patients had postural instability, and seven had rigidity. Two patients initially presented with Parkinsonian gait and asymmetrical bradykinesia. These two patients and two others exhibited bradykinesia, rigidity, postural instability, and tremor (two with resting) early in the course of the illness. Levodopa/carbidopa therapy in these four patients provided no benefit, and the remaining 12 patients were not treated. The mean age of onset for all patients was about 45 years (range, 18-71) and the mean disease duration was approximately six years (range, 3-11).
We also reviewed HDLS patients published prior to the CSF1R discovery for the presence of Parkinsonian features. Out of 50 patients, 37 had gait impairments, 8 rigidity, 7 bradykinesia, and 5 resting tremor. Our report emphasizes the presence of atypical Parkinsonism in HDLS due to CSF1R mutations.
Keywords: HDLS, CSF1R mutation, Parkinsonism, Autosomal dominant, White matter disorders
INTRODUCTION
Atypical Parkinsonism associated with white matter (WM) disorders has been recognized since 1929 [1]. However, it is unclear how the interruption of WM function leads to atypical Parkinsonism. Nonetheless, Parkinsonian features occur in patients of vascular origin[2-4], in patients with traumatic etiology [5], in certain osmotic demyelinating syndromes such as central pontine and extrapontine myelinolysis [6], in various leukoencephalopathies including leukodystrophies and [7, 8], mitochondrial disorders [9], occasionally in neuroinflammatory diseases like multiple sclerosis [10], and in certain rare hereditary neurodegenerative conditions such as tremor-ataxia syndrome [11], spinocerebellar ataxia[12] and others.[13]
WM lesions with accumulation of axonal spheroids is the pathological hallmark of hereditary diffuse leukoencephalopathy with spheroids (HDLS), a rare, progressive adult-onset neurodegenerative disease that was initially identified in a Swedish family [14]. Subsequently, HDLS was described in additional kindreds and sporadic patients from diverse Caucasian populations worldwide [15, 16]. HDLS is characterized by symptoms including memory decline, depression, executive dysfunction, pyramidal signs, and atypical Parkinsonism[16]. WM lesions are evident on routine brain magnetic resonance imaging (MRI) studies [16]. Recently, the genetic cause of HDLS was discovered in a subset of families, and was due to mutations in the colony stimulating factor 1 receptor (CSF1R) gene located on chromosome 5[17].
The aim of this study is to describe the Parkinsonian features in genetically and pathologically confirmed patients with HDLS due to CSF1R mutations and to review the literature for the presence of Parkinsonian features in previously reported HDLS patients.
METHODS
Human subjects
The patients were collected through the International Consortium and were either prospectively studied (alive) or retrospectively (deceased) reviewed (Table 1). If medical records were incomplete or if the living patients were not seen by one of the investigators of this study for over a year, phone calls were made to immediate family members or the research subjects to obtain current information (Cases 10 and 11). Sixteen HDLS patients, all CSF1R mutation carriers with neuropathological confirmation, from 10 independent families were identified from the US (8), Norway (1), and Germany (1)[17]. Eleven patients had autopsies, and five had brain biopsies. All patients had pathologically confirmed WM abnormalities with axonal spheroids consistent with the diagnosis of HDLS. MRI scans were performed locally for diagnostic purposes. All studies used standard MRI techniques; 1.5 Tesla scanners with 5 mm thickness and 5 mm spacing. Axial and Sagittal T1 and T2 weighted images and fluid attenuated inversion recovery (FLAIR) images were used to detect the location of WM lesions, atrophy, and structural abnormalities in the brain. Contrast enhancement had been performed at least once in all 16 patients. Additionally, this study was approved by the institutional review boards of Mayo Clinic and the participating institutions.
Table 1.
Clinical characteristics of HDLS cases
| Family | US 1 | US 2 | Norwegian | US 3 | US 4 | US 5 | US 6 | Germany | US 7 | US 8 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSF1R mutation | Met766Thr | Ile775Asn | Gly585_Lys619delinsAla | Glu633Lys | Cys774_Asn814del |
Cys774_Asn814delinsQG LQSHVGPSLPSSSPQAQ |
Ile794Thr | Ala770Pro | Met875Thr | Gly589Glu | ||||||
| Case # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| Age of onset | 18 | 43 | 27 | 48 | 38 | 36 | 42 | 67 | 50 | 23 | 35 | 57 | 52 | 58 | 71 | 58 |
| Disease duration | Alive | 6 | 4 | Alive | 3 | 4 | 4 | 7 | 5 | Alive | Alive | 6 | 11 | 8 | Alive | 3 |
| Initial symptom(s) | Depression | Depression/Cognitive impairments |
Depression | Left leg stiffness and gait problems |
Depression/Cognitive impairments |
Depression Cognitive impairments |
Impaired dexterity in hand and fingers and bradykinesia |
Cognitive impairments/ Personality changes |
Cognitive impairments/ Memory problems |
Spastic gait | Cognitive impairments |
Depression, Personality changes |
Cognitive impairments |
Cognitive impairments/ Personality changes |
Cognitive impairments |
Cognitive impairments |
| Bradykinesia | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 1 |
| Rigidity | 0 | 2 | 1 | 2 | 0 | 0 | 2 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Tremor | 0 | 2 | 1 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 |
| Resting tremor | - | + | - | - | - | - | - | + | - | - | - | - | - | - | - | - |
| Kinetic or postural tremor | - | + | + | + | - | - | + | + | - | - | - | + | + | - | - | - |
| Postural instability | 0 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| Gait impairments | 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 1 | 1 | 2 |
| Shuffling gait | - | + | - | + | - | - | + | + | - | - | - | na | - | - | na | + |
| Freezing | - | + | - | + | - | - | + | + | - | - | - | na | - | - | na | - |
| Reduced strides | - | - | + | + | - | - | + | + | - | - | + | na | + | - | na | + |
| Stooped posture | - | - | - | + | - | + | - | - | - | - | - | na | - | - | na | - |
| Reduced arm swing | + | + | + | + | na | + | + | - | - | + | + | na | - | - | na | - |
| Spastic gait | - | - | - | - | - | - | + | - | - | + | - | na | - | - | na | - |
| Hypomimia | 1 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Hypophonia | 0 | 2 | 1 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Dysarthria | 0 | 2 | 2 | 2 | 0 | 0 | 2 | 2 | 0 | 2 | 0 | 0 | 1 | na | na | 1 |
| Other speech problems¥ | 0 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| Dyskinesia | 0 | 0 | 1פ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2۵ | 0 | 0 | 0 | 1פ |
| Dystonia | 0 | 2 | 1 | 0 | 0 | 2 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 |
| Myoclonus | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 1 |
| Chorea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ataxia | 0 | na | 2L | na | 1L | 1L | 1L | na | na | 2SAD | na | na | 1L | na | 0 | na |
| Spasticity | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | na | 0 | 0 | na | 0 |
| Pyramidal weakness; Initially | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | na | 0 |
| Pyramidal weakness; Advanced | 0 | 2 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 1 |
| Gegenhalten | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | na | 0 | 1 | na | 1 |
| Cognitive impairments/memory problems | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Apraxia | 0 | 1a | 2b | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | na | 2 | 1 | 2 |
| Depression | 2 | 2 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| Executive dysfunction | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Aggressiveness | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| Seizure | - | - | - | - | +β | +β | - | - | - | - | - | +∞ | - | - | - | +β |
| Daytime sleepiness | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | na | 1 | 1 | 1 | 1 | 1 | 1 |
| Nighttime sleep disturbance | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2μ | 1 | 1 | 1 | 2μ |
| Eye movement abnormalities | 0 | na | 21 | 22 | 21 | 21 | na | 13 | na | na | 13 | na | 14 | na | na | 22 |
| Orthostatic hypotension | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Urinary/fecal incontinent | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0 | 1 | 2 | 2 | 2 | 0 | 2 |
| Clinical diagnosis | LD | LD | LD | PD vs. PSP vs. MS | MS | MS | CBS | CBS | FTD vs. AD vs CADASIL | LD | CADASIL | AD | FTD | FTD vs. AD | FTD | FTD |
| Criteria for parkinsonism | - | D | D | D | Ps | Ps | D | D | D | Ps | Ps | Ps | Ps | Ps | Ps | D |
AD: Alzheimer’s disease; CADASIL: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy; CBS: Corticobasal syndrome; D: Definite; FTD: Frontotemporal dementia; L: Limb; LD: Leukodystrophy of unknown origin; MS: Multiple sclerosis; na: Not available; Ps: Possible; PSP: Progressive supranuclear palsy; SAD: Spastic, ataxic dysarthria;
Defined as aphasia and mutism at the advanced stage of the disease;
Restless leg syndrome, the other cases had unspecific sleep problems;
Generalized tonic clonic seizure;
Complexed partial seizure;
Homonymous hemianopsia to either right or left side;
Occulomotor apraxia;
Poor eye pursuit movements;
Vertical gaze palsy;
Limb apraxia;
Ideomotor and constructional apraxia;
Limb, ideomotor, and constructional apraxia;
Tremor of tongue;
Grimacing and tics;
Akathisia.
Regarding Parkinsonian features, we looked for bradykinesia; rigidity of both axial and appendicular type; resting tremor; postural instability; gait impairments including shuffling gait, freezing while walking, reduced strides; stooped posture; reduced arm swing; hypomimia; symmetry of signs; and response to dopaminergic treatments. We analyzed speech and language impairments such as dysarthria, hypophonia, and dysphasia. The presence/absence of other movement disorder features were also analyzed, including dyskinesias, akathisia, tics, dystonia, myoclonus, chorea, ataxia, and kinetic or postural tremor. We assessed medical histories for administration of medications known to produce secondary Parkinsonism, such as first-generation neuroleptics, antidepressants, and calcium channel-blocking agents. We also analyzed the data related to pyramidal signs, including spasticity, pyramidal weakness, spastic gait, gegenhalten, cognitive impairments/memory problems, apraxia, depression, executive dysfunction, aggressiveness, seizures, daytime sleepiness and nighttime sleep disturbances, eye movement dysfunction, orthostatic hypotension, and urinary and fecal incontinence. We looked for the clinical diagnosis assigned. All available data was tabulated.
We used the UK Parkinson’s Disease Society Brain Bank’s clinical criteria for the diagnosis of probable Parkinson’s disease (PD) [18, 19], and we used the criteria for Parkinsonism as summarized and defined in Fahn 2011 [20].
Review of published HDLS families/patients
We also conducted a review of the English language literature for any data on Parkinsonism and other movement disorders in published HDLS families/patients beginning with papers published in 1984. The National Institute of Health’s PubMed database was searched for families and patients, using the keywords: HDLS; leukoencephalopathy with spheroids; neuroaxonal spheroids; and white matter disease with spheroids. The data was then tabulated.
Neuropathological assessment
Neuropathological evaluations of the eight patients (Cases 2, 3, 5, 7, 8, 9, 12, and 13) were performed at the brain bank of Mayo Clinic in Jacksonville, Florida. The left hemibrains of three patients (Cases 2, 7, and 8) were fixed in formalin and sampled for histology according to a standardized protocol. Previously autopsied brain tissues were available on the other patients. Tissue sections were embedded in paraffin and 5 μm thick sections were mounted on glass slides for histological studies and immunohistochemistry. The areas sampled were neocortices, amygdala, basal nucleus of Meynert, caudate nucleus, putamen, thalamus, subthalamic nucleus, midbrain, pons, medulla, cerebellum, and spinal cord. The basal ganglia, including the amygdala and basal nucleus of Meynert, were not available on Case 12. The medulla was not available on Cases 2 and 5, and the spinal cord was available on Cases 3, 5, and 13. Paraffin-embedded sections were stained with hematoxylin, eosin, and thioflavin S. Sections were also processed for immunohistochemistry for α-synuclein (NACP, rabbit polyclonal; 1:3,000) on Cases 2, 7, 8, 12, and 13. The Braak PD stage was determined according to distribution of Lewy-related pathology [21]. The density and distribution of neurofibrillary tangles (NFT) were used to assign a Braak NFT stage [22].
RESULTS
The clinical data and CSF1R mutation status of each case are summarized in Table 1. Of the 16 patients in the study, all were Caucasian (9 males). The mean age of onset was about 45 years (range, 18-71 years); the mean duration of disease from symptom onset to death was approximately six years (range, 3-11), and the mean age of death was 54 years (range, 31-74). None of the patients had been given an initial clinical diagnosis of HDLS.
Parkinsonism in HDLS
The initial symptoms in 13 patients (81 %, 8 males) were depression, cognitive problems, and personality changes. These initial symptoms could be seen alone or in combination. One woman initially developed spastic gait impairment (Case 10). Parkinsonian features were the initial symptoms in two patients (13 %, 1 male), manifested by a shuffling gait (Case 4) or bradykinesia and impaired hand and finger dexterity (Case 7). The mean age of onset of Parkinsonism for these two patients was 45 years (range 42-48). The deceased patient (Case 7) had disease duration of four years. The second patient (Case 4) is still alive; the current disease duration is seven years. Details of these two patients were reported previously [16, 23].
Two additional patients (Cases 2 and 8) displayed Parkinsonism as their main phenotype a few months after the initial symptoms of cognitive impairment and/or depression. Parkinsonian features were characterized by bradykinesia and shuffling gait in both. The mean age of onset for these two patients was 55 years (range, 43-67) and the disease duration for the deceased patient (Case 8) was seven years. The disease duration is currently four years for the living patient.
With disease progression, the Parkinsonian features in these four patients (Cases 2, 4, 7, and 8) evolved, leading to bilateral, severe bradykinesia (all slightly asymmetrical, except Case 7, which had substantial asymmetry), rigidity, shuffling gait, freezing, hesitancy on turning, and postural instability with falls. Hypophonia and eventually dysarthria were present in all four patients. Two patients (Cases 2 and 8) exhibited resting and kinetic tremors, and the two others (Cases 4 and 7) only had kinetic tremor. Dystonia was observed in three patients (Cases 2, 7, and 8), myoclonus in two (Cases 2 and 8) patients, and ataxia in one patient (Case 7). None of these cases benefited from levodopa/carbidopa therapy. They were not treated with neuroleptics or other mediations associated with secondary Parkinsonism.
The 12 remaining patients (Cases 1, 3, 5, 6, 9 - 16) developed asymmetrical bradykinesia, which was minimal in the advanced stages of the illness. Patients also developed hypomimia. with an average of two years into the disease course. Gait impairments were present in all patients. Rigidity was evident in three patients (Cases 3, 9, and 16), which progressed and led to limb contractures. Three patients (Cases 3, 12, and 13) had kinetic tremor in their hands, one also had intention tremor. Myoclonus affecting the fingers was seen in one patient (Case 13), and myoclonus affecting the legs and foot was seen in three patients (Cases 2, 3 and 16). Four patients (Cases 2, 6, 8, and 16) had severe dystonia affecting the hand or foot on one side of the body only. Two patients (Cases 3 and 16) exhibited facial dyskinesias (grimacing and eye tics in both). One patient (Case 12) was described as having akathisia. Three patients (Case 3, 5, 6 and 13) had limb ataxia, and one (Case 10) had ataxic dysarthria. Therapy with levodopa/carbidopa was not employed in any of these 12 patients; they were also not treated with neuroleptics. Some of these patients had been given small doses of selective serotonin re-uptake inhibitors in the initial stage of the illness, usually to treat depression, but as the disease progressed and no response was noted, these medications were discontinued (Cases 1, 5, 6, and 12). Other medications with known Parkinsonism as a side-effect were not given. Chorea was not observed in any of the 16 patients.
Other clinical features in HDLS
Apart from Parkinsonian features, all of our patients exhibited additional clinical features such as cognitive impairment, memory problems, personality changes, apraxia, dysphasia, pyramidal signs, and urinary and fecal incontinence. These additional features occurred with different severity, at different stages of the illness, and in different combinations throughout the course of the disease. Urinary and fecal incontinence were consistently late features. Vertical gaze abnormalities were noted in only one patient (Case 13), and poor pursuit was noted in another two (Cases 8 and 11). Additional clinical signs were similar to those reported in the literature. In previous reports, visual disturbances and Parkinsonism were not emphasized.
Neuroimaging features in HDLS
All of our 16 patients demonstrated WM lesions with a frontal predominance (bifrontal or bifrontoparietal) involving the periventricular, deep, and subcortical cerebral WM (Figure 1). There was no evidence of grey matter, brainstem or cerebellar pathology, and there was no enhancement. Involvement of the corticospinal tracts was noted on MRI (T2 and FLAIR) later in the disease course of some patients. Similar features have been reported previously [15].
Figure 1.
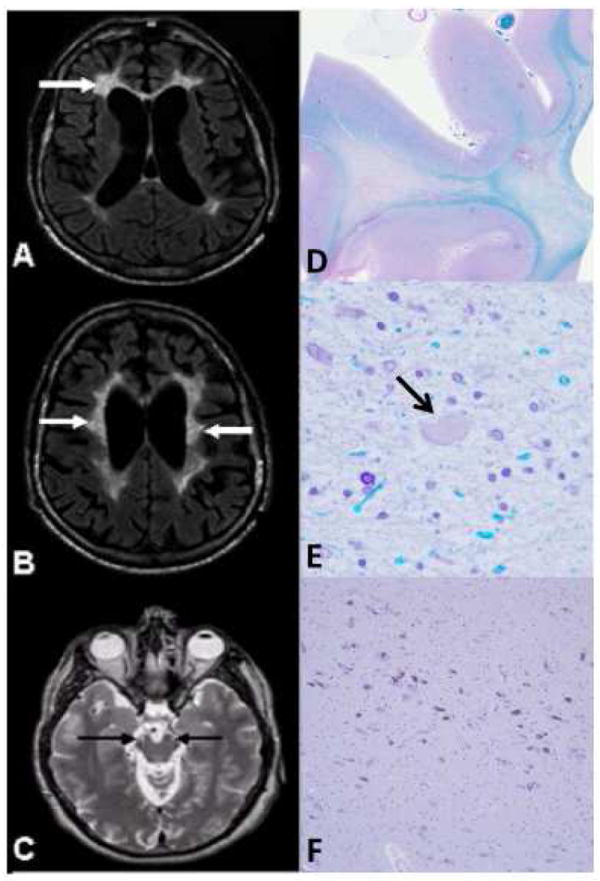
MRI (A-C) and neuropathology (D-F) of HDLS cases with CSF1R mutations
A, B) Axial FLAIR MRI images; C) Axial T2-weighted MRI image. A) Localized, periventricular, deep, and subcortical white matter lesions (WML), more severe on the left side (arrows). B) Bilateral frontoparietal WML involving periventricular, deep, and subcortical areas with U-fiber sparing (arrows). C) Involvement of the corticospinal tracts bilaterally (arrows) at the level of the mesencephalon. D, E) Luxol fast blue; F) Hematoxylin-eosin. D) Myelin loss in the white matter of the superior frontal lobe. E) Tissue vacuolation with axonal spheroids (arrow) (magnification: ×400). F) The ventrolateral part of substantia nigra has minimal focal neuronal loss with extraneuronal neuromelanin (magnification: ×100).
Pathologic findings in HDLS
Eleven of our patients had a brain autopsy that demonstrated the presence of WM lesions in a distribution similar to that seen on MRI studies. The pathological hallmark in all of our autopsies was myelin damage and the presence of axonal loss and spheroids (Figure 1), and similar pathology was also present in five patients in which only biopsy material was available for study. Microscopic characteristics of the CSF1R mutation carriers are summarized in Table 2. The neuropathological features of our HDLS patients are similar to those reported in the literature
Table 2.
Neuropathological features in eight HDLS cases with CSF1R mutation
| Case | Case 2 | Case 3 | Case 5 | Case 7 | Case 8 | Case 9 | Case 12 | Case 13 |
|---|---|---|---|---|---|---|---|---|
| Ventricular enlargement | +++ | na | na | +++ | +++ | na | na | na |
| White matter changes | ||||||||
| Frontal lobe | + | +++ | +++ | ++ | +++ | +++ | +++ | +++ |
| Parietal lobe | + | ++ | ++ | + | + | ++ | na | + |
| Temporal lobe | - | +++ | - | - | - | na | ++ | ± |
| Occipital lobe | - | + | na | - | - | + | na | ± |
| Corticospinal tracts degeneration | ||||||||
| Motor cortex | +++ | na | na | +++ | + | na | na | na |
| Posterior limb of internal capsule | +++ | ++ | +++ | + | - | ++ | + | +++ |
| Cerebral peduncle | +++ | ++ | +++ | ++ | - | + | + | ++ |
| Pontine base | ++ | ++ | +++ | + | - | - | ± | + |
| Medullary pyramids | na | + | na | ± | - | - | - | ± |
| Anterior corticospinal tracts | na | ± | - | na | na | na | na | + |
| Posterior corticospinal tracts | na | ± | +++ | na | na | na | na | + |
| Neuronal loss in the substantia nigra | + | - | - | + | - | - | ±¥ | - |
| Gliosis in the basal ganglia | ±£ | -€ | -€ | + | - | +© | na | + |
| Braak PD stage | 0 | -* | -* | 0 | 0 | -* | 1† | 3‡ |
| Braak NFT stage | 0 | 0 | 0 | 0 | II | I | 0 | III |
Only autopsy cases with multiple sections, including neocortexes, basal ganglia, thalamus, midbrain, pons, and medulla available are summarized in the table. CSF1R: Colony stimulating factor 1 receptor; HDLS: Hereditary diffuse leukoencephalopathy with spheroids; na: Not available; NFT: Neurofibrillary tangle; PD: Parkinson’s disease; -: None, ±: Minimal; +: Mild; ++: Moderate; +++: Severe;
Only a small part of the substantia nigra was available;
Only an anterior part of the putamen was available;
Only posterior and lateral parts of putamen were available;
Only a lateral part of putamen was available;
Lewy bodies were not seen in the substantia nigra with H&E staining;
A few neurites were seen in medullary raphe abscurus;
Incidental Lewy body disease (there is no neural loss in the substantia nigra and dorsal motor nucleus of the vagus, and minimal neuronal loss in the locus ceruleus)
Clinical features of HDLS in a review of literature
We identified a total of 64 HDLS patients published in the English literature since 1984 (Supplementary e-Table). Five of these patients were CSF1R mutation carriers [17], and we re-analyzed additional clinical data that became available after the initial publications. Therefore, these five previously published patients (Cases 7, 10 - 12 and 14) [23-26] are included in our report of the 16 patients. We also excluded nine additional patients who had either no descriptional data or limited clinical data [25, 27-29]. The remaining 50 patients analyzed included 37 patients from 12 kindreds and 13 sporadic patients. Rigidity was reported in a total of eight patients (16%); seven familial patients (from five kindreds) and one sporadic patient. Two of these familial patients also had bradykinesia. Five additional patients, three familial from three different kindreds and two sporadic, were described as having bradykinesia alone. Significant gait impairments were reported in 37 patients, where five were specifically described with a Parkinsonian gait. The other 32 (22 familial and 10 sporadic) patients had less well-characterized gait problems, but in most, it was noted to be broad-based and spastic. Postural instability was noted in 20 (40%) patients, resting tremor in five (10%), intension tremor in one, myoclonic tremor in another one, and unclassified tremor in additional two patients. Five patients (10%) had dystonia. Myoclonus was described in four (8%). Dyskinesia was present in three (6%); two patients from a Swedish family had oral dyskinesia attributed to therapy with neuroleptics. One had both akathisia and facial grimacing. Chorea was present in two patients (4%). Hypophonia was reported in one patient, and dysarthria was noted in nine patients (18%). Ataxia was described in 14 patients (28%). Levodopa/carbidopa therapy was used in one patient in doses up to 1000 mg per day for at least five months, without any benefit. Dopamine agonists were not used as a treatment. Only in the Swedish kindred could the presence of Parkinsonian features be potentially attributed to treatment with neuroleptics. For the other patients, exposure to neuroleptics or other drugs known to produce secondary Parkinsonism were not reported.
DISCUSSION
The etiology of Parkinsonism is complicated by the lack of in vivo biological markers for precise diagnosis. Consequently, current diagnosis requires reliance on clinical and pathological criteria. None of our 16 HDLS patients, all CSF1R mutation carriers, fulfill the strict diagnostic criteria for Parkinson’s disease [18, 19], but seven fulfill definite clinical diagnostic criteria, and eight fulfill the possible clinical diagnostic criteria for atypical Parkinsonism [20]. Bradykinesia was present in all 16 patients. Bradykinesia is defined as a reduction in spontaneous movements, and a slowness in the initiation and execution of a movement, without significant muscle weakness [30]. Patients with pyramidal tract involvement can clinically show bradykinesia, but it is usually accompanied by muscle weakness, including pyramidal weakness. Eleven patients (Cases 2-6, 8-11, 14 and 16) showed bradykinesia without muscle weakness during earlier stages of their illness, and they developed pyramidal weakness later in the course of the disease. Rigidity was described in seven patients (44%). There are several other distinctive types of increased muscle tone, such as spasticity and gegenhalten. Spasticity is caused by pyramidal tract involvement and is usually accompanied by muscle weakness. Gegenhalten is seen in patients with frontal lobe involvement with deterioration of attention. Two patients (Cases 8 and 9) had severe rigidity during the course of illness. Two patients (Case 2 and 8) had resting tremor in combination with kinetic tremor. All patients had gait impairment and fifteen showed postural instability. Parkinsonian gait impairment accompanied by postural instability was evident in four patients in an early disease stage; these patients later developed broad-based gait that was mixed with spastic features. The remaining 11 patients had gait impairment later in the course of the illness, which was characterized by reduced arm swing, reduced strides, stooped posture, shuffling gait, and some spastic features. Several patients had features of a Parkinsonian gait, which was characterized by reduced stride, shuffling gait, freezing, stooped posture and others. Righting reflexes are also impaired in patients with Parkinsonism. Gait abnormalities in some of the patients may have been related to a spastic paraplegic gait, also referred to as a “scissor gait,” which is characterized by slow, regular, and short steps with restricted motion of hips and knee [30] Spastic gait is usually accompanied by weakness of affected muscle. Patients with frontal lobe involvement show reduced and hesitant steps and shuffling gait without stooped posture. Three patients (Cases 2, 4, and 8) clearly showed only Parkinsonism, including Parkinsonian gait impairment in the early stage of illness. Our HDLS patients eventually manifested complex clinical phenotypes mixed with Parkinsonism, pyramidal signs, frontal lobe signs, and ataxia; however, longitudinal clinical observations allowed us to detect Parkinsonian features in HDLS patients.
In the group of 50 published HDLS patients included in our review, 18 had some of the criteria for Parkinsonism, six patients were clinically definite, four were clinically probable, and eight were clinically possible for atypical Parkinsonism [20] In four patients from the original Swedish family, Parkinsonian features could be potentially attributed to treatment with first-generation of neuroleptics; however, one of these four patients, Case III:19, had “Parkinsonian” tremor and gait disturbances before any exposure to neuroleptic therapy [14]. There is no information provided for the other patients from the literature regarding possible treatment with first-generation neuroleptics. Out of the six patients meeting definite criteria, one had all cardinal signs of Parkinsonism, including rigidity, bradykinesia, gait impairment, postural instability, and resting tremor. Two had rigidity, resting tremor, and gait impairment with additional postural instability in one, and bradykinesia in the other. Two additional patients had rigidity, postural instability, and gait impairment. One patient had resting tremor and postural instability. Of the four patients that met the probable diagnostic criteria for Parkinsonism, two had rigidity and gait impairments, one had resting tremor and gait impairment, and one had only rigidity. Of the eight patients meeting the possible criteria for Parkinsonism, three had bradykinesia, postural instability, and gait impairments, two had gait impairments with additional bradykinesia in one and postural instability in the other. Three patients had gait impairments defined as “Parkinsonian gait”.
Sixteen (32%) of the reviewed published HDLS patients were described as having gait impairment alone without any additional Parkinsonian features. The gait impairment in these patients is not well characterized. Nevertheless, gait impairment is a significant clinical problem in HDLS, occurring in 37 (74%) of the reported literature patients and in all 16 (100%) of our pathologically-assessed HDLS patients due to CSF1R mutations.
The small number of patients in our study does not allow for correlating phenotypes with genotypes; however, all four patients with a predominant Parkinsonian phenotype included in our study, had amino acid point mutations in the CSF1R protein. In contrast, patients with deletion mutations tended to have more pyramidal features. We analyzed the DNA specimens from the original Swedish family and three unrelated US family patients, but no CSF1R mutations have been identified in these kindreds. We do not have access to the DNA specimens from other reported patients. It is possible that at least some of these patients are carriers of CSF1R mutations; however, based on limited genetic data from the Swedish and US families, it is possible to speculate that other genes for HDLS will be found in the future.
Besides the parkinsonian features, all of our CSF1R mutation carrier patients and the patients from our literature review presented with additional clinical features that define the Parkinsonism seen in HDLS as an atypical Parkinsonian syndrome. None of our four patients with a predominant Parkinsonian phenotype responded to levodopa/carbidopa therapy when given in large doses for a prolonged time. The remaining 12 patients in our study were not treated with levodopa/carbidopa or dopamine agonists. There may be several explanations for this, including the masking of Parkinsonian signs by other features of this illness, such as severe cognitive impairments and pyramidal signs, which are particularly seen in the advanced stages of the illness. In the later stages of the illness, when the patients were immobile, wheelchair- or bed- confined, the neurological evaluations were not always performed, and therefore, the presence of Parkinsonian signs could have been missed by primary physicians. The patients from our literature review included only one patient who was treated with levodopa/carbidopa (dose and duration of therapy not provided), but without any benefit.
All of our 16 CSF1R mutation carriers demonstrated WM lesions as their main pathological finding on brain MRI studies [31]. This was also the case for patients ascertained from our literature review [15]. The pathological hallmark in all of the patients was myelin damage and loss with the presence of axonal spheroids. As Table 2 demonstrates, the substantia nigra was not affected, or was only mildly affected in our autopsy patients, including one with a predominant Parkinsonian phenotype. The basal ganglia were only minimally or mildly affected in four of the patients; Lewy body pathology and Alzheimer type pathology were not prominent in any of the autopsied patients. Of patients ascertained from our literature review, 42 had autopsies, and seven of those had minimal involvement of substantia nigra, caudate nucleus, and putamen. In many of these patients, details regarding pathological examination of basal ganglia are missing. Patient III:19 from the Swedish family presented with resting tremor and gait disturbance prior to the exposure to neuroleptics was found to have a “pale” substantia nigra and slight atrophy of caudate nucleus and thalamus [14]. The details for microscopic examination of substantia nigra in this patient were not provided. Accordingly, the Parkinsonian features seen in HDLS are most likely due to the disruption of connections between basal ganglia nuclei and cerebral cortex rather than major pathology affecting the basal ganglia. In our HDLS patients with the CSF1R mutations, the cortical gray matter was only minimally or mildly affected on MRI and autopsy studies. Therefore, severe cognitive impairment in HDLS is disproportionate to the mild cortical gray matter abnormalities, thus supporting the notion that the disruption of connections among different cortical structures may lead to dementia in HDLS. Involvement of corticospinal tracts evident on MRI and autopsy studies may explain the spasticity and other pyramidal signs demonstrated in HDLS patients
The association of WM lesions and motor impairments such as bradykinesia, rigidity, gait and postural disturbances, and even dystonia has been noted in small vessel diseases (SVD) such as CADASIL, and Binswanger disease [2, 3, 32-35], osmotic demyelinating syndrome [6], neurodegenerative disorders [36], and others [13]. It has been hypothesized that the mechanism linking WM lesions to Parkinsonism is that WM lesions structurally or functionally disconnect fiber tracts in the brain that are critical for performing movements [32-34, 37]. Subcortical and periventricular WM lesions might interfere with ascending thalamocortical and descending corticospinal fibers. The confluent WM lesions can interfere with striato-pallido-thalamic fibers in the same way as SVD and larger strokes, respectively [37, 38]. Further support for this view comes from Snijders and colleagues [39] who demonstrated that patients with freezing of gait tend to have reduced activity in the supplementary motor cortex. This finding has also been found in association with hypokinesia in PD [40].
De Laat and colleagues [34] demonstrated that gait disturbance in SVD is, to some extent, due to widespread disruption of the WM, even in normally-appearing WM. It may be that with newer neuroimaging techniques, such as diffusion tensor imaging, these subtle changes will be demonstrated in early stages of HDLS. The topographical location of WM lesions is critical for development of clinical symptoms rather than the overall burden of WM lesions [41]. The development of antibodies to CSF1R protein will help to delineate the extent of damage seen in the substantia nigra and other basal ganglia nuclei.
A major strength of our report is that all of the patients in our series had pathologically confirmed HDLS, and all had CSF1R mutations. As HDLS is a rare condition, our collection of 16 patients is substantial. A major limitation of our study is that the data was collected retrospectively for the majority of patients; no systematic and quantitative analysis was performed, and no consensus of definitions and terms was obtained. Therefore, our results need to be confirmed with larger samples. There is also significant limitation in regard to the analysis of literature patients. Many of them had only limited clinical, neuroimaging, and neuropathological descriptions, and they do not have genetic confirmation of CSF1R mutations.
In conclusion, our study highlights the presence of atypical Parkinsonism in HDLS despite the pathologic focus in the cerebral WM. We demonstrated that Parkinsonism can be the initial symptom in HDLS with CSF1R mutations and can dominate the clinical picture during the progression of the disease. The mechanism of the atypical Parkinsonism remains enigmatic. Molecular studies on HDLS might provide novel insights into potential mechanisms underlying Parkinsonism and thereby pave the way for future targeted therapies aimed at disease prevention and cure.
Supplementary Material
Acknowledgments
Work was partially supported by the NIH/NINDS P50NS072187 and P50NS072187-01S2; Mayo Clinic Florida (MCF) Research Committee CR program (MCF #90052030); and Dystonia Medical Research FoundationCS was sponsored by The Swedish and Gothenburg Societies for the Neurologically Disabled and The Gothenburg Foundation for Neurological Research, “Rune och Ulla Amlövs” Foundation for Neurological research, Capio Research Foundation, Thuréus Foundation for Geriatric research, Stiftelsen för Gamla Tjänarinnor, the Swedish Society of Medicine Gothenburg, and the Anna - Lisa and Bror Bjornssons Foundation for neurological research 2012/2013. AMN was partially supported by the Association for Frontotemporal Degeneration Postdoctoral Fellowship.
We would like to thank Kelly Viola of the Mayo Clinic for her editorial assistance.
Finally, we would also like to acknowledge the patients and their families for their participation. This research would not have been possible without their consistent support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crithely M. Arteriosclerotic parkinsonism. Brain. 1929;52:23–83. [Google Scholar]
- 2.Demirkiran M, Bozdemir H, Sarica Y. Vascular parkinsonism: a distinct, heterogeneous clinical entity. Acta Neurol Scand. 2001;104:63–7. doi: 10.1034/j.1600-0404.2001.104002063.x. [DOI] [PubMed] [Google Scholar]
- 3.Wegner F, Strecker K, Schwarz J, Wagner A, Heinritz W, Sommerer F, et al. Vascular parkinsonism in a CADASIL case with an intact nigrostriatal dopaminergic system. J Neurol. 2007;254:1743–5. doi: 10.1007/s00415-007-0529-4. [DOI] [PubMed] [Google Scholar]
- 4.Sibon I, Guyot M, Allard M, Tison F. Parkinsonism following anterior choroidal artery stroke. Eur J Neurol. 2004;11:283–4. doi: 10.1046/j.1468-1331.2003.00751.x. [DOI] [PubMed] [Google Scholar]
- 5.Netravathi M, Pal PK, Indira Devi B. A clinical profile of 103 patients with secondary movement disorders: correlation of etiology with phenomenology. Eur J Neurol. 2012;19:226–33. doi: 10.1111/j.1468-1331.2011.03469.x. [DOI] [PubMed] [Google Scholar]
- 6.Brown WD. Osmotic demyelination disorders: central pontine and extrapontine myelinolysis. Curr Opin Neurol. 2000;13:691–7. doi: 10.1097/00019052-200012000-00014. [DOI] [PubMed] [Google Scholar]
- 7.Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131:2321–31. doi: 10.1093/brain/awn178. [DOI] [PubMed] [Google Scholar]
- 8.Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 2011;26:1593–604. doi: 10.1002/mds.23774. [DOI] [PubMed] [Google Scholar]
- 9.Finsterer J. Parkinson’s syndrome and Parkinson’s disease in mitochondrial disorders. Mov Disord. 2011;26:784–91. doi: 10.1002/mds.23651. [DOI] [PubMed] [Google Scholar]
- 10.Saidha S, Mok TH, Butler M, Fanning N, Harrington H. Multiple sclerosis exceptionally presenting as parkinsonism responds to intravenous methylprednisolone. J Clin Neurosci. 2010;17:654–5. doi: 10.1016/j.jocn.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 11.Raske C, Hagerman PJ. Molecular pathogenesis of fragile X-associated tremor/ataxia syndrome. J Investig Med. 2009;57:825–9. doi: 10.231/JIM.0b013e3181be329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perlman SL. Spinocerebellar degenerations. Handb Clin Neurol. 2011;100:113–40. doi: 10.1016/B978-0-444-52014-2.00006-9. [DOI] [PubMed] [Google Scholar]
- 13.Loesch DZ, Kotschet K, Trost N, Greco CM, Kinsella G, Slater HR, et al. White matter changes in basis pontis in small expansion FMR1 allele carriers with parkinsonism. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:502–6. doi: 10.1002/ajmg.b.31189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Axelsson R, Roytta M, Sourander P, Akesson HO, Andersen O. Hereditary diffuse leucoencephalopathy with spheroids. Acta psychiatrica Scandinavica. 1984;314:1–65. [PubMed] [Google Scholar]
- 15.Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology. 2009;72:1953–9. doi: 10.1212/WNL.0b013e3181a826c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundal C, F S, Van Gerpen JA, Wider C, Shuster EA, Aasly J, Ghetti B, Roeber S, Garbern J, Tselis A, Swerdlow RH, Miller BB, Rademakers R, Dickson DW, Wszolek ZK. Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids (HDLS): A Misdiagnosed Disease Entity. the Journal of Neurological Sciences. 2011 doi: 10.1016/j.jns.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rademakers R, Baker M, Nicholson AN, Rutherford NJ, Finch N, Soto A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) cause hereditary diffuse leukoencephalopathy with spheroids. Nature Genetics. 2011 doi: 10.1038/ng.1027. In print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry. 1992;55:181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol. 1993;50:140–8. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 20.Fahn S, Jankovic J, Hallett M. Principles and practice of movement disorders. Second. Elsevier Saunders; 2011. [Google Scholar]
- 21.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 23.Baba Y, Ghetti B, Baker MC, Uitti RJ, Hutton ML, Yamaguchi K, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol (Berl) 2006;111:300–11. doi: 10.1007/s00401-006-0046-z. [DOI] [PubMed] [Google Scholar]
- 24.Van Gerpen JA, Wider C, Broderick DF, Dickson DW, Brown LA, Wszolek ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology. 2008;71:925–9. doi: 10.1212/01.wnl.0000325916.30701.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swerdlow RH, Miller BB, Lopes MB, Mandell JW, Wooten GF, Damgaard P, et al. Autosomal dominant subcortical gliosis presenting as frontotemporal dementia. Neurology. 2009;72:260–7. doi: 10.1212/01.wnl.0000339484.61490.a4. [DOI] [PubMed] [Google Scholar]
- 26.Mateen FJ, Keegan BM, Krecke K, Parisi JE, Trenerry MR, Pittock SJ. Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study. Journal of neurology, neurosurgery, and psychiatry. 2010;81:619–22. doi: 10.1136/jnnp.2008.169243. [DOI] [PubMed] [Google Scholar]
- 27.Ali ZS, Van Der Voorn JP, Powers JM. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia--a role for oxidative damage. J Neuropathol Exp Neurol. 2007;66:660–72. doi: 10.1097/nen.0b013e3180986247. [DOI] [PubMed] [Google Scholar]
- 28.Goodman LE, DW D. Nonhereditary diffuse leukoencephalopathy with spheroids presenting as early-onset rapidly progressive dementia. J Neuropathol Exp Neurol. 1995;54:471. [Google Scholar]
- 29.Mayer B, Oelschlaeger C, Keyvani K, Niederstadt T. Two cases of LENAS: diagnosis by MRI and biopsy. J Neurol. 2007;254:1453–4. doi: 10.1007/s00415-007-0541-8. [DOI] [PubMed] [Google Scholar]
- 30.Ropper AH, Adams RD, Victor M, Samuels MA. Adams and Victor’s principles of neurology. 9. New York: McGraw-Hill Medical; 2009. [Google Scholar]
- 31.Sundal C, Wider C, Van Gerpen JA, Lash J, Garbern JY, Schweitzer KJ, et al. MRI in HDLS shows a unique mechanism of neuroaxonal degeneration. American Neurological Association. 2011 Sep;(S204):33. 2011 Book of Abstracts. [Google Scholar]
- 32.Bonilha L, de Vries PM, Hurd MW, Rorden C, Morgan PS, Besenski N, et al. Disrupted thalamic prefrontal pathways in patients with idiopathic dystonia. Parkinsonism & related disorders. 2009;15:64–7. doi: 10.1016/j.parkreldis.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Whitwell JL, Avula R, Master A, Vemuri P, Senjem ML, Jones DT, et al. Disrupted thalamocortical connectivity in PSP: A resting-state fMRI, DTI, and VBM study. Parkinsonism & related disorders. 2011;17:599–605. doi: 10.1016/j.parkreldis.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Laat KF, Tuladhar AM, van Norden AG, Norris DG, Zwiers MP, de Leeuw FE. Loss of white matter integrity is associated with gait disorders in cerebral small vessel disease. Brain. 2011;134:73–83. doi: 10.1093/brain/awq343. [DOI] [PubMed] [Google Scholar]
- 35.Bohnen NI, Muller ML, Zarzhevsky N, Koeppe RA, Bogan CW, Kilbourn MR, et al. Leucoaraiosis, nigrostriatal denervation and motor symptoms in Parkinson’s disease. Brain. 2011;134:2358–65. doi: 10.1093/brain/awr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary Parkinsonisms. Mov Disord. 2011;26:1083–95. doi: 10.1002/mds.23713. [DOI] [PubMed] [Google Scholar]
- 37.Bohnen NI, Albin RL. White matter lesions in Parkinson disease. Nat Rev Neurol. 2011;7:229–36. doi: 10.1038/nrneurol.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nutt JG, Bloem BR, Giladi N, Hallett M, Horak FB, Nieuwboer A. Freezing of gait: moving forward on a mysterious clinical phenomenon. Lancet neurology. 2011;10:734–44. doi: 10.1016/S1474-4422(11)70143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snijders AH, Leunissen I, Bakker M, Overeem S, Helmich RC, Bloem BR, et al. Gait-related cerebral alterations in patients with Parkinson’s disease with freezing of gait. Brain. 2011;134:59–72. doi: 10.1093/brain/awq324. [DOI] [PubMed] [Google Scholar]
- 40.Nachev P, Kennard C, Husain M. Functional role of the supplementary and pre-supplementary motor areas. Nat Rev Neurosci. 2008;9:856–69. doi: 10.1038/nrn2478. [DOI] [PubMed] [Google Scholar]
- 41.Preziosa P, Rocca MA, Mesaros S, Pagani E, Stosic-Opincal T, Kacar K, et al. Intrinsic damage to the major white matter tracts in patients with different clinical phenotypes of multiple sclerosis: a voxelwise diffusion-tensor MR study. Radiology. 2011;260:541–50. doi: 10.1148/radiol.11110315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.