

Abstract
Cardiorespiratory complications are frequent symptoms of Duchenne muscular dystrophy, a neuromuscular disorder caused by primary abnormalities in the dystrophin gene. Loss of cardiac dystrophin initially leads to changes in dystrophin-associated glycoproteins and subsequently triggers secondarily sarcolemmal disintegration, fibre necrosis, fibrosis, fatty tissue replacement, and interstitial inflammation. This results in progressive cardiac disease, which is the cause of death in a considerable number of patients afflicted with X-linked muscular dystrophy. In order to better define the molecular pathogenesis of this type of cardiomyopathy, several studies have applied mass spectrometry-based proteomics to determine proteome-wide alterations in dystrophinopathy-associated cardiomyopathy. Proteomic studies included both gel-based and label-free mass spectrometric surveys of dystrophin-deficient heart muscle from the established mdx animal model of dystrophinopathy. Comparative cardiac proteomics revealed novel changes in proteins associated with mitochondrial energy metabolism, glycolysis, signaling, iron binding, antibody response, fibre contraction, basal lamina stabilisation, and cytoskeletal organisation. This review summarizes the importance of studying cardiomyopathy within the field of muscular dystrophy research, outlines key features of the mdx heart and its suitability as a model system for studying cardiac pathogenesis, and discusses the impact of recent proteomic findings for exploring molecular and cellular aspects of cardiac abnormalities in inherited muscular dystrophies.
1. Introduction
Primary genetic abnormalities in the dystrophin gene result in the early-onset and debilitating muscle wasting disease Duchenne muscular dystrophy or the delayed-onset and milder disorder Becker muscular dystrophy [1–3]. In addition, mutations in cardiac dystrophin are linked to X-linked dilated cardiomyopathy in teenage men [4–6]. A variety of primary or secondary abnormalities in dystrophin-associated proteins are involved in several forms of limb-girdle muscular dystrophy, congenital muscular dystrophy, and dystroglycanopathy [7–9]. The Duchenne type of muscular dystrophy is the most frequently inherited neuromuscular disorder of childhood [10]. It occurs in approximately 1 in 3,500 live born males with substantial regional and national differences in disease frequency [11–13]. Early symptoms of muscular weakness are usually present before 5 years of age and drastically increased levels of serum creatine kinase, pyruvate kinase, and carbonic anhydrase isoform CA3 are characteristic for this type of inherited muscle disease [14–16]. The highly progressive nature of symmetrical muscle wasting often causes a loss of unassisted ambulation around 12 years of age.
Muscle biopsies show an abnormal variation in fibre diameter, large numbers of fibres with central nucleation, necrosis, and a certain degree of regenerating fibres, as well as a progressive increase in fat and connective tissue [10, 20, 21]. In muscle biopsy specimens from Duchenne patients, dystrophin isoform Dp427 is completely or almost completely absent from contractile fibres [22]. In some cases, rare reverting mutants may account for a small percentage of dystrophin-positive muscle fibres [23]. Besides effects on skeletal muscle integrity, abnormalities in dystrophin are also linked to nonprogressive forms of mental retardation [24, 25], scoliosis [26, 27], impaired respiratory function [28, 29], and cardiomyopathic complications [30, 31]. The fact that respiratory care of Duchenne patients has greatly improved over the years gives the treatment of dystrophinopathy-associated cardiomyopathic side effects a more prominent role in the overall therapy of Duchenne muscular dystrophy [32–34].
This review briefly outlines the pathophysiological significance of cardiomyopathic complications in dystrophinopathies and then focuses on the scientific impact of recent mass spectrometry-based studies of cardiac abnormalities in X-linked muscular dystrophy. Below sections summarize the clinical cardiac symptoms of dystrophinopathy and the pathoanatomical, pathophysiological, and pathobiochemical aspects of the mdx mouse heart model of Duchenne muscular dystrophy. Following a brief introduction into the principles of cardiac proteomics as a major biomarker discovery tool for improving our general understanding of cardiac disease mechanisms, recent findings from gel-based proteomic analyses of dystrophin-deficient cardiac tissue and label-free mass spectrometric studies of the aging mdx heart are discussed. The considerable influence of cardiac proteomics on the field of muscular dystrophy research and the usefulness of newly discovered proteomic biomarkers for improving diagnostic procedures, prognosis of cardiomyopathic complications in dystrophinopathies, and the evaluation of novel pharmacological or cell-based treatment strategies is examined.
2. Cardiac Dystrophin-Glycoprotein Complex
For a full comprehension of the molecular and cellular complexity of dystrophinopathy, it is important to point out that dystrophin does not exist in isolation within the subsarcolemmal membrane cytoskeleton. Although its overall protein structure and sequence similarity to members of the spectrin-like superfamily of proteins suggest that it possibly forms an intertwined lattice of dystrophin molecules underneath the sarcolemma [35], the linkage to nondystrophin molecules appears to be absolutely vital for sarcolemmal integrity and proper muscle functioning [36–38]. It is well established that the full-length protein product of the dystrophin gene with an apparent molecular mass of 427 kDa forms a supramolecular protein complex at the plasmalemma of both skeletal and cardiac muscle fibres. The core element of the dystrophin-glycoprotein complex consists of the integral glycoprotein β-dystroglycan of 43 kDa that directly interacts on the one hand with the actin-binding protein dystrophin in the subsarcolemmal domain and on the other hand with the extracellular laminin-receptor α-dystroglycan [39]. This large assembly of surface proteins forms a stabilizing linkage between the basal lamina on the outside of muscle fibres and the actin membrane cytoskeleton in the inside of contractile cells [40]. In addition to the core α/β-dystroglycan complex, a large number of additional dystrophin-associated proteins exist, including sarcoglycans, sarcospan, dystrobrevins, and syntrophins [41–44].
Differences exist between the dystrophin-associated glycoprotein complex from skeletal muscle and heart with respect to subcellular localization and protein composition. While the muscle complex is highly enriched in the sarcolemma [45] and at the neuromuscular junction [46], in coexistence with the utrophin-glycoprotein complex [47], the cardiac dystrophin complex is also present in the transverse tubular system [48, 49]. The cardiac dystrophin-glycoprotein complex partially associates with costameric vinculin, suggesting a mechanical role in the maintenance of surface membrane integrity and membrane domain organization [50, 51]. Of note, the recent proteomic analysis of the cardiac dystrophin complex suggests a different range of indirectly associated proteins as compared to skeletal muscle fibres. The cardiac complex appears to lack an interaction with the signaling enzyme nNOS, has a differential composition of syntrophins and dystrobrevins, and displays additional binding partners, including Cavin-1, Ahnak-1, Cypher, and Cryab [52].
3. Dystrophinopathy-Associated Cardiomyopathy
Although dystrophinopathies are primarily categorised as disorders of the neuromuscular system [10], heart disease also plays a crucial role in the etiology of X-linked muscular dystrophy [53]. Almost all patients afflicted with Duchenne muscular dystrophy show clinical cardiac symptoms, especially during the second decade of life [54]. These cardiac abnormalities may include arrhythmias, cardiomyopathy, and regional wall abnormalities [55–59]. A gradual replacement of contractile cardiac fibres by noncontracting cell populations, such as connective and fatty tissue, causes a critical loss of cellular function in the heart of Duchenne patients [55]. The highly progressive decline in the cardiomyocyte population is probably closely connected to the limited regenerative capacity of dystrophin-deficient heart fibres. In contrast to dystrophic skeletal muscles, the heart does not undergo extensive cycles of fibre degeneration and regeneration in dystrophinopathy. In a large number of Duchenne cases, serious cardiac complications result in death [54], warranting special attention to the pathophysiological role of cardiac dystrophin and its associated glycoprotein complex. The primary loss of cardiac dystrophin results initially in changes in dystrophin-associated glycoproteins which in turn triggers a plethora of secondary cellular abnormalities, including sarcolemmal disintegration, necrosis, fibrosis, fatty tissue replacement, and interstitial inflammation. Cellular degeneration leads to progressive cardiac disease and thus fatal complications in Duchenne muscular dystrophy [60].
4. The Cardiac mdx Model of Dystrophinopathy
The pathological status of the mdx mouse model of Duchenne muscular dystrophy is based on a point mutation in exon 23 of the dystrophin gene, resulting in a truncated protein product that is quickly degraded in dystrophic fibres [61]. Interestingly, different types of muscle exhibit greatly varying degrees of tissue degeneration. While laryngeal, extraocular, and interosseus muscles show a relatively mild phenotype [62–64] and leg muscles such as soleus, gastrocnemius, extensor digitalis longus, or tibialis anterior are moderately weakened by segmental necrosis [65–67], the diaphragm represents the most severely disturbed skeletal muscle type [68, 69] in the mdx mouse. Besides the skeletal musculature, the mdx heart is also affected by a large number of cellular, physiological, and biochemical abnormalities, as recently discussed in several extensive reviews on the cardiac phenotype of dystrophinopathy [70–72]. Thus, if one takes into account the biological limitations of genetic mouse models as surrogates for human disorders, the mdx mouse can be employed as an excellent model system to study basic pathophysiological mechanisms of muscular dystrophy [73].
The dystrophin-deficient heart from mdx mice clearly exhibits abnormal histological features, including necrosis, fibrosis, and inflammation [74]. On the subcellular level, a considerable disorganization of the cardiac membrane surface and disruption of the transverse tubular network were revealed by scanning ion conductance microscopy [75]. Signs of overt cardiomyopathy are more pronounced in aged mdx mice as compared to milder cardiac alterations in young animals [76, 77]. Aged mdx mice showed a widespread and patchy increase in ventricular wall fibrosis [78], whereby the basal region exhibited a greater degree of fibrotic changes than the apex of the dystrophic heart [79]. The onset of fibrosis in the mdx heart was found to be associated with an increased expression of collagen and the connective tissue growth factor CTGF [80]. At a later stage of fibrosis, a drastic increase in connective tissue volume was accompanied by the activation of key profibrotic genes, including the heart-specific induction of the Nox4 gene [81]. Coronary endothelial cells are implicated in mediating cardiac fibrosis via transmural TGF-β signaling mechanisms [82]. Interestingly, physical exercise was shown to accelerate the cardiomyopathic process [83, 84]. Exercised mdx hearts were characterized by an increase in inflammatory cell infiltration, elevated levels of interstitial fibrosis, and a higher degree of adipose tissue deposition [83]. In the absence of the membrane cytoskeletal protein dystrophin, cardiomyocyte injury was increased considerably by workload-induced cell damage or an acute elevation of mechanical stress [85].
Histopathological features of the mdx heart correlate well with the assessment of functional deficits in cardiac output. The dystrophin-deficient heart showed an abnormal electrocardiogram [86] with significant tachycardia and decreased heart rate variability [87]. In vivo cardiac MRI studies demonstrated larger right ventricular end-diastolic and end-systolic volumes and lower right ventricular ejection fractions in mdx mice [88]. High-resolution doppler echocardiography confirmed that the extent of changes in posterior wall thickness and left ventricular mass are dependent on the age of mdx mice [89]. The contractile properties of the mdx heart are markedly altered with a reduced force amplitude [90] and considerably prolonged half-relaxation time [91]. The pathophysiological basis of these functional abnormalities is associated with hypersensitive excitation-contraction coupling [92], increased ion fluxes through the fragile plasmalemma [93–95], elevated Ca2+-levels in the cytosol [96, 97], impaired cytosolic and luminal Ca2+-handling [98, 99], enhanced intracellular Ca2+-responses to mechanical challenges [97], an altered mitochondrial redox state, and an increased production of reactive oxygen species [97, 100]. Deficiency in cardiac dystrophin is postulated to cause plasmalemmal fragility, which in turn alters ion fluxes and signaling events at the surface membrane ultimately leading to a pathophysiologically elevated cytosolic Ca2+-concentration [101]. The Ca2+-dependent activation of proteolytic processes and mitochondrial dysfunction probably act as the starting point for the formation of fibrotic patches in the dystrophic heart, as recently reviewed by Shirokova and Niggli [72].
Besides dysregulation of excitation-contraction coupling and Ca2+-handling due to membrane perturbation, metabolic disturbances may predispose the Dp427-deficient heart to contractile dysfunction [102]. Pathobiochemically, the primary loss in cardiac dystrophin isoform Dp427 appears to affect the dystrophin-associated glycoprotein complex in a less severe way as compared to skeletal muscle, possibly due to the upregulation of the dystrophin homologue utrophin [47]. In normal heart, the cardiac-specific dystrophin-glycoprotein complex localizes to the sarcolemma and transverse tubules [48, 49, 103] and probably functions as a membrane-stabilizing linker during excitation-contraction-relaxation cycles in a similar way as the skeletal muscle complex [50, 51], although differences in its composition suggest additional functions [52]. In dystrophy-related cardiomyopathy, both the abundance and glycosylation of α-dystroglycan were shown to be altered in dystrophin-deficient heart muscle [104, 105]. In order to study global changes downstream from the primary defect in dystrophin and secondary alterations in the dystroglycan complex, mass spectrometry-based proteomics was employed for the large-scale analysis of the dystrophic heart.
5. Cardiac Proteomics
Over the last few years, mass-spectrometry-based proteomics has been widely applied to studying cardiac tissues in health and disease. A variety of extensive reviews have been published that summarize and discuss the underlying objectives of cardioproteomic strategies [106, 107], the usefulness of proteomic biomarker research for improving diagnostic, prognostic and therapeutic approaches [108–110], the application of clinical proteomics in the study of cardiovascular diseases [111–113], the evaluation of post-translational modifications in cardiac proteins [114, 115], and technological advances in the field of mass spectrometry and cardiac proteomics [106, 116]. Mass spectrometry-based proteomics was instrumental in the cataloging of the protein constellation of normal heart tissue [117–121], the global assessment of changes in the cardiac proteome during development [122], the determination of functional adaptations following exercise [123–125], and the establishment of protein changes during the natural aging process [126–130], as well as the biomedical analysis of a variety of heart diseases in patients or animal models of heart disease, including dilated cardiomyopathy, atrial fibrillation, the diabetic heart, and cardiac failure [131–136]. The total number of proteins belonging to cardiac tissues is not known, since no one proteomic method can completely separate and accurately identify all proteins within a complex tissue that exhibits a wide dynamic concentration range. Most likely, the cardiac proteome consists of several thousand different protein species with a wide range of posttranslational modifications [117–121]. For a comprehensive analysis of changes in cardiac proteins with greatly differing physicochemical properties with respect to size, charge, and hydrophobicity, a combination of various proteomic techniques is often advantageous.
Diverse proteomic approaches and methods have been applied in global studies of the heart. For the initial large-scale separation of distinct protein populations, both gel-based and/or liquid chromatography-focused techniques have been employed. Labeling methodology or label-free applications were routinely used for the high-throughput identification of cardiac proteins. Proteomic methods that involve gel electrophoresis are highly suitable for the analysis of contractile proteins, regulatory proteins, metabolic enzymes, metabolite transporters, and molecular chaperones [118]. Two-dimensional gel electrophoresis can conveniently separate cardiac proteins in the range of approximately 10 kDa to 200 kDa and isoelectric points ranging from pH3 to pH11 [117, 118, 120]. Combinations of isoelectric focusing with narrow- or wide-range immobilised pH gradients, native gel electrophoresis, nonreducing gel electrophoresis, and reducing gel electrophoresis can be used for various two-dimensional applications [137–140]. While post-electrophoretic staining with protein dyes is relatively cheap and fast, the differential pre-electrophoretic labeling with fluorescent CyDyes usually results in a larger number of identified cardiac proteins and greatly reduces gel-to-gel variations [141, 142]. One-dimensional gradient gels, in combination with on-membrane digestion protocols, can also cover the separation of high-molecular-mass proteins following detergent solubilization [143]. However, low-abundance proteins, hydrophobic proteins, and components with extreme pI-values are difficult to study using routine gel electrophoretic methods [137, 140].
The usefulness of alternative gel-free proteomic labeling methods, such as iTRAQ (isobaric tags for relative and absolute quantitation) or SILAC (stable isotope labeling by amino acids in cell culture), which have been successfully applied to studying cardiac cells [144, 145], has been described in recent reviews [106, 107]. One of the most advanced proteomic approaches involves label-free mass spectrometry. The advantages of this method are that it (i) requires only very small amounts of protein samples, (ii) has broad applicability, (iii) detects a large range of cardiac protein species, and, most importantly, (iv) does not require protein labeling [146]. Thus, in order to overcome some of the problems associated with gel-based methods in cardiac proteomics, label-free mass spectrometry has recently been applied to investigate cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy [18].
Figure 1 gives an overview of the key methods employed in comparative cardioproteomic studies and illustrates typical findings from a gel-based analysis of the dystrophic heart proteome. Shown are two-dimensional gels representing the urea-soluble proteome from the young versus the aged mdx heart, post-electrophoretically labeled with the fluorescent dye RuBPs (ruthenium II tris bathophenantroline disulfonate) [147]. Fluorescent labeling with RuBPs dye is an excellent and cheap alternative to the more labor-intensive 2D-DIGE approach with its relatively expensive CyDyes [148]. The individual analytical steps performed to achieve the two-dimensional gel image depicted in Figure 1 have been previously described in detail by our laboratory [17].
Figure 1.
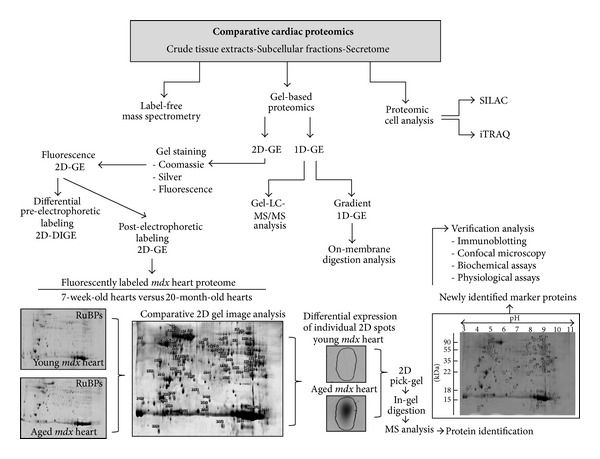
Overview of proteomic methods used in comparative studies of the heart. Shown is a flowchart of the various techniques used to identify changes in the cardiac proteome, including label-free mass spectrometry, gel-based methods (GE, gel electrophoresis), and cellular analyses (SILAC, stable isotope labeling by amino acids in cell culture; iTRAQ, isobaric tags for relative and absolute quantitation). To illustrate the typical work flow of a gel-based analysis of the dystrophic heart proteome, two-dimensional gels representing the urea-soluble proteome from the young versus the aged mdx heart are shown. The post-electrophoretic labeling of cardiac proteins with the fluorescent dye RuBPs (ruthenium II tris bathophenantroline disulfonate) was carried out by standard methodology [17].
6. Gel-Based Analysis of Cardiac Changes in Dystrophinopathy
Prior to the development of the proteomic concept and the streamlining of established biochemical techniques for the large-scale analysis of entire protein populations, protein biochemical studies of the dystrophic mdx heart have mostly focused on individual proteins, protein complexes, specific pathways, or signalling cascades. Such focused protein chemical approaches, also highly informative about specific aspects of a disease process, inevitably generate biomedical data sets with limited scope. Hence, in order to better complement findings from detailed physiological, cell biological, and histological studies of cardiomyopathic changes, mass spectrometry-based proteomics was used to establish proteome-wide alterations in mdx preparations. The parallel analysis of hundreds of cardiac proteins promised to swiftly determine their molecular fate in dystrophin-deficient heart tissues and thus decisively improve our understanding of the molecular pathogenesis of dystrophy-associated cardiomyopathy. Initially, comparative proteomic studies used gel-based surveys of the mdx heart muscle and revealed novel changes in proteins mostly associated with mitochondrial energy metabolism, the contractile apparatus, the cytoskeleton, and the cellular stress response [141, 142]. Both studies used fluorescence two-dimensional difference in-gel electrophoresis (2D-DIGE) for the analysis of the dystrophic heart.
The 2D-DIGE technique is an extremely powerful preelectrophoretic labeling approach that can swiftly determine potential changes in the concentration of thousands of proteins in large analytical gel systems [149–151] and has proven to be an excellent biomarker discovery tool for comparative studies of contractile fibres [152]. The 2D-DIGE method has been widely applied to studying various subtypes of muscle in animal models of Duchenne muscular dystrophy [153–158]. It is one of the key techniques in comparative gel-based proteomics and is employed with fluorescent 2-CyDye [159] or 3-CyDye [160] labeling systems for the differential tagging of proteins from dissimilar mixtures prior to two-dimensional gel electrophoresis [149]. The optimized analysis of 2D-DIGE images with advanced 2D software analysis tools [161–163] can highly accurately quantitate multiple protein samples on the same two-dimensional gel [164, 165]. Importantly, the completion of reverse DIGE labeling controls is not usually necessary, since selective labeling artifacts were shown not to play a significant role in the analysis of soluble proteins [166], which considerably lowers the overall time and costs involved in large-scale 2D-DIGE studies. The analysis of the murine heart proteome with the 2-CyDye labeling system and the combination of pH 4–7 and pH 6–11 gels resulted in the identification of 2,509 distinct protein spots [142], illustrating the powerful separation and labeling capabilities of the 2D-DIGE technique within the field of gel-based comparative cardiac proteomics [106].
The proteomic profiling of 1-to-9-month-old mdx heart extracts by Gulston et al. [141] revealed differential expression patterns for ATP synthase, glyceraldehyde-3-phosphate dehydrogenase, serine proteinase inhibitor, trifunctional enzyme, and hemoglobin. Additional metabolomic analyses suggest metabolic disturbances in the dystrophic heart, agreeing with the altered concentration of key mitochondrial and glycolytic enzymes [141]. Since abnormal heart function was shown to be prominent at 9 months of age [81], a detailed 2D-DIGE analysis of potential changes in the concentration of distinct proteins was carried out with cardiac proteins at this age of mdx mice [142]. Electrospray ionization MS/MS analysis identified 26 proteins with a decreased abundance, including various myosin light chains, tropomyosin, actin, adenylate kinase, creatine kinase, vimentin, fatty acid binding protein isoform FABP3, isocitrate dehydrogenase, NADH dehydrogenase, myozenin, porin, and peroxiredoxin. In contrast, 3 heart-associated proteins were found to be significantly increased, including lamin and nucleoside diphosphate kinase. An independent verification of the DIGE analysis was performed by immunoblotting and confocal microscopy of a select group of cardiac proteins. The comparative immunoblot analysis showed a drastic decrease in the enzyme adenylate kinase, the fatty acid binding protein FABP3, isocitrate dehydrogenase, and mitochondrial porin in 9-month-old mdx heart tissue [142]. The decreased abundance of the AK1 isoform of adenylate kinase did not correspond with a previous combined metabolomic and proteomic analysis of the mdx heart [141] but agrees with several comprehensive proteomic surveys of dystrophin-deficient muscle preparations [152, 153, 167–169]. Since the proteomic result was independently confirmed by immunoblotting, it appears that cardiac nucleotide metabolism that involves adenylate kinase and creatine kinase is perturbed in the dystrophin-deficient heart.
Mitochondrial dysfunction and accompanied oxidative stress have been linked to various cardiac pathologies, including cardiomyopathy, congestive heart failure, and ischaemia reperfusion injury [170], conveying considerable importance to the results from the proteomic profiling of the mdx heart with respect to explaining abnormal mitochondrial function in dystrophy-associated cardiomyopathy [97]. The mitochondrial proteome from heart tissue has been well catalogued and studied using proteomic techniques, focusing especially on the role of mitochondrial proteins in bioenergetics, pathology, and the natural aging process [171–173]. The proteomic finding that a variety of mitochondrial proteins exhibit an altered concentration in the mdx heart [141, 142] necessitated microscopical studies in order to evaluate whether these protein alterations were due to a reduced number of organelles in cardiomyopathic tissue or based on internal changes within the mitochondrial proteome. A microscopical survey using the fluorescent labeling of mitochondria with the MitoTracker dye CMXRos, staining of nuclei with the DNA binding dye DAPI, and immunofluorescence staining of cardiac marker proteins revealed no statistically significant differences in mitochondrial content, the number of nuclei, and the subcellular localization of key mitochondrial enzymes between normal and dystrophic heart [142]. Thus, the overall isoform complement of mitochondrial enzymes is not majorly altered, but certain subspecies of distinct cardiac protein isoforms are changed due to the deficiency in dystrophin. Since cardiac mitochondria are the primary site for energy generation via oxidative phosphorylation, even subtle changes in the protein population responsible for oxidative phosphorylation complexes, the citric acid cycle, and metabolite transport can be assumed to have an extensive effect on the bioenergetic status of the mdx heart. Besides energy metabolism, cardiac mitochondria are also involved in calcium signaling, the regulation of apoptosis, cell cycle progression, and the production of heme and iron-sulfur clusters [170]. Therefore, alterations in the mitochondrial proteome may affect these crucial cellular functions and render the mdx heart more susceptible to damage pathways and ultimately to extensive fibrosis.
7. Label-Free MS Analysis of Cardiac Changes in Dystrophinopathy
Based on the above outlined findings from gel-based proteomic analyses of the dystrophic heart, it was concluded that changes in proteins involved in fibre contraction, nucleotide metabolism, the cellular stress response, mitochondrial bioenergetics, and fatty acid transportation play a central role in the progressive loss of cardiac function in the mdx model of Duchenne muscular dystrophy [141, 142]. However, since two-dimensional gel electrophoresis does not properly display very large proteins, these analyses did not produce any information on a key member of the wider network of the cardiac dystrophin-glycoprotein complex, namely, the basal lamina protein laminin. In skeletal muscle, the concentration of laminin is unexpectedly not altered in dystrophin-deficient fibres [40, 152, 174], so it was of considerable interest to determine its molecular fate in cardiac tissue and evaluate whether differences exist in the extracellular matrix of both types of contractile mdx tissues. Label-free mass spectrometry suggested itself as an ideal analytical way to study high-molecular-mass cardiac proteins and was therefore applied to determine global downstream effects due to dystrophin deficiency within the cardiac system.
Prior to the proteomic profiling of age-related changes in the mdx heart, a label-free LC-MS/MS analysis of 7-week-old dystrophic versus age-matched normal mice was carried out to initially establish potential differences between unaffected and dystrophic heart tissue at an age prior to the occurrence of extensive cardiomyopathic changes [18]. Comparative proteomics established moderate changes in 20 cardiac proteins, which clearly agrees with the relatively mild pathological phenotype in young mdx mice. A differential expression pattern was shown for various mitochondrial enzymes, including succinyl-CoA ligase, methylmalonate-semialdehyde dehydrogenase, 3-hydroxyacyl-CoA dehydrogenase, 2,4-dienoyl-CoA reductase, 3-ketoacyl-CoA thiolase, glutamate dehydrogenase, succinyl-CoA: 3-ketoacid-coenzyme A transferase, 2-oxoglutarate dehydrogenase, and isocitrate dehydrogenase.
The detailed proteomic profiling of the aging process in 7-week-old to 20-month-old mdx hearts by label-free mass spectrometry demonstrated that aged dystrophic hearts exhibit a generally perturbed expression pattern of key cardiac proteins involved in the stabilization of the basal lamina, the organization of the cytoskeletal network, cellular iron homeostasis, antibody response, fibre contraction, and energy metabolism [18]. Age-related changes were found in 67 cardiac protein species, of which 39 proteins were shown to be increased and 28 proteins were identified as being decreased in their concentration. Of note, the most drastic alterations were increases in transferrin and various immunoglobulin chains and decreases in laminin, nidogen, and annexin. Thus, the collapse of the dystrophin network in the heart and resulting sarcolemmal fragility appears to trigger serious secondary alterations, including the disintegration of the basal lamina structure and cytoskeletal network, an increased level of antibodies in a potential autoimmune reaction of the degenerating heart, and the compensatory binding of excess iron in dystrophinopathy-related cardiomyopathy. Figure 2 shows the bioinformatic STRING analysis of the proteomic data from the recent label-free mass spectrometric study of the aging mdx heart. For the evaluation of protein-protein interactions of the mass spectrometrically identified proteins with a changed abundance in the dystrophic mdx heart, bioinformatic analysis was carried out with the publically available STRING (http://string-db.org/; version 9.1) database of known and predicted protein interactions that include direct physical and indirect functional protein associations [19]. The interaction map illustrates the enormous complexity of potential protein interactions, especially with respect to mitochondrial components.
Figure 2.
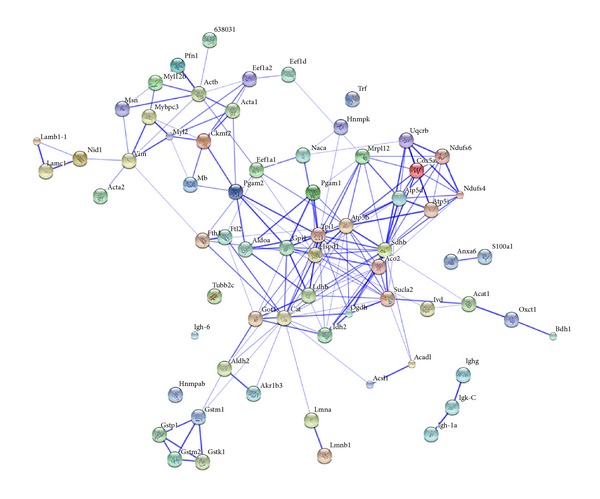
Bioinformatic STRING analysis of the proteomic data from the label-free mass spectrometric study of the aged mdx heart. For the evaluation of protein-protein interactions of the mass spectrometrically identified proteins with a changed abundance in the dystrophic mdx heart [18], bioinformatic analysis was carried out with the publically available STRING (http:// http://string-db.org/; version 9.1) database of known and predicted protein interactions that include direct physical and indirect functional protein associations [19]. The interaction map of cardiac proteins with a changed abundance in the dystrophic mdx heart illustrates the enormous complexity of potential protein interactions, especially with respect to mitochondrial components.
Functional analyses, confocal microscopy, and/or immunoblotting are routinely used to independently verify proteomic data. A comprehensive immunoblot analysis of young and senescent wild type versus mdx hearts has verified key proteomic results and clarified differences in protein changes due to natural aging versus muscular dystrophy [18]. While antibody decoration demonstrated that the concentration of laminin, nidogen, and annexin increased during the natural aging process, a drastic decrease in the expression levels of these 3 cardiac proteins was observed in the aged dystrophin-deficient heart. Both, the proteomic data and their confirmation by immunoblotting strongly suggest that the maintenance and architecture of the extracellular matrix, basement membrane, and cytoskeletal network are severely impaired in the aged mdx heart. The loss of cardiac dystrophin seems to indirectly affect the essential laminin component of the basement membrane [175] via alterations in the dystroglycan subcomplex. The reduced levels of laminin in turn appear to lower the concentration of nidogen, a sulfated glycoprotein present in many specialized basement membranes [176] and annexin, which is crucial for the maintenance of the cytoskeleton and the extracellular matrix, as well as cardiac Ca2+-homeostasis [177]. This loss in surface integrity of the dystrophin-deficient heart could be one of the major triggering factors that induce progressive fibrosis in dystrophinopathy-associated cardiomyopathy.
The main findings from recent proteomic studies that have focused on the cardiac dystrophin-glycoprotein complex and dystrophin-deficient mdx heart tissues are listed in Table 1. The overall emphasis of the individual studies, the main technological approach, and major findings with respect to novel proteomic biomarker candidates of dystrophinopathy-associated cardiomyopathy are displayed. In addition, the flowchart in Figure 3 summarizes the variety of biochemical, physiological, and cellular abnormalities that result in cardiac fibrosis and progressive functional decline of the cardiovascular system.
Table 1.
Proteomic profiling of the dystrophin-deficient mdx heart.
| Proteomic study | Methods | Major findings | References |
|---|---|---|---|
| Proteomic analysis of the cardiac-specific dystrophin complex | IP-based copurification, LC-MS/MS, IB, CM | Confirmation of main dystrophin-associated proteins: dystroglycans, sarcoglycans, dystrobrevins, sarcospan, and syntrophins; plus identification of novel dystrophin-associated proteins: Cavin-1, Ahnak-1, Cypher, and Cryab | Johnson et al., 2012 [52] |
| Comparative proteomic study of 1-month to 9-month-old mdx hearts versus age-matched normal hearts | 2D-DIGE, LC-MS/MS | Differential expression of ATP synthase, serine proteinase inhibitor, glyceraldehyde-3-phosphate dehydrogenase, trifunctional enzyme, and hemoglobin | Gulston et al., 2008 [141] |
| Comparative proteomic analysis of 9-month-old mdx hearts versus age-matched normal hearts | 2D-DIGE, LC-MS/MS, IB, CM | Increased levels of lamin and nucleoside diphosphate kinase; drastic decrease in myosin light chains, tropomyosin, actin, adenylate kinase, creatine kinase, vimentin, fatty acid binding protein FABP3, isocitrate dehydrogenase, NADH dehydrogenase, myozenin, porin, and peroxiredoxin. | Lewis et al., 2010 [142] |
| Comparative proteomic analysis of 7-week-old mdx hearts versus age-matched normal hearts | Label-free MS analysis, IB | Moderate changes in young mdx hearts: actin, biglycan, troponin, protein disulphide isomerase, succinyl-CoA ligase | Holland et al., 2013 [18] |
| Proteomic analysis of the aging process in 7-week to 20-month-old mdx hearts | Label-free MS analysis, IB | Severe changes in aged mdx hearts: drastic reduction in laminin, nidogen, annexin, vimentin, ATP synthase, cytochromes, NADH dehydrogenase; increases in various IgG molecules, hydroxybutyrate dehydrogenase, ferritin, transferrin, catalase, glutathione transferase | Holland et al., 2013 [18] |
Listed are major findings from recent proteomic studies that have focused on the cardiac dystrophin-glycoprotein complex and dystrophin-deficient mdx heart tissues. Abbreviations used: 2D-DIGE: two-dimensional difference in-gel electrophoresis; CM: confocal microscopy; IB: immunoblotting; IP: immunoprecipitation; LC: liquid chromatography; MS: mass spectrometry.
Figure 3.
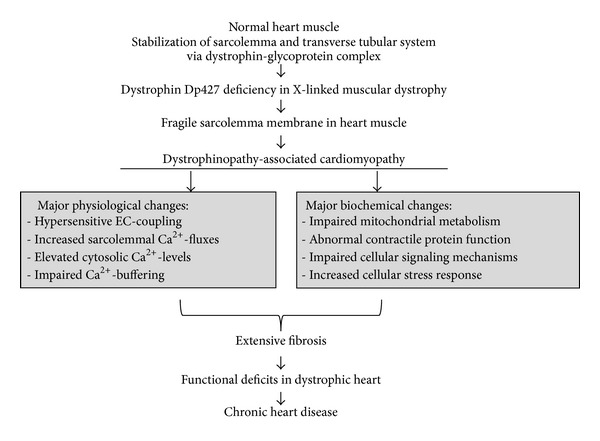
Molecular pathogenesis of muscular dystrophy-associated cardiomyopathy. Shown is a flowchart of major pathophysiological and pathobiochemical changes that render the dystrophin-deficient heart more susceptible to fibre degeneration and fibrosis, which eventually triggers chronic heart disease in dystrophinopathy. Key changes in the physiological regulation of the dystrophic heart are associated with abnormal calcium handling and hypersensitive excitation-contraction (EC) coupling.
8. Conclusions
Heart disease is a common clinical manifestation of X-linked muscular dystrophies. Hence, future approaches to treating the overall medical complications present in dystrophinopathy have to take into account the remodeling of incapacitating cardiac fibrosis and resulting functional abnormalities in the dystrophin-deficient heart. As recently reported by Wasala et al. [178], the exclusive correction of abnormalities in the dystrophic skeletal musculature unfortunately does not modulate cardiac pathogenesis in the aged mdx model of Duchenne cardiomyopathy. To address this biomedical issue and the fact that a high frequency of cardiomyopathy exists in teenage patients suffering from inherited X-linked muscular dystrophy, a large and diverse number of novel therapeutic approaches are currently tested to specifically address cardiac symptoms in dystrophinopathy. This includes various forms of gene therapy [179–182], exon-skipping therapy [183], and a large number of experimental drug treatments [184–192]. This in turn makes the availability of both a substantial array of reliable proteomic biomarkers and established animal models of muscular dystrophy an important prerequisite for the high-throughput and large-scale testing of new therapeutic options. In order to evaluate the long-term usefulness and potential cytotoxic side effects of gene therapy, exon-skipping, stem cell therapy, and/or pharmacological interventions, simple, cost-effective, and reliable assays with significant protein biomarkers are needed [193].
As outlined in this review, mass spectrometry-based proteomic profiling studies have clearly established the mdx mouse as a suitable animal model for exploring molecular and cellular aspects of cardiac pathogenesis and the aged mdx heart as a highly appropriate organ system for studying the progressive aspects of muscular dystrophy-associated cardiomyopathy. Most importantly, the application of comparative proteomics has identified a large number of new changes in cardiac proteins associated with cellular signaling mechanisms, mitochondrial energy metabolism, glycolysis, antibody response, iron binding, the contraction-relaxation cycle, basal lamina stabilisation, and cytoskeletal organisation. These novel protein marker candidates can now be used for the systematic screening of the cardiac mdx heart following experimental therapeutic interventions. The combined utilization of both label-free mass spectrometry and gel-based techniques promises the most comprehensive coverage of the cardiac proteome, including highly hydrophobic components, low-abundance elements, proteins with extreme isoelectric points, and proteins with extensive posttranslational modifications.
Acknowledgments
Research was supported by project grants from the Higher Education Authority (BioAT Programme of PRTLI Cycle 5) and Muscular Dystrophy Ireland. The authors would like to thank Professor Dieter Swandulla and Margit Zweyer (University of Bonn, Germany) for their continued support of our muscular dystrophy research initiative.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Koening M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 3.Koening M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus becker muscular dystrophy: correlation of severity with type of deletion. The American Journal of Human Genetics. 1989;45(4):498–506. [PMC free article] [PubMed] [Google Scholar]
- 4.Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy: molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87(6):1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz-Lopez R, Li H, Su J, Goytia V, Towbin JA. Evidence for a dystrophin missense mutation as a cause of X-linked dilated cardiomyopathy. Circulation. 1997;95(10):2434–2440. doi: 10.1161/01.cir.95.10.2434. [DOI] [PubMed] [Google Scholar]
- 6.Diegoli M, Grasso M, Favalli V, et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. Journal of the American College of Cardiology. 2011;58(9):925–934. doi: 10.1016/j.jacc.2011.01.072. [DOI] [PubMed] [Google Scholar]
- 7.Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nature Genetics. 1995;11(2):216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- 8.Nigro V, de Sa Moreira E, Piluso G, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nature Genetics. 1996;14(2):195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- 9.Godfrey C, Clement E, Mein R, et al. Refining genotype-phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130(10):2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 10.Emery AEH. The muscular dystrophies. The Lancet. 2002;359(9307):687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 11.Hauser E, Toifl K, Mad A, Bittner R. The incidence of Duchenne muscular dystrophy in eastern Austria. The controversy regarding CK screening. Wiener Klinische Wochenschrift. 1993;105(15):433–436. [PubMed] [Google Scholar]
- 12.Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle and Nerve. 2013;48(1):21–26. doi: 10.1002/mus.23810. [DOI] [PubMed] [Google Scholar]
- 13.Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK) European Journal of Human Genetics. 2013;21(10):1049–1053. doi: 10.1038/ejhg.2012.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zatz M, Rapaport D, Vainzof M, et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. Journal of the Neurological Sciences. 1991;102(2):190–196. doi: 10.1016/0022-510x(91)90068-i. [DOI] [PubMed] [Google Scholar]
- 15.Ohta M, Itagaki Y, Itoh N, Hayashi K, Nishitani H, Ohta K. Carbonic anhydrase III in serum in muscular dystrophy and other neurological disorders: relationship with creatine kinase. Clinical Chemistry. 1991;37(1):36–39. [PubMed] [Google Scholar]
- 16.Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Annals of Neurology. 2012;71(3):304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 17.Carberry S, Ohlendieck K. Gel electrophoresis-based proteomics of senescent tissues. (Methods in Molecular Biology).Biological Aging. 2013;1048:229–246. doi: 10.1007/978-1-62703-556-9_17. [DOI] [PubMed] [Google Scholar]
- 18.Holland A, Dowling P, Zweyer M, et al. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics. 2013;13(15):2312–2323. doi: 10.1002/pmic.201200578. [DOI] [PubMed] [Google Scholar]
- 19.Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy—part 1: diagnosis, and pharmacological and psychosocial management. The Lancet Neurology. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 21.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy—part 2: implementation of multidisciplinary care. The Lancet Neurology. 2010;9(2):177–189. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. The New England Journal of Medicine. 1988;318(21):1363–1368. doi: 10.1056/NEJM198805263182104. [DOI] [PubMed] [Google Scholar]
- 23.Thanh LT, Man NT, Helliwell TR, Morris GE. Characterization of revertant muscle fibers in Duchenne muscular dystrophy, using exon-specific monoclonal antibodies against dystrophin. The American Journal of Human Genetics. 1995;56(3):725–731. [PMC free article] [PubMed] [Google Scholar]
- 24.Kozicka A, Prot J, Wasilewski R. Mental retardation in patients with Duchenne progressive muscular dystrophy. Journal of the Neurological Sciences. 1971;14(2):209–213. doi: 10.1016/0022-510x(71)90090-6. [DOI] [PubMed] [Google Scholar]
- 25.Pane M, Lombardo ME, Alfieri P, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. Journal of Pediatrics. 2012;161(4):705–709. doi: 10.1016/j.jpeds.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Kinali M, Messina S, Mercuri E, et al. Management of scoliosis in Duchenne muscular dystrophy: a large 10-year retrospective study. Developmental Medicine and Child Neurology. 2006;48(6):513–518. doi: 10.1017/S0012162206001083. [DOI] [PubMed] [Google Scholar]
- 27.Hsu JD, Quinlivan R. Scoliosis in Duchenne muscular dystrophy (DMD) Neuromuscular Disorders. 2013;23(8):611–617. doi: 10.1016/j.nmd.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Gayraud J, Ramonatxo M, Rivier F, Humberclaude V, Petrof B, Matecki S. Ventilatory parameters and maximal respiratory pressure changes with age in duchenne muscular dystrophy patients. Pediatric Pulmonology. 2010;45(6):552–559. doi: 10.1002/ppul.21204. [DOI] [PubMed] [Google Scholar]
- 29.Khirani S, Ramirez A, Aubertin G, et al. Respiratory muscle decline in duchenne muscular dystrophy. Pediatric Pulmonology. 2013 doi: 10.1002/ppul.22847. [DOI] [PubMed] [Google Scholar]
- 30.Connuck DM, Sleeper LA, Colan SD, et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the pediatric cardiomyopathy registry. The American Heart Journal. 2008;155(6):998–1005. doi: 10.1016/j.ahj.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romfh A, McNally EM. Cardiac assessment in duchenne and becker muscular dystrophies. Current Heart Failure Reports. 2010;7(4):212–218. doi: 10.1007/s11897-010-0028-2. [DOI] [PubMed] [Google Scholar]
- 32.Bach JR, Martinez D. Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respiratory Care. 2011;56(6):744–750. doi: 10.4187/respcare.00831. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa Y, Miura T, Ishikawa Y, et al. Duchenne muscular dystrophy: survival by cardio-respiratory interventions. Neuromuscular Disorders. 2011;21(1):47–51. doi: 10.1016/j.nmd.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myologica. 2012;31(2):121–125. [PMC free article] [PubMed] [Google Scholar]
- 35.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. The Journal of Biological Chemistry. 1990;265(8):4560–4566. [PubMed] [Google Scholar]
- 36.Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338(6212):259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. Journal of Biochemistry. 1990;108(5):748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 38.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345(6273):315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 39.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. Journal of Cell Biology. 1993;122(4):809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. European Journal of Cell Biology. 1996;69(1):1–10. [PubMed] [Google Scholar]
- 41.Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. Journal of Neuroscience. 1998;18(1):128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida M, Noguchi S, Wakabayashi E, et al. The fourth component of the sarcoglycan complex. FEBS Letters. 1997;403(2):143–148. doi: 10.1016/s0014-5793(97)00040-9. [DOI] [PubMed] [Google Scholar]
- 43.Peters MF, O’Brien KF, Sadoulet-Puccio HM, Kunkel LM, Adams ME, Froehner SC. β-dystrobrevin, a new member of the dystrophin family: identification, cloning, and protein associations. The Journal of Biological Chemistry. 1997;272(50):31561–31569. doi: 10.1074/jbc.272.50.31561. [DOI] [PubMed] [Google Scholar]
- 44.Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. The Journal of Biological Chemistry. 1997;272(50):31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- 45.Ohlendieck K, Ervasti JM, Snook JB, Campbell KP. Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. Journal of Cell Biology. 1991;112(1):135–148. doi: 10.1083/jcb.112.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohlendieck K, Ervasti JM, Matsumura K, Kahl SD, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7(3):499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- 47.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360(6404):588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 48.Klietsch R, Ervasti JM, Arnold W, Campbell KP, Jorgensen AO. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circulation Research. 1993;72(2):349–360. doi: 10.1161/01.res.72.2.349. [DOI] [PubMed] [Google Scholar]
- 49.Kaprielian RR, Stevenson S, Rothery SM, Cullen MJ, Severs NJ. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation. 2000;101(22):2586–2594. doi: 10.1161/01.cir.101.22.2586. [DOI] [PubMed] [Google Scholar]
- 50.Kaprielian RR, Severs NJ. Dystrophin and the cardiomyocyte membrane cytoskeleton in the healthy and failing heart. Heart Failure Reviews. 2000;5(3):221–238. doi: 10.1023/A:1009805419285. [DOI] [PubMed] [Google Scholar]
- 51.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circulation Research. 2004;94(8):1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 52.Johnson EK, Zhang L, Adams ME, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0043515.e43515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox GF, Kunkel LM. Dystrophies and heart disease. Current Opinion in Cardiology. 1997;12(3):329–343. [PubMed] [Google Scholar]
- 54.Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology. 2003;99(1):1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- 55.Frankel KA, Rosser RJ. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Human Pathology. 1976;7(4):375–386. doi: 10.1016/s0046-8177(76)80053-6. [DOI] [PubMed] [Google Scholar]
- 56.Miyoshi K. Echocardiographic evaluation of fibrous replacement in the myocardium of patients with Duchenne muscular dystrophy. The British Heart Journal. 1991;66(6):452–455. doi: 10.1136/hrt.66.6.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonald CM, Abresch RT, Carter GT, et al. Duchenne muscular dystrophy. The American Journal of Physical Medicine and Rehabilitation. 1995;74(5, supplement):S70–S92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 58.Lanza GA, Russo AD, Giglio V, et al. Impairment of cardiac autonomic function in patients with Duchenne muscular dystrophy: relationship to myocardial and respiratory function. The American Heart Journal. 2001;141(5):808–812. doi: 10.1067/mhj.2001.114804. [DOI] [PubMed] [Google Scholar]
- 59.Groh WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm. 2012;9(11):1890–1895. doi: 10.1016/j.hrthm.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 60.Muntoni F. Cardiomyopathy in muscular dystrophies. Current Opinion in Neurology. 2003;16(5):577–583. doi: 10.1097/01.wco.0000093100.34793.81. [DOI] [PubMed] [Google Scholar]
- 61.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 62.Dowling P, Lohan J, Ohlendieck K. Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of β-dystroglycan. European Journal of Cell Biology. 2003;82(5):222–230. doi: 10.1078/0171-9335-00315. [DOI] [PubMed] [Google Scholar]
- 63.Marques MJ, Ferretti R, Vomero VU, Minatel E, Neto HS. Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy. Muscle and Nerve. 2007;35(3):349–353. doi: 10.1002/mus.20697. [DOI] [PubMed] [Google Scholar]
- 64.Carberry S, Brinkmeier H, Zhang Y, Winkler CK, Ohlendieck K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscles from the mdx mouse model of Duchenne muscular dystrophy. International Journal of Molecular Medicine. 2013;32(3):544–556. doi: 10.3892/ijmm.2013.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torres LFB, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110(2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- 66.Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. Journal of the Neurological Sciences. 1987;80(1):39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- 67.Coulton GR, Morgan JE, Partridge TA, Sloper JC. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathology and Applied Neurobiology. 1988;14(1):53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 68.Stedman HH, Sweeney HL, Shrager JB, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352(6335):536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 69.Louboutin JP, Fichter-Gagnepain V, Thaon E, Fardeau M. Morphometric analysis of mdx diaphragm muscle fibres. Comparison with hindlimb muscles. Neuromuscular Disorders. 1993;3(5-6):463–469. doi: 10.1016/0960-8966(93)90098-5. [DOI] [PubMed] [Google Scholar]
- 70.Ameen V, Robson LG. Experimental models of Duchenne muscular dystrophy: relationship with cardiovascular disease. Open Cardiovascular Medicine Journal. 2010;4(2):265–277. doi: 10.2174/1874192401004010265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mosqueira M, Zeiger U, Förderer M, Brinkmeier H, Fink RH. Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Medicinial Research Reviews. 2013;33(5):1174–1213. doi: 10.1002/med.21279. [DOI] [PubMed] [Google Scholar]
- 72.Shirokova N, Niggli E. Cardiac phenotype of Duchenne muscular dystrophy: insights from cellular studies. Journal of Molecular and Cellular Cardiology. 2013;58(1):217–224. doi: 10.1016/j.yjmcc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. Federation of European Biochemical Societies Journal. 2013;280(17):4177–4186. doi: 10.1111/febs.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bridges LR. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of mdx mice. Journal of the Neurological Sciences. 1986;72(2-3):147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 75.Lorin C, Gueffier M, Bois P, Faivre JF, Cognard C, Sebille S. Ultrastructural and functional alterations of EC coupling elements in mdx cardiomyocytes: an analysis from membrane surface to depth. Cell Biochemistry and Biophysics. 2013;66(3):723–736. doi: 10.1007/s12013-013-9517-8. [DOI] [PubMed] [Google Scholar]
- 76.van Erp C, Loch D, Laws N, Trebbin A, Hoey AJ. Timeline of cardiac dystrophy in 3–18-month-old mdx mice. Muscle and Nerve. 2010;42(4):504–513. doi: 10.1002/mus.21716. [DOI] [PubMed] [Google Scholar]
- 77.Verhaart IEC, van Duijn RJM, den Adel B, et al. Assessment of cardiac function in three mouse dystrophinopathies by magnetic resonance imaging. Neuromuscular Disorders. 2012;22(5):418–426. doi: 10.1016/j.nmd.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 78.Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscular Disorders. 2004;14(8-9):491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 79.Cheng YJ, Lang D, Caruthers SD, Efimov IR, Chen J, Wickline SA. Focal but reversible diastolic sheet dysfunction reflects regional calcium mishandling in dystrophic mdx mouse hearts. The American Journal of Physiology—Heart and Circulatory Physiology. 2012;303(5):H559–H568. doi: 10.1152/ajpheart.00321.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Au CG, Butler TL, Sherwood MC, Egan JR, North KN, Winlaw DS. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. International Journal of Experimental Pathology. 2011;92(1):57–65. doi: 10.1111/j.1365-2613.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spurney CF, Knoblach S, Pistilli EE, Nagaraju K, Martin GR, Hoffman EP. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscular Disorders. 2008;18(5):371–381. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ieronimakis N, Hays AL, Janebodin K, et al. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFβ1 signaling in the mdx mouse model of Duchenne muscular dystrophy. Journal of Molecular and Cellular Cardiology. 2013;63(1):122–134. doi: 10.1016/j.yjmcc.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Letters. 2002;520(1–3):18–24. doi: 10.1016/s0014-5793(02)02739-4. [DOI] [PubMed] [Google Scholar]
- 84.Danialou G, Comtois AS, Dudley R, et al. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. The FASEB Journal. 2001;15(9):1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 85.Hourdé C, Joanne P, Medja F, et al. Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. The American Journal of Pathology. 2013;182(5):1509–1518. doi: 10.1016/j.ajpath.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 86.Bia BL, Cassidy PJ, Young ME, et al. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of duchenne muscular dystrophy. Journal of Molecular and Cellular Cardiology. 1999;31(10):1857–1862. doi: 10.1006/jmcc.1999.1018. [DOI] [PubMed] [Google Scholar]
- 87.Chu V, Otero JM, Lopez O, et al. Electrocardiographic findings in mdx mice: a cardiac phenotype of Duchenne muscular dystrophy. Muscle and Nerve. 2002;26(4):513–519. doi: 10.1002/mus.10223. [DOI] [PubMed] [Google Scholar]
- 88.Zhang W, Hove MT, Schneider JE, et al. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: an MRI and MRS study. Journal of Molecular and Cellular Cardiology. 2008;45(6):754–760. doi: 10.1016/j.yjmcc.2008.09.125. [DOI] [PubMed] [Google Scholar]
- 89.Fayssoil A, Renault G, Guerchet N, Marchiol-Fournigault C, Fougerousse F, Richard I. Cardiac characterization of mdx mice using high-resolution doppler echocardiography. Journal of Ultrasound in Medicine. 2013;32(5):757–761. doi: 10.7863/ultra.32.5.757. [DOI] [PubMed] [Google Scholar]
- 90.Wagner S, Knipp S, Weber C, et al. The heart in Duchenne muscular dystrophy: early detection of contractile performance alteration. Journal of Cellular and Molecular Medicine. 2012;16(12):3028–3036. doi: 10.1111/j.1582-4934.2012.01630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sapp JL, Bobet J, Howlett SE. Contractile properties of myocardium are altered in dystrophin-deficient mdx mice. Journal of the Neurological Sciences. 1996;142(1-2):17–24. doi: 10.1016/0022-510x(96)00167-0. [DOI] [PubMed] [Google Scholar]
- 92.Ullrich ND, Fanchaouy M, Gusev K, Shirokova N, Niggli E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. The American Journal of Physiology—Heart and Circulatory Physiology. 2009;297(6):H1992–H2003. doi: 10.1152/ajpheart.00602.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Woolf PJ, Lu S, Cornford-Nairn R, et al. Alterations in dihydropyridine receptors in dystrophin-deficient cardiac muscle. The American Journal of Physiology—Heart and Circulatory Physiology. 2006;290(6):H2439–H2445. doi: 10.1152/ajpheart.00844.2005. [DOI] [PubMed] [Google Scholar]
- 94.Koenig X, Dysek S, Kimbacher S, et al. Voltage-gated ion channel dysfunction precedes cardiomyopathy development in the dystrophic heart. PLoS ONE. 2011;6(5) doi: 10.1371/journal.pone.0020300.e20300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Viola HM, Davies SM, Filipovska A, Hool LC. L-type Ca2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. The American Journal of Physiology—Heart and Circulatory Physiology. 2013;304(6):H767–H775. doi: 10.1152/ajpheart.00700.2012. [DOI] [PubMed] [Google Scholar]
- 96.Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. The American Journal of Physiology—Heart and Circulatory Physiology. 2007;292(2):H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- 97.Jung C, Martins AS, Niggli E, Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovascular Research. 2008;77(4):766–773. doi: 10.1093/cvr/cvm089. [DOI] [PubMed] [Google Scholar]
- 98.Lohan J, Ohlendieck K. Drastic reduction in the luminal Ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2004;1689(3):252–258. doi: 10.1016/j.bbadis.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 99.Doran P, Dowling P, Donoghue P, Buffini M, Ohlendieck K. Reduced expression of regucalcin in young and aged mdx diaphragm indicates abnormal cytosolic calcium handling in dystrophin-deficient muscle. Biochimica et Biophysica Acta—Proteins and Proteomics. 2006;1764(4):773–785. doi: 10.1016/j.bbapap.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 100.Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. The American Journal of Physiology—Heart and Circulatory Physiology. 2007;293(3):H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- 101.Fanchaouy M, Polakova E, Jung C, Ogrodnik J, Shirokova N, Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46(2):114–121. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khairallah M, Khairallah R, Young ME, Dyck JRB, Petrof BJ, des Rosiers C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2007;43(2):119–129. doi: 10.1016/j.yjmcc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 103.Masubuchi N, Shidoh Y, Kondo S, Takatoh J, Hanaoka K. Subcellular localization of dystrophin isoforms in cardiomyocytes and phenotypic analysis of dystrophin-deficient mice reveal cardiac myopathy is predominantly caused by a deficiency in full-length dystrophin. Experimental Animals. 2013;62(3):211–217. doi: 10.1538/expanim.62.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lohan J, Culligan K, Ohlendieck K. Deficiency in cardiac dystrophin affects the abundance of the α-/β-dystroglycan complex. Journal of Biomedicine and Biotechnology. 2005;2005(1):28–36. doi: 10.1155/S111072430440103X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sharpe KM, Premsukh MD, Townsend D. Alterations of dystrophin-associated glycoproteins in the heart lacking dystrophin or dystrophin and utrophin. Journal of Muscle Research and Cell Motility. 2013;34(5-6):395–405. doi: 10.1007/s10974-013-9362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Langley SR, Dwyer J, Drozdov I, Yin X, Mayr M. Proteomics: from single molecules to biological pathways. Cardiovascular Research. 2013;97(4):612–622. doi: 10.1093/cvr/cvs346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharma P, Cosme J, Gramolini AO. Recent advances in cardiovascular proteomics. Journal of Proteomics. 2013;81(1):3–14. doi: 10.1016/j.jprot.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.White MY, van Eyk JE. Cardiovascular proteomics: past, present, and future. Molecular Diagnosis and Therapy. 2007;11(2):83–95. doi: 10.1007/BF03256227. [DOI] [PubMed] [Google Scholar]
- 109.Edwards AVG, White MY, Cordwell SJ. The role of proteomics in clinical cardiovascular biomarker discovery. Molecular and Cellular Proteomics. 2008;7(10):1824–1837. doi: 10.1074/mcp.R800007-MCP200. [DOI] [PubMed] [Google Scholar]
- 110.Doehner W. Diagnostic biomarkers in cardiovascular disease: the proteomics approach. European Heart Journal. 2012;33(18):2249–2251. doi: 10.1093/eurheartj/ehs187. [DOI] [PubMed] [Google Scholar]
- 111.McGregor E, Dunn MJ. Proteomics of the heart: unraveling disease. Circulation Research. 2006;98(3):309–321. doi: 10.1161/01.RES.0000201280.20709.26. [DOI] [PubMed] [Google Scholar]
- 112.Cui Z, Dewey S, Gomes AV. Cardioproteomics: advancing the discovery of signaling mechanisms involved in cardiovascular diseases. The American Journal of Cardiovascular Disease. 2011;1(3):274–292. [PMC free article] [PubMed] [Google Scholar]
- 113.Cieniewski-Bernard C, Acosta A, Dubois E, et al. Proteomic analysis in cardiovascular diseases. Clinical and Experimental Pharmacology and Physiology. 2008;35(4):362–366. doi: 10.1111/j.1440-1681.2008.04878.x. [DOI] [PubMed] [Google Scholar]
- 114.Sun Z, Hamilton KL, Reardon KF. Phosphoproteomics and molecular cardiology: techniques, applications and challenges. Journal of Molecular and Cellular Cardiology. 2012;53(3):354–368. doi: 10.1016/j.yjmcc.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 115.Liddy KA, White MY, Cordwell SJ. Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome Medicine. 2013;5(2, article 20) doi: 10.1186/gm424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vivanco F, editor. (Methods in Molecular Biology).Heart Proteomics. 2013;1005 [Google Scholar]
- 117.Westbrook JA, Wheeler JX, Wait R, Welson SY, Dunn MJ. The human heart proteome: two-dimensional maps using narrow-range immobilised pH gradients. Electrophoresis. 2006;27(8):1547–1555. doi: 10.1002/elps.200500777. [DOI] [PubMed] [Google Scholar]
- 118.Raddatz K, Albrecht D, Hochgräfe F, Hecker M, Gotthardt M. A proteome map of murine heart and skeletal muscle. Proteomics. 2008;8(9):1885–1897. doi: 10.1002/pmic.200700902. [DOI] [PubMed] [Google Scholar]
- 119.Bousette N, Kislinger T, Fong V, et al. Large-scale characterization and analysis of the murine cardiac proteome. Journal of Proteome Research. 2009;8(4):1887–1901. doi: 10.1021/pr800845a. [DOI] [PubMed] [Google Scholar]
- 120.Polden J, Mcmanus CA, dos Remedios C, Dunn MJ. A 2-D gel reference map of the basic human heart proteome. Proteomics. 2011;11(17):3582–3586. doi: 10.1002/pmic.201000182. [DOI] [PubMed] [Google Scholar]
- 121.Cosme J, Emili A, Gramolini AO. Large-scale characterization of the murine cardiac proteome. (Methods in Molecular Biology).Heart Proteomics. 2013;1005:1–10. doi: 10.1007/978-1-62703-386-2_1. [DOI] [PubMed] [Google Scholar]
- 122.Bon E, Steegers R, Steegers EAP, et al. Proteomic analyses of the developing chicken cardiovascular system. Journal of Proteome Research. 2010;9(1):268–274. doi: 10.1021/pr900614w. [DOI] [PubMed] [Google Scholar]
- 123.Burniston JG. Adaptation of the rat cardiac proteome in response to intensity-controlled endurance exercise. Proteomics. 2009;9(1):106–115. doi: 10.1002/pmic.200800268. [DOI] [PubMed] [Google Scholar]
- 124.Burniston JG, Kenyani J, Wastling JM, et al. Proteomic analysis reveals perturbed energy metabolism and elevated oxidative stress in hearts of rats with inborn low aerobic capacity. Proteomics. 2011;11(16):3369–3379. doi: 10.1002/pmic.201000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rocha LA, Petriz BA, Borges DH, et al. High molecular mass proteomics analyses of left ventricle from rats subjected to differential swimming training. Biomedcentral Physiology. 2012;12(1, article 11) doi: 10.1186/1472-6793-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chakravarti B, Oseguera M, Dalal N, et al. Proteomic profiling of aging in the mouse heart: altered expression of mitochondrial proteins. Archives of Biochemistry and Biophysics. 2008;474(1):22–31. doi: 10.1016/j.abb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 127.Grant JE, Bradshaw AD, Schwacke JH, Baicu CF, Zile MR, Schey KL. Quantification of protein expression changes in the aging left ventricle of Rattus norvegicus . Journal of Proteome Research. 2009;8(9):4252–4263. doi: 10.1021/pr900297f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dai Q, Escobar GP, Hakala KW, Lambert JM, Weintraub ST, Lindsey ML. The left ventricle proteome differentiates middle-aged and old left ventricles in mice. Journal of Proteome Research. 2008;7(2):756–765. doi: 10.1021/pr700685e. [DOI] [PubMed] [Google Scholar]
- 129.Richardson MR, Lai X, Mason SB, Miller SJ, Witzmann FA. Differential protein expression during aging in ventricular myocardium of Fischer 344 × Brown Norway hybrid rats. Experimental Gerontology. 2008;43(10):909–918. doi: 10.1016/j.exger.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nishtala K, Phong TQ, Steil L, et al. Proteomic analyses of age related changes in A.BY/SnJ mouse hearts. Proteome Science. 2013;11(1, article 29) doi: 10.1186/1477-5956-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dabkowski ER, Baseler WA, Williamson CL, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. The American Journal of Physiology—Heart and Circulatory Physiology. 2010;299(2):H529–H540. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hammer E, Goritzka M, Ameling S, et al. Characterization of the human myocardial proteome in inflammatory dilated cardiomyopathy by label-free quantitative shotgun proteomics of heart biopsies. Journal of Proteome Research. 2011;10(5):2161–2171. doi: 10.1021/pr1008042. [DOI] [PubMed] [Google Scholar]
- 133.Goudarzi M, Ross MM, Zhou W, et al. Development of a novel proteomic approach for mitochondrial proteomics from cardiac tissue from patients with atrial fibrillation. Journal of Proteome Research. 2011;10(8):3484–3492. doi: 10.1021/pr200108m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang P, Wang W, Wang X, et al. Protein analysis of atrial fibrosis via label-free proteomics in chronic atrial fibrillation patients with mitral valve disease. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0060210.e60210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fessart D, Martin-Negrier ML, Claverol S, et al. Proteomic remodeling of proteasome in right heart failure. Journal of Molecular and Cellular Cardiology. 2014;66:41–52. doi: 10.1016/j.yjmcc.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 136.Dai DF, Hsieh EJ, Chen T, et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circulation: Heart Failure. 2013;6(5):1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rabilloud T, Vaezzadeh AR, Potier N, Lelong C, Leize-Wagner E, Chevallet M. Power and limitations of electrophoretic separations in proteomics strategies. Mass Spectrometry Reviews. 2009;28(5):816–843. doi: 10.1002/mas.20204. [DOI] [PubMed] [Google Scholar]
- 138.Weiss W, Görg A. High-resolution two-dimensional electrophoresis. (Methods in Molecular Biology).Proteomics. 2009;564:13–32. doi: 10.1007/978-1-60761-157-8_2. [DOI] [PubMed] [Google Scholar]
- 139.Friedman DB, Hoving S, Westermeier R. Chapter 30 isoelectric focusing and two-dimensional gel electrophoresis. Methods in Enzymology. 2009;463:515–540. doi: 10.1016/S0076-6879(09)63030-5. [DOI] [PubMed] [Google Scholar]
- 140.Rabilloud T, Chevallet M, Luche S, Lelong C. Two-dimensional gel electrophoresis in proteomics: past, present and future. Journal of Proteomics. 2010;73(11):2064–2077. doi: 10.1016/j.jprot.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 141.Gulston MK, Rubtsov DV, Atherton HJ, et al. A combined metabolomic and proteomic investigation of the effects of a failure to express dystrophin in the mouse heart. Journal of Proteome Research. 2008;7(5):2069–2077. doi: 10.1021/pr800070p. [DOI] [PubMed] [Google Scholar]
- 142.Lewis C, Jockusch H, Ohlendieck K. Proteomic profiling of the dystrophin-deficient MDX heart reveals drastically altered levels of key metabolic and contractile proteins. Journal of Biomedicine and Biotechnology. 2010;2010:20 pages. doi: 10.1155/2010/648501.648501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ohlendieck K. On-membrane digestion technology for muscle proteomics. Journal of Membrane and Separation Technology. 2013;2(1):1–12. [Google Scholar]
- 144.Parker BL, Palmisano G, Edwards AVG, et al. Quantitative N-linked glycoproteomics of myocardial ischemia and reperfusion injury reveals early remodeling in the extracellular environment. Molecular and Cellular Proteomics. 2011;10(8) doi: 10.1074/mcp.M110.006833.M110.006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Scholten A, Mohammed S, Low TY, et al. In-depth quantitative cardiac proteomics combining electron transfer dissociation and the metalloendopeptidase Lys-N with the SILAC mouse. Molecular and Cellular Proteomics. 2011;10(10) doi: 10.1074/mcp.O111.008474.O111.008474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Angel TE, Aryal UK, Hengel SM, et al. Mass spectrometry-based proteomics: existing capabilities and future directions. Chemical Society Reviews. 2012;41(10):3912–3928. doi: 10.1039/c2cs15331a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rabilloud T, Strub J, Luche S, van Dorsselaer A, Lunardi J. A comparison between Sypro Ruby and ruthenium ii tris (bathophenanthroline disulfonate) as fluorescent stains for protein detection in gels. Proteomics. 2001;1(5):699–704. doi: 10.1002/1615-9861(200104)1:5<699::AID-PROT699>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 148.Aude-Garcia C, Collin-Faure V, Luche S, Rabilloud T. Improvements and simplifications in in-gel fluorescent detection of proteins using ruthenium II tris-(bathophenanthroline disulfonate): the poor man’s fluorescent detection method. Proteomics. 2011;11(2):324–328. doi: 10.1002/pmic.201000370. [DOI] [PubMed] [Google Scholar]
- 149.Viswanathan S, Ünlü M, Minden JS. Two-dimensional difference gel electrophoresis. Nature Protocols. 2006;1(3):1351–1358. doi: 10.1038/nprot.2006.234. [DOI] [PubMed] [Google Scholar]
- 150.Timms JF, Cramer R. Difference gel electrophoresis. Proteomics. 2008;8(23-24):4886–4897. doi: 10.1002/pmic.200800298. [DOI] [PubMed] [Google Scholar]
- 151.Minden JS. Two-dimensional difference gel electrophoresis. (Methods in Molecular Biology).Protein Electrophoresis. 2012;869:287–304. doi: 10.1007/978-1-61779-821-4_24. [DOI] [PubMed] [Google Scholar]
- 152.Carberry S, Zweyer M, Swandulla D, Ohlendieck K. Application of fluorescence two-dimensional difference in-gel electrophoresis as a proteomic biomarker discovery tool in muscular dystrophy research. Biology. 2013;2(4):1438–1464. doi: 10.3390/biology2041438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Doran P, Martin G, Dowling P, Jockusch H, Ohlendieck K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics. 2006;6(16):4610–4621. doi: 10.1002/pmic.200600082. [DOI] [PubMed] [Google Scholar]
- 154.Doran P, Wilton SD, Fletcher S, Ohlendieck K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics. 2009;9(3):671–685. doi: 10.1002/pmic.200800441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lewis C, Ohlendieck K. Proteomic profiling of naturally protected extraocular muscles from the dystrophin-deficient mdx mouse. Biochemical and Biophysical Research Communications. 2010;396(4):1024–1029. doi: 10.1016/j.bbrc.2010.05.052. [DOI] [PubMed] [Google Scholar]
- 156.Gardan-Salmon D, Dixon JM, Lonergan SM, Selsby JT. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. European Journal of Applied Physiology. 2011;111(11):2763–2773. doi: 10.1007/s00421-011-1906-3. [DOI] [PubMed] [Google Scholar]
- 157.Lewis C, Doran P, Ohlendieck K. Proteomic analysis of dystrophic muscle. (Methods in Molecular Biology).Myogenesis. 2012;798:357–369. doi: 10.1007/978-1-61779-343-1_20. [DOI] [PubMed] [Google Scholar]
- 158.Carberry S, Zweyer M, Swandulla D, Ohlendieck K. Comparative proteomic analysis of the contractile protein-depleted fraction from normal versus dystrophic skeletal muscle. Analytical Biochemistry. 2014;446(1):108–115. doi: 10.1016/j.ab.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 159.Ünlü M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 160.Alban A, David SO, Bjorkesten L, et al. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3(1):36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 161.Tonge R, Shaw J, Middleton B, et al. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1(3):377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 162.Marouga R, David S, Hawkins E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Analytical and Bioanalytical Chemistry. 2005;382(3):669–678. doi: 10.1007/s00216-005-3126-3. [DOI] [PubMed] [Google Scholar]
- 163.Kang Y, Techanukul T, Mantalaris A, Nagy JM. Comparison of three commercially available DIGE analysis software packages: minimal user intervention in gel-based proteomics. Journal of Proteome Research. 2009;8(2):1077–1084. doi: 10.1021/pr800588f. [DOI] [PubMed] [Google Scholar]
- 164.Karp NA, Kreil DP, Lilley KS. Determining a significant change in protein expression with DeCyder™ during a pair-wise comparison using two-dimensional difference gel electrophoresis. Proteomics. 2004;4(5):1421–1432. doi: 10.1002/pmic.200300681. [DOI] [PubMed] [Google Scholar]
- 165.Karp NA, Lilley KS. Maximising sensitivity for detecting changes in protein expression: experimental design using minimal CyDyes. Proteomics. 2005;5(12):3105–3115. doi: 10.1002/pmic.200500083. [DOI] [PubMed] [Google Scholar]
- 166.Corzett TH, Fodor IK, Choi MW, et al. Statistical analysis of the experimental variation in the proteomic characterization of human plasma by two-dimensional difference gel electrophoresis. Journal of Proteome Research. 2006;5(10):2611–2619. doi: 10.1021/pr060100p. [DOI] [PubMed] [Google Scholar]
- 167.Ge Y, Molloy MP, Chamberlain JS, Andrews PC. Proteomic analysis of mdx skeletal muscle: great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics. 2003;3(10):1895–1903. doi: 10.1002/pmic.200300561. [DOI] [PubMed] [Google Scholar]
- 168.Doran P, Dowling P, Lohan J, McDonnell K, Poetsch S, Ohlendieck K. Subproteomics analysis of Ca2+-binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. European Journal of Biochemistry. 2004;271(19):3943–3952. doi: 10.1111/j.1432-1033.2004.04332.x. [DOI] [PubMed] [Google Scholar]
- 169.Holland A, Carberry S, Ohlendieck K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Current Protein and Peptide Science. 2013;14(8):680–897. doi: 10.2174/13892037113146660083. [DOI] [PubMed] [Google Scholar]
- 170.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovascular Research. 2008;77(2):334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 171.Taylor SW, Fahy E, Zhang B, et al. Characterization of the human heart mitochondrial proteome. Nature Biotechnology. 2003;21(3):281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 172.Gaucher SP, Taylor SW, Fahy E, et al. Expanded coverage of the human heart mitochondrial proteome using multidimensional liquid chromatography coupled with tandem mass spectrometry. Journal of Proteome Research. 2004;3(3):495–505. doi: 10.1021/pr034102a. [DOI] [PubMed] [Google Scholar]
- 173.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Molecular and Cellular Proteomics. 2006;5(4):608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 174.Culligan KG, Mackey AJ, Finn DM, Maguire PB, Ohlendieck K. Role of dystrophin isoforms and associated proteins in muscular dystrophy (review) International Journal of Molecular Medicine. 1998;2(6):639–648. doi: 10.3892/ijmm.2.6.639. [DOI] [PubMed] [Google Scholar]
- 175.Domogatskaya A, Rodin S, Tryggvason K. Functional diversity of laminins. Annual Review of Cell and Developmental Biology. 2012;28(1):523–553. doi: 10.1146/annurev-cellbio-101011-155750. [DOI] [PubMed] [Google Scholar]
- 176.Ho MS, Böse K, Mokkapati S, Nischt R, Smyth N. Nidogens-extracellular matrix linker molecules. Microscopy Research and Technique. 2008;71(5):387–395. doi: 10.1002/jemt.20567. [DOI] [PubMed] [Google Scholar]
- 177.Camors E, Monceau V, Charlemagne D. Annexins and Ca2+ handling in the heart. Cardiovascular Research. 2005;65(4):793–802. doi: 10.1016/j.cardiores.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 178.Wasala NB, Bostick B, Yue Y, Duan D. Exclusive skeletal muscle correction does not modulate dystrophic heart disease in the aged mdx model of Duchenne cardiomyopathy. Human Molecular Genetics. 2013;22(13):2634–2641. doi: 10.1093/hmg/ddt112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108(13):1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Human Molecular Genetics. 2005;14(14):1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- 181.Shin JH, Nitahara-Kasahara Y, Hayashita-Kinoh H, et al. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Therapy. 2011;18(9):910–919. doi: 10.1038/gt.2011.36. [DOI] [PubMed] [Google Scholar]
- 182.Bostick B, Shin JH, Yue Y, Wasala NB, Lai Y, Duan D. AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2012;53(2):217–222. doi: 10.1016/j.yjmcc.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wu B, Moulton HM, Iversen PL, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(39):14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Buyse GM, van der Mieren G, Erb M, et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. European Heart Journal. 2009;30(1):116–124. doi: 10.1093/eurheartj/ehn406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Spurney CF, Sali A, Guerron AD, et al. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. Journal of Cardiovascular Pharmacology and Therapeutics. 2011;16(1):87–95. doi: 10.1177/1074248410381757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hori YS, Kuno A, Hosoda R, et al. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Journal of Pharmacology and Experimental Therapeutics. 2011;338(3):784–794. doi: 10.1124/jpet.111.183210. [DOI] [PubMed] [Google Scholar]
- 187.Delfín DA, Xu Y, Schill KE, et al. Sustaining cardiac claudin-5 levels prevents functional hallmarks of cardiomyopathy in a muscular dystrophy mouse model. Molecular Therapy. 2012;20(7):1378–1383. doi: 10.1038/mt.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Blain A, Greally E, Laval S, Blamire A, Straub V, MacGowan GA. Beta-blockers, left and right ventricular function, and in-vivo calcium influx in muscular dystrophy cardiomyopathy. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0057260.e57260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vianello S, Yu H, Voisin V, et al. Arginine butyrate: a therapeutic candidate for Duchenne muscular dystrophy. Federation of American Societies for Experimental Biology Journal. 2013;27(6):2256–2269. doi: 10.1096/fj.12-215723. [DOI] [PubMed] [Google Scholar]
- 190.Ather S, Wang W, Wang Q, Li N, Anderson ME, Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm. 2013;10(4):592–599. doi: 10.1016/j.hrthm.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Berry SE, Andruszkiewicz P, Chun JL, Hong J. Nestin expression in end-stage disease in dystrophin-deficient heart: implications for regeneration from endogenous cardiac stem cells. Stem Cells Translational Medicine. 2013;2(11):848–861. doi: 10.5966/sctm.2012-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sciorati C, Staszewsky L, Zambelli V, et al. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacological Research. 2013;73(1):35–43. doi: 10.1016/j.phrs.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 193.Ohlendieck K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomarkers in Medicine. 2013;7(1):169–186. doi: 10.2217/bmm.12.96. [DOI] [PubMed] [Google Scholar]