

Abstract
Phage-assisted continuous evolution (PACE) uses a modified filamentous bacteriophage life cycle to dramatically accelerate laboratory evolution experiments. In this work we expand the scope and capabilities of the PACE method with two key advances that enable the evolution of biomolecules with radically altered or highly specific new activities. First, we implemented small molecule-controlled modulation of selection stringency that enables otherwise inaccessible activities to be evolved directly from inactive starting libraries through a period of evolutionary drift. Second, we developed a general negative selection that enables continuous counter-selection against undesired activities. We integrated these developments to continuously evolve mutant T7 RNA polymerase enzymes with ∼10,000-fold altered, rather than merely broadened, substrate specificities during a single three-day PACE experiment. The evolved enzymes exhibit specificity for their target substrate that exceeds that of wild-type RNA polymerases for their cognate substrates, while maintaining wild-type-like levels of activity.
Introduction
During phage-assisted continuous evolution (PACE), a population of bacteriophage, each encoding a library member of an evolving gene, selectively replicates in a “lagoon” of continuously replenished E. coli host cells in a manner that depends on an activity of interest (Figure 1).1 These selection phage (SP) lack gene III, which has been moved from the phage genome to an accessory plasmid (AP) carried by the host cells. Gene III encodes the phage protein pIII, which is essential for infectious phage production.2 Expression of pIII from the AP is made dependent on the activity of interest, such that only SP encoding active library members induce expression of pIII and the production of infectious progeny phage. A mutagenesis plasmid (MP) enables small-molecule control over the mutation rate during phage replication. Infectious progeny phage can infect fresh host cells flowing into the lagoon and thereby continue to undergo additional cycles of selection, replication, and mutation. Because one complete cycle of phage replication can be as brief as 10 minutes,3 PACE enables hundreds of rounds of directed evolution to take place in less than a week with minimal researcher intervention, representing a ∼100-fold increase in speed over traditional protein evolution methods.4,5 We previously used PACE to evolve T7 RNA polymerase (RNAP) mutants that initiate transcription at the T3 promoter (PT3), which differs from the T7 promoter (PT7) at six of 17 base positions.1,6,7 Though successful, our early PACE efforts highlighted two major needs.
Figure 1. Overview of PACE with real-time monitoring.

During PACE, host E. coli cells flow continuously into a fixed-volume vessel (a “lagoon”) containing a population of filamentous bacteriophages that encode a library of evolving proteins. The lagoon is continuously drained to a waste container after passing through an in-line luminescence monitor that measures expression from a geneIII-luciferase cassette on the AP. Dilution occurs faster than cell division but slower than phage replication. Each phage carries a protein-encoding gene to be evolved instead of a phage gene (gene III) that is required for infection. Phage encoding active library members trigger expression of gene III on the AP in proportion to the desired activity and consequently produce infectious progeny. Phage encoding less active library members produce fewer infectious progeny and are lost by dilution.
First, when we challenged wild-type T7 RNAP to evolve activity directly on PT3, active mutants did not emerge and the phage in the lagoon washed out, indicating that the stringency of this initial selection was too high.1,6,7 The successful evolution of activity on PT3 instead required initial selection on a hybrid T7/T3 promoter (PT7/T3) that served as an evolutionary stepping-stone toward the PT3 selection. For many potential applications of PACE, suitable intermediate substrates may not be obvious or accessible. To address this limitation, we sought to develop a general modulator of selection stringency that can be tuned simply by adding a small molecule to a lagoon. Initiating PACE under reduced selection stringency in principle can allow weakly active or inactive variants to access favorable mutations that enable propagation under subsequent higher-stringency selection conditions.
Second, the T7 RNAPs evolved to recognize PT3 retained most or all of their activity on PT7. In the absence of explicit negative selection against undesired activity, the evolution of novel substrate recognition often results in enzymes with broadened, rather than truly altered specificity.8,9 For many potential targets of PACE, including proteases, binding proteins, and genome engineering enzymes, evolved proteins will require exceptional substrate specificity to function in complex cellular environments containing many potential off-target substrates. We therefore sought to develop a general negative selection for PACE in which SPs encoding variants with undesired activities, such as off-target substrate recognition, incur a replicative penalty.
In this study we developed robust and general solutions that address both of these needs. Integrating these advances, we directly evolved in a single PACE experiment T7 RNAP mutants that have wild-type like activity on PT3 and remarkable specificity for PT3 over PT7. This evolved degree of specificity exceeds that of wild-type T7 RNAP for PT7 over PT3, as well as that of wild-type T3 RNAP for PT3 over PT7. The combination of stringency modulation and negative selection enabled the evolution of polymerases with ∼10,000-fold changes in specificity in a total time of only three days. Together, these developments bring to PACE two capabilities recognized to be highly enabling for laboratory evolution efforts. These capabilities also expand the scope of PACE to include the evolution of biomolecules that possess radically altered and highly specific new activities.
Results
Small Molecule-Controlled Selection Stringency Modulation
We hypothesized that selection stringency during PACE can be varied by providing host cells with carefully regulated amounts of pIII in a manner independent of the desired evolving activity. To create this capability, we placed expression of gene III on an AP under the control of the small molecule-inducible TetA promoter (Ptet) and observed anhydrotetracycline (ATc) concentration-dependent gene III expression and phage production in a discrete host cell culture (Supplementary Results, Supplementary Figure 1). To test propagation in a streamlined PACE format, we modified our original host cell culture system to operate as a chemostat. Compared to host cell cultures maintained at constant turbidity (turbidostats), cultures maintained at a constant nutrient flow rate (chemostats) are simpler to set up and do not require constant turbidity monitoring. We found that chemostats using E. coli S1030 cells support PACE comparably to turbidostats cultures (data not shown), and therefore used chemostats for the PACE experiments in this study. Although an AP containing Ptet-gene III supported ATc-dependent phage production in a discrete host cell culture, this AP did not support robust phage propagation in a PACE format when ATc was added to a lagoon (Figure 2a). Because even low levels of pIII render cells resistant to filamentous phage infection,10 we hypothesized that these host cells, which begin producing pIII soon after entering a lagoon, become phage infection-resistant prior to encountering phage, thereby preventing phage propagation (Supplementary Figure 1).
Figure 2. Drift cassette enables ATc-dependent, activity-independent phage propagation.

(a-c) Cells harboring APs with the indicated gene III-luxAB expression cassettes served as recipients for phage propagation experiments using SP-T7WT. Data show representative single measurements of phage concentrations (n = 1). (a) Using Ptet to induce gene III expression with ATc prior to phage infection inhibits infection and results in minimal phage propagation and low phage titers. (b) Using Ppsp to express gene III only after infection takes place results in robust activity-independent phage propagation and high phage titers. (c) Infection- and ATc-dependent gene III expression using Ppsp-tet enables robust, activity-independent propagation. (d) Recipient cells carrying a drift plasmid (DP) and a PT7-gene III AP were used to propagate a mixture of SP-T7WT (wild-type T7 RNAP, high activity) and SP-T7Dead (D812G mutant T7 RNAP,14 no activity) at a ratio of 1:102 (“Mix” in (e)). (e) In the absence of ATc, SP-T7WT (WT) is rapidly enriched over the inactive D812G mutant polymerase (D) and a rapid increase in luciferase signal is observed. (f) At an intermediate ATc concentration (150 ng/mL), SP-T7WT is enriched at a slower rate, concurrent with a slower rise in luciferase signal. (g) At the highest ATc concentration (400 ng/mL), SP-T7WT is not enriched and baseline luciferase signal is observed. After ending ATc supplementation at t = 8 h, SP-T7WT is rapidly enriched and the luciferase signal rapidly rises.
To create a system in which activity-independent pIII expression requires both ATc and prior phage infection, we used the previously described E. coli phage shock promoter (Ppsp), which is induced by infection with filamentous phage via a pIV-dependent signaling cascade.11 Transcription from Ppsp can also be induced by overexpression of a plasmid-encoded phage pIV gene.12 Phage lacking gene III are known to form plaques on cells containing a Ppsp-gene III cassette.13 We confirmed and extended this observation by showing that an AP with this cassette supports robust propagation in a PACE format (Figure 2b).
To make phage propagation ATc-dependent, we examined the gene expression properties of Ppsp variants with TetR operators installed at positions intended to disrupt either PspF or E. coli RNA polymerase binding (Supplementary Figure 1). We found that placing an operator adjacent to the +1 transcription initiation site creates a promoter (Ppsp-tet) that is induced only with the combination of phage infection and ATc (Supplementary Figure 1). Importantly, this AP supported robust ATc-dependent propagation of a SP without any activity-dependent gene III expression (Figure 2c). High Ppsp-tet-gene III induction (200 ng/mL ATc) supported propagation with dilution rates of 2.5 vol/h, corresponding to an average of 30 phage generations per 24 hours. These results collectively demonstrate that SPs can propagate in an ATc-dependent, activity-independent manner using the Ppsp-tet-gene III AP, thus enabling selection stringency to be altered during PACE in a small molecule-regulated manner.
Tuning Selection Stringency During PACE
Next we examined how the Ppsp-tet-gene III cassette influences the enrichment of active library members over inactive library members in the context of an actual PACE selection in which an additional copy of gene III is controlled by an activity-dependent promoter. At saturating concentrations of ATc, the Ppsp-tet-gene III cassette should provide sufficient pIII to maximize phage propagation regardless of SP-encoded activity, enabling genetic drift of the SP. At intermediate concentrations of ATc, SPs encoding active library members should enjoy a replicative advantage over a SP encoding an inactive variant by inducing additional pIII expression from an activity-dependent promoter. This advantage, and therefore selection stringency, should be inversely proportional to the concentration of ATc provided. In the absence of any added ATc, selection stringency should be fully determined by the activity-dependent AP components with no assistance from the Ppsp-tet-gene III cassette.
To characterize the relationship between ATc concentration and selection stringency, we combined the Ppsp-tet-gene III cassette with the arabinose-inducible mutagenesis cassette1 onto a single “drift plasmid” continuously flowing host cells that contained both this DP and an AP with a PT7-gene III cassette (Figure 2d). We then seeded lagoons with a mixture of two SPs encoding either the highly active wild-type T7 RNAP (SP-T7WT) or the catalytic dead mutant D812G (SP-T7Dead).14 These phage were added in a ratio of 1:100 SP-T7WT:SP-T7Dead to each of three lagoons receiving either 0, 150, or 400 ng/mL ATc. The phage populations were followed over time using a combination of restriction endonuclease digests and real-time measurements of luminescence monitoring of PT7 transcriptional activity.
As expected, under the highest stringency conditions (0 ng/mL ATc), SP-T7Dead washed out of the lagoon and the highly active SP-T7WT was quickly enriched (Figure 2e). At an intermediate concentration of ATc (150 ng/mL), SP-T7Dead again washed out in favor of SP-T7WT, but at a slower rate than in the absence of ATc (Figure 2f). At the highest concentration of ATc (400 ng/mL), SP-T7Dead was able to propagate and no substantial enrichment of the active SP-T7WT was observed (Figure 2g). When we stopped ATc supplementation to this lagoon, we observed very rapid enrichment of SP-T7WT consistent with the expected increase in stringency (Figure 2e). Taken together, these results establish that Ppsp-tet-gene III supports propagation of inactive starting libraries in the presence of high ATc concentrations, and supports the selective enrichment of active mutants at a rate inversely proportional to the concentration of ATc added to the lagoon.
Development of a PACE Negative Selection
An ideal PACE negative selection inhibits infectious phage production in a manner that is tunable and proportional to the ratio of undesired (off-target) to desired (on-target) activity, rather that simply reflecting the absolute level of undesired activity. We therefore sought to develop a negative selection in which undesired activity induces expression of a protein that antagonizes the wild-type pIII induced in a positive selection.
The pIII protein consists of three domains, N1, N2, and C, that mediate initial attachment of the phage to the E. coli F-pilus (N2 domain), subsequent docking with the E. coli TolA cell-surface receptor (N1 domain), and unlocking of the particle for genome entry (C domain).15,16 During progeny phage synthesis, five pIII molecules are attached by the C-domain to the end of each nascent phage particle.17,18 A presumed conformational change in this C-domain then catalyzes detachment of the nascent phage from the inner membrane, thereby releasing the phage from the host cell.16
One pIII mutant described by Rakonjac and co-workers, N-C83, contains intact N1 and N2 domains but has a mutant C domain with an internal deletion of 70 amino acids.16 The residual C domain is sufficient to mediate attachment to a phage particle, but cannot catalyze detachment of nascent phage from the host cell membrane.16 When co-expressed with a pIII variant containing only the intact C domain (which can mediate nascent phage detachment), phage particle production was found to be normal, but the infectivity of these particles was drastically reduced.16 These observations raised the possibility that N-C83 may function as a dominant negative mutant of wild-type pIII.
To test the suitability of N-C83 as the basis for a PACE negative selection, we created a host cell line containing an AP with a Ptet-gene III cassette induced by ATc, a second accessory plasmid (APneg) with N-C83 driven by an IPTG-inducible promoter (Plac). Discrete host cell cultures were grown in the presence or absence of ATc and IPTG, and culture broths were assayed for infectious phage titer. As expected, the addition of ATc induced expression of wild-type pIII and stimulated phage production (Supplementary Figure 2). The simultaneous addition of IPTG, which induces N-C83 expression, dramatically reduced the titer of infectious phage. This result suggests that N-C83 can act as a dominant negative form of pIII. Alternative truncation candidates of pIII did not diminish infectious phage titer to the same degree as N-C83 (Supplementary Figure 2). We also observed evidence to suggest that N-C83 may inhibit infectious phage production by blocking the release of phage from the host cell, rather than by reducing the infectivity of the released phage particles (Supplementary Figure 3). These results reveal that expression of N-C83, hereafter referred to as pIII-neg, can form the basis of a potent negative selection.
PACE Negative Selection Enriches Substrate-Specific RNAPs
Next we tested if this negative selection could enrich a substrate-specific T7 RNAP over a promiscuous RNAP in the PACE format. For this competition experiment, we used SPs encoding two RNAP variants that we recently described: L2-48.3 (specific for PT7, expressed from SP-T7Spec) and L1-192.2 (active on both PT7 and PT3, expressed from SP-T7Prom).1 We prepared a host cell strain containing both a positive selection AP (PT7-gene III AP) and a negative selection AP (PT3-gene III-neg APneg) in which pIII-neg translation is controlled by a theophylline-activated riboswitch (Figure 3a).19 During PACE with this host cell strain, the propagation of SP-T7Prom should be impaired, relative to SP-T7Spec, in the presence of theophylline.
Figure 3. Dominant negative pIII-neg is a potent inhibitor of phage propagation.

(a) Recipient cells carrying a PT7-gene III AP and a PT3-gene III-neg APneg in which the theophylline riboswitch controls gene III-neg expression were used to propagate a 1:106 mixture (“mix” in (b)) of SP-T7Spec (specific for PT7, “spec”) and SP-T7Prom (promiscuous on both PT7 and PT3, “prom”), respectively. (b) At a high theophylline concentration (1000 μM), the promiscuous T7 RNAP SP is rapidly depleted and the specific T7 RNAP SP quickly takes over the lagoon, concomitant with a sharp rise in luciferase signal from PT7. (c) At an intermediate theophylline concentration, the promiscuous T7 RNAP SP slowly washes out and is gradually replaced by the specific T7 RNAP SP. (d) In the absence of theophylline, the promiscuous T7 RNAP SP propagates unhindered and the lagoon maintains the starting ratio of the inoculated phage. Upon addition of high concentrations of theophylline to this lagoon at t = 12 h, a rapid washout of the promiscuous T7 RNAP SP takes place, with a rebound in luciferase signal consistent with specific T7 RNAP SP enrichment. In (b)-(d), data show representative single measurements (n = 1).
Three lagoons containing these host cells were each seeded with a 106:1 ratio of SP-T7Prom:SP-T7Spec and allowed to equilibrate for 4.5 hours. We treated the lagoons with 0, 75, or 1000 μM theophylline and followed the concentrations of the two SP species over time. In the presence of 1000 μM theophylline, SP-T7Prom was rapidly depleted from the lagoon and the SP-T7Spec population expanded dramatically into the predominant species after only 6 hours of PACE (Figure 3b). Furthermore, the rate of enrichment was dependent on the concentration of theophylline added to the lagoon, where lower concentrations of theophylline resulted in slower enrichment rates (Figures 3c and 3d). These results demonstrate that pIII-neg can serve as the basis of a potent and dose-dependent negative selection that can effect the rapid enrichment of substrate-selective enzymes in a PACE format.
PACE of RNAPs with Altered Promoter Specificities
We integrated both the tunable selection stringency and the negative selection developments described above to rapidly evolve enzymes with dramatically altered substrate specificity without the use of intermediate substrates. We prepared two lagoons with host cells containing a PT3-gene III AP and the DP containing the Ppsp-tet-gene III cassette, and seeded these lagoons with SP-T7WT, which has negligible starting activity on PT3. To one lagoon, we added no ATc and observed that phage quickly washed out (data not shown), as expected.1 To the second lagoon, we added 200 ng/mL ATc, which allowed even inactive phage to propagate. After 12 hours (t = 12 h), the phage in this lagoon increased in concentration dramatically, but still encoded RNAPs with negligible activity on PT3. We then reduced the concentration of ATc to 20 ng/mL, thus increasing the selection stringency for recognition of PT3 (Figure 4).
Figure 4. Continuous evolution of T3-specific RNAP variants.

PACE of T7 RNAP variants that recognize PT3 and reject PT7 was performed in three contiguous stages of differing stringency (top). The PT3 and PT7 bars conceptually represent amounts of gene III (red) or gene III-neg (blue) expressed for a given amount of polymerase activity. At t = 0, host cells were E. coli containing the DP and a PT3-gene III AP. For the first 12 hours, 200 ng/mL ATc was added to the lagoon to reduce selection stringency to zero, enabling drift. At t = 12 h, the concentration of ATc was reduced to 20 ng/mL to increase the stringency of positive selection. At t = 28 h, host cells were switched to E. coli harboring an MP, a PT3-gene III AP and a riboswitch-controlled PT7-gene III-neg APneg. At t = 32 h, 1 mM theophylline was added to increase negative selection stringency. At t = 52 h, host cells were switched to E. coli containing an MP, a reduced RBS PT3-gene III AP, and an enhanced RBS/increased origin copy number PT7-gene III-neg APneg to further increase negative and positive selection stringency. The in-line luminescence monitor was used throughout to infer population fitness (center). Gene expression activities on the T7 and T3 promoters of randomly chosen clones from each stage were measured (bottom). Gene expression data show mean values ± s.e. for two replicates. See Supplementary Figure 4 for mutations present in each clone. N = no RNAP; T7 = wild-type T7 RNAP.
After an additional six hours of propagation in 20 ng/mL ATc (t = 18 h), a PT3-active population overtook the lagoon as indicated by an increase in signal from the in-line luminescence monitor. This signal continued to increase over the next 10 hours. RNAP clones isolated from the 28-hour time point evolved an ∼100-fold increase in activity on PT3 (Figure 4) and converged on two mutations, E222K and N748D, that we1 and others20 previously found to broaden the substrate scope of T7 RNAP (Supplementary Figure 4). These results demonstrate that selection stringency modulation can be used to directly evolve a novel activity from an inactive starting gene without the use of intermediate substrates as evolutionary stepping-stones, as was previously necessary to evolve T3 promoter recognition without selection stringency modulation.1
As expected, the RNAPs recovered from this stage of the selection still retained high activity on PT7 (Figure 4). To initiate negative selection against recognition of PT7, we used host cells containing the same PT3-gene III AP, the MP (lacking the drift cassette), and a PT7-gene III-neg APneg in which pIII-neg expression is driven by PT7 transcription, and controlled by a theophylline-dependent riboswitch (Figure 4). The 28-hour lagoon described above was divided into two new lagoons, each of which received these host cells for 4 hours. Theophylline was added to the first lagoon to activate the negative selection (t = 32 h), whereas the second lagoon did not receive any theophylline. After 24 hours (t = 52 h), several RNAPs isolated from the first lagoon showed clear improvements in specificity for PT3 (Figure 4) and contained a variety of mutations (Supplementary Figure 4) at residues proximal to the promoter in the initiation complex and known to be relevant to substrate selectivity. In contrast, clones isolated from the second lagoon, which did not receive theophylline, did not show clear changes in specificity.
In an effort to further enhance the specificity of the evolving RNAPs, we increased the stringency of the negative selection in two ways. First, we weakened the ribosome binding site driving translation of pIII from the AP, with the expectation that evolved RNAPs would compensate with increased (but still PT3-selective) transcriptional activity. Second, we installed a high-copy pUC origin into APneg, and replaced the theophylline riboswitch with a very strong ribosome-binding site driving pIII-neg translation (Figure 4). Together, these modifications should increase the stringency of the negative selection by increasing the ratio of pIII-neg to pIII produced for a given ratio of PT7:PT3 activity.
The phage population from the previous stage (t = 52 h) was transferred to lagoons fed by these high-stringency host cells and a substantial decline and rebound of the phage population was observed (Figure 4). Evolved RNAPs isolated following this recovery (t = 70 h) exhibited dramatically improved specificity for PT3 over PT7 of up to 100-fold and strongly converged on mutations observed in the previous stage (R96L, K98R, E207K, P759S/L) (Figure 4 and Supplementary Figure 4). A forward mutational analysis of these mutations in the E222K/N748D background highlighted the role of each of these changes in conferring strong specificity for PT3 (Figure 5). The degree of specificity evolved using stringency control and negative selection PACE rivals or exceeds that of naturally occurring wild-type T7 or T3 RNA polymerase enzymes; for example, wild-type T7 RNAP exhibits a PT7:PT3 activity ratio of ∼100-fold, and wild-type T3 RNAP exhibits a PT3:PT7 activity ratio of ∼20-fold (Figure 5). Compared to the wild-type T7 RNAP enzyme, the most PT3-specific evolved RNAP clones exhibited a net ∼10,000-fold change in specificity for PT3. Importantly, this remarkable degree of specificity did not evolve at the expense of activity, and all assayed clones retained levels of transcriptional activity on PT3 that are comparable to or higher than that of wild-type T7 RNAP on its cognate T7 promoter (Figure 4). Collectively, these results establish that the PACE negative selection can be used to explicitly evolve enzymes with truly altered, rather than broadened, specificity.
Figure 5. Analysis of evolved T7 RNAP mutations that confer PT3 specificity.
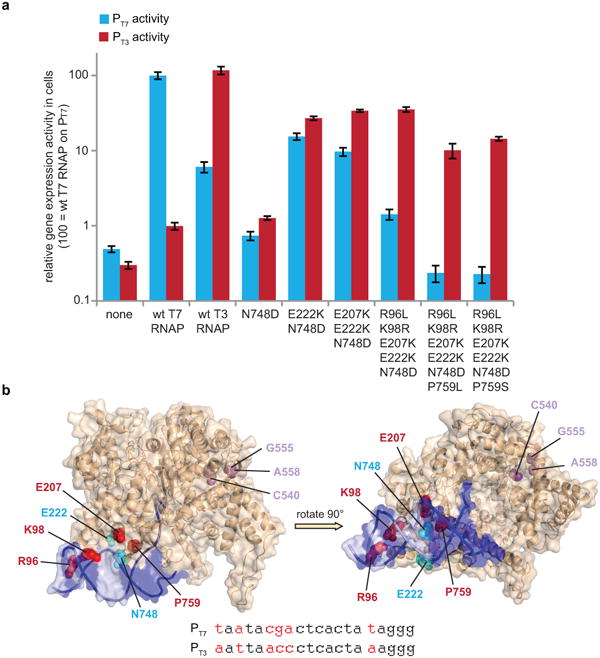
(a) The gene expression activity in cells of T7 RNAP variants containing subsets of mutations found in clones described in Fig. 5 are shown on the T7 promoter (blue bars) and the T3 promoter (red bars). Gene expression data represent mean values ± s.e. for two replicates. (b) Location of evolved mutations.21 The T7 promoter DNA is rendered as dark blue and light purple surfaces, with light purple denoting nucleotide differences between the T7 and T3 promoters. Cyan spheres identify evolved mutations that enable PT3 recognition. Red spheres identify mutations that evolved during negative selection that contribute to specific recognition of PT3 over PT7. Magenta spheres represent a conserved cluster of mutually exclusive mutations evolved in clones following negative selection. The sequences of the T7 and T3 promoters are shown at the bottom, with the differences in red.
Discussion
Traditional directed evolution techniques, while valuable and successful, require frequent researcher intervention throughout each round of mutation, screening or selection, and replication to access biomolecules with desired properties. By mapping the essential components of the directed evolution cycle onto the very rapid M13 filamentous bacteriophage lifecycle, PACE can dramatically accelerate laboratory evolution. In this work we have significantly expanded the capabilities of PACE by developing a general strategy to modulate selection stringency—to zero if needed—with a small molecule, and by developing a negative selection that links undesired activities to the inhibition of phage propagation. Together, these capabilities were used to evolve RNA polymerase variants with ∼10,000-fold altered promoter specificity in a PACE experiment lasting only three days.
T7 RNA polymerases that evolved the ability to accept the T3 promoter and reject the T7 promoter acquired a suite of highly conserved mutations, including R96L, K98R, E207K, E222K, N748D, and P759S/L. The mutations at positions 98 and 748 change each residue to the amino acid found in wild-type T3 RNAP, and the R96L mutation is at a position that also differs between wild-type T7 and T3 RNAPs (although it is K96 in T3 RNAP). These residues are predicted to be proximal to the promoter bases (Figure 5b), and their role in promoter recognition has been previously appreciated.21 The occurrence of these mutations in our selections suggests that the evolved mutants may use a mode of PT3-selective promoter recognition similar to that of wild-type T3 RNAP at these positions. In contrast, residues 207, 222, and 759 are conserved between T7 and T3 RNAPs, and the significant role of mutations at these residues in conferring selectivity (particularly P759S/L, Figure 5a) suggests the evolution of some novel determinants of promoter recognition that may not be used by the native enzymes. E207 modestly increases selectivity for PT3 (Figure 5a) and is within 4 Å of the RNAP specificity loop (residues 739-770), a major determinant of promoter specificity22 that includes P759.
The negative selection uses a dominant negative variant of pIII, the protein that is the basis of the positive selection of PACE, and does not rely on any property of the gene or gene product being evolved. Therefore, we anticipate that the negative selection will be general in its compatibility with a variety of activities that can be evolved using PACE, provided that the selection scheme can link undesired activity to expression of pIII-neg (and not pIII). For many potential proteins that can be evolved with PACE, including DNA-binding proteins, recombinases, protein-protein interfaces, proteases, and other enzymes, a suitable dual selection scheme can be created by localizing undesired substrates upstream of gene III-neg.1 In most cases, such a negative selection can operate simultaneously with pIII-mediated positive selection. Likewise, since the method to modulate selection stringency developed here relies on providing pIII in a regulated manner that is also independent of the evolving gene, this stringency modulation capability should also be applicable to other PACE experiments.
Methods
Bacterial strains
All DNA cloning was performed with Mach1 cells (Invitrogen) or NEB Turbo cells (New England Biolabs). All discrete infection assays, plaque assays and PACE experiments were performed with E. coli S1030. This strain was derived from E. coli S1091 and was modified using the Lambda Red method23 as follows: 1) scarless mutation to rpoZ to introduce a frameshift mutation to enable n-hybrid schemes;24 2) integration of lacI and tetR overexpression cassettes onto the F plasmid to enable small-molecule regulated transcription of various genes; 3) integration of luxCDE onto the F plasmid for the production of decanal to facilitate luciferase monitoring experiments;25 4) deletion of flu/pgaC/csgABCDEFG to dramatically reduce biofilm formation in chemostat PACE experiments;26-28 and 5) mutation of the chromosomal, low-affinity high-capacity AraE promoter to a constitutive promoter for titratable arabinose induction of the PBAD promoter on the mutagenesis plasmid.29 The complete genotype of the resulting strain is F'proA+B+ Δ(lacIZY) zzf∷Tn10(TetR) lacIQ1 PN25-tetR luxCDE/endA1 recA1 galE15 galK16 nupG rpsL(StrR) ΔlacIZYA araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) proBA∷pir116 araE201 ΔrpoZ Δflu ΔcsgABCDEFG ΔpgaC λ−.
Chemostat growth media
Media for all chemostat PACE experiments was prepared by autoclaving a 20 L carboy of MilliQ water containing a custom Davis rich media formulation: anhydrous K2HPO4 (140 g), KH2PO4 (40 g), (NH4)2SO4 (20 g), NaCl (58 g), casamino acids (100 g), Tween-80 (20 mL), L-cysteine (1 g), L-tryptophan (0.5 g), adenine (0.5 g), guanine (0.5 g), cytosine (0.5 g), uracil (0.5 g), CaCl2 (0.5 μM final). This was allowed to cool overnight, and supplemented with a 500 mL filter-sterilized solution of MilliQ water containing: NaHCO3 (16.8g), glucose (90 g), sodium citrate (10 g), MgSO4 (1 g), FeSO4 (56 mg), thiamine (134 mg), calcium pantothenate (94 mg), para-aminobenzoic acid (54 mg), para-hydroxybenzoic acid (54 mg), 2,3-dihydroxybenzoic acid (62 mg), (NH4)6Mo7 (3 nM final), H3BO3 (400 nM final), CoCl2 (30 nM), CuSO4 (10 nM final), MnCl2 (80 nM final), and ZnSO4 (10 nM final). Cultures were supplemented with the appropriate antibiotics in the following final concentrations: carbenicillin (AP; 50 μg/mL), spectinomycin (APneg; 50 μg/mL), chloramphenicol (MP; 40 μg/mL). Streptomycin (for selection of S1030 cells carrying the rpsL marker) and tetracycline (for selection of the F plasmid) were not routinely included in culture media.
Real-time luminescence monitor
The in-line bioluminescence measurements were made by a modified luminometer. A TD-20e luminometer (Turner Designs) was installed inside a dark box in continuous reading mode. While in this mode, the luminometer outputs a DC voltage that varies between 0 and 4 V depending on the light level received by the photomultiplier fitted in the instrument (the range was set to the most sensitive one). This signal was sent to an Arduino Mega (Arduino) prototyping board through the analog input port. The Arduino board was controlled via Matlab (Mathworks), using a custom Graphical User Interface (GUI). Measurements were taken at around 100 Hz and then integrated to 1 s before being written by the software into a text file. To allow for multichannel recordings, the system was fitted with a custom tube holder that could hold up to four flow-through tubes with minimal cross talk between channels. Because the holder allows for only one tube to be observed at a time, it is mounted on a stepper motor that is in turn controlled using the Matlab GUI via the Arduino board and a Pololu A4988 stepper motor driver (Pololu R&E). The system waits until a measurement is complete before rotating the sample holder until the next tube is in position. The cycle repeats until all channels are covered, then unwinds to start a new cycle. The time between measurements of a particular channel is ∼30 s. Data analysis comprised temporal binning and low-pass Fourier filtering of the raw traces.
Assay for infection-competence of recipient cells
Recipient cultures were grown to mid-log phase, and 100 μL of culture was mixed by pipetting with 2 μL of pJC126b phagemid prep (encodes R6K origin, f1 origin, pLac-YFP) totaling ∼109 cfu. Reactions were incubated for 2 min and then 1 μL of the reaction was diluted into 1 mL of fresh 2xYT media. Cells were pelleted, resuspended in 1 mL fresh 2xYT, and an aliquot was diluted 10-fold into fresh media. 20 μL of the resulting media was plated on 2xYT-agar plates with 1 mM IPTG and grown overnight. Colonies were scanned on a Typhoon laser imager (488 nm laser, 520/40 filter).
Discrete assay for phage production
Cultures with candidate drift plasmids contained: pT7 (pJC173b), pTet (pJC148i2), psp (pJC175e), psp-tet (pJC175g/h/k/m/n), psp-tet on MP (pJC184d5), psp-tet on colE1-spect plasmid (pJC184d-Sp). Cultures were grown to mid-log phase in Davis rich media and infected with an excess of SP-T7 for 10 min at 37°C. Cells were pelleted and washed to remove residual excess phage. Cells were re-inoculated into fresh Davis rich media and grown to OD600 ∼0.8, pelleted, and supernatants were saved for titering by plaque assay on S1030 E. coli cells carrying a PT7-gene III AP (pJC173b).. OD600 values were measured to normalize phage titers.
Cultures with candidate negative selection plasmids contained PTet-gene III (pJC148i2) and either Plac-C-domain (pJC156a2), Plac-C83 (pJC156c2), Plac-N1-N2-C83 (pJC156j2), Plac-N2-C83 (pJC156m2), or Plac-N1*-N2-C83 (pJC156o2). Cells were treated with SP-T7 phage as described above and then cultured in 2xYT media with either 4 ng/mL or 20 ng/mL ATc and in the presence or absence of 2 mM IPTG. Phage titers were assayed as described above.
Cultures for theophylline-dependent negative selection contained PT7-gene III (pJC173c) and PT3-theophylline riboswitch-gene III-neg (pJC174c-R5). Cells were treated with either SP-T7192.2 or SP-T748.3 and then cultured with varying concentrations of theophylline. Phage titers were assayed as described above.
Continuous flow experiment of PTet-gene III drift cassette
A chemostat culture of S1030 cells carrying an AP encoding PTet-gene III (pJC148i2) was prepared and growth was equilibrated overnight at a flow rate of 400 mL/h in a chemostat volume of 250 mL. The next morning, two lagoons (40 mL each, flow rate from the chemostat of 100 mL/h) were seeded with 107 pfu of SP-T7. Lagoons received supplements delivered by syringe pump (flow rate 1 mL/h) consisting of either 20 μg/mL ATc (resulting in 200 ng/mL final concentration) or water only. At 4-hour intervals, 0.6-mL samples were taken from lagoons, measured discretely for luminescence, and centrifuged to remove cells. Supernatants were combined with an equal volume of 50% glycerol and stored in the freezer. Phage titers of supernatants were measured on S1030 cells containing an AP encoding PT7-gene III (pJC173b).
Continuous flow experiment of Ppsp-gene III drift cassette
A chemostat culture of S1030 cells carrying an AP encoding Ppsp-gene III (pJC175e) was prepared and equilibrated overnight. The flow rate was 400 mL/h in a chemostat volume of 250 mL. The next morning, a lagoon (40 mL each, flow rate 100 mL/h) was started and seeded with 107 pfu of SP-T7WT. At 4-hour intervals, samples were taken and processed as described above.
Continuous flow experiment of Ppsp-tet-gene III drift cassette
A chemostat culture of S1030 cells carrying plasmid Ppsp-tet-gene III (pJC175h) was prepared equilibrated overnight. The flow rate was 400 mL/h in a chemostat volume of 250 mL. The next morning, two lagoons (40 mL each, flow rate 100 mL/h) were started and seeded with 107 pfu of SP-T7WT. Lagoons received supplements delivered by syringe pump (flow rate 1 mL/h) consisting of either 20 μg/mL ATc (resulting in 200 ng/mL final concentration) or water only. At 4-hour intervals, samples were taken and processed as described above.
Continuous flow experiment of Ppsp-tet-gene III drift cassette with ATc dose-dependent stringency
A chemostat culture of S1030 cells carrying a DP encoding Ppsp-tet-gene III with a mutagenesis cassette (pJC184d5) and an AP encoding PT7-6xHisTag-gene III (pJC173c) was prepared and equilibrated overnight. The flow rate was 400 mL/h in a chemostat volume of 250 mL. The next morning, three lagoons (40 mL each, flow rate 100 mL/h) were started, with their waste lines diverted through the in-line luminescence monitor. Lagoons were seeded with a mixture containing 109 pfu SP-T7Dead (D812G) and 107 pfu SP-T7WT, along with ATc in an amount that brought the 40 mL lagoon to the target final ATc concentration for each lagoon. Lagoons received supplements delivered by syringe pump (flow rate 1 mL/h) consisting of either 15 μg/mL ATc (to make 150 ng/mL final concentration), 40 μg/mL ATc (to make 400 ng/mL final concentration), or no ATc (water only). At t = 8 h, the lagoon receiving 40 μg/mL ATc was switched to receive water only. At timepoints t = 2, 4, 6, 8, and 16 h, 0.6-mL samples were taken from the lagoon, measured individually for luminescence, and centrifuged to remove cells. Supernatants were combined with an equal volume of 50% glycerol and stored in the freezer.
Supernatants were analyzed for relative phage ratios using a PCR and analytical restriction digestion. 1 μL of each supernatant was added to a 20 μL qPCR reaction (iQ SYBR Green Mastermix, Biorad) with primers JC1480 (5′-TCCAGCACTTCTCCGCGATGCTC-3′) and JC1481 (5′-GAAGTCCGACTCTAAGATGT CACGGAGGTTCAAG-3′) and amplified by PCR with SYBR Green fluorescence monitored following each amplification cycle. Samples were removed individually from the PCR block after exceeding a pre-determined fluorescence threshold, and placed on ice. Samples were then quantitated again by SYBR Green fluorescence in a plate reader, and approximately equal amounts of DNA from the reactions (normalized based on the fluorescence readings) were added to restriction digestion reactions. The digestion reactions used enzymes EcoRV and HindIII in buffer NEB2 with BSA and were performed following the manufacturer's instructions in 20 μL total volumes. Digestions were inactivated with heat, combined with 0.2 volumes of 5× loading dye containing Orange G and xylene cyanol, and analyzed by agarose gel electrophoresis (1% agarose, 0.5× TBE, 120V, 80 min). The electrophoresed gel was stained with SYBR Gold (Invitrogen) and imaged on a Typhoon laser scanner (excitation 488, emission 520/40 nm).
Continuous flow experiment of dose-dependent negative selection
A chemostat of S1030 host cells containing an AP encoding PT7-gene III (pJC173c) and an APneg encoding PT3-theophylline riboswitch-gene III-neg (pJC174c-R5) was prepared and equilibrated overnight at a flow rate of 400 mL/h in a chemostat volume of 250 mL. The next morning, three lagoons (40 mL each, flow rate 100 mL/h) were started, with their waste lines diverted through the in-line luminescence monitor. Lagoons were seeded with a mixture containing 1010 pfu SP-T7Prom and 104 pfu SP-T7Spec and allowed to equilibrate for 4.5 h; during this time the lagoons received 0.1 M NaOH at 0.5 mL/h by syringe pump to equilibrate the lagoons to the pH of the theophylline vehicle. After equilibration (t = 0 h), lagoons received supplements delivered by syringe pump (flow rate 0.5 mL/h) consisting of either 200 mM theophylline (to make 1 mM final concentration), 15 mM theophylline (to make 75 μM final concentration), or vehicle only (0.1 M NaOH). After 16 h, the lagoon receiving vehicle only was switched to receive 200 mM theophylline (1 mM final concentration). At timepoints t = 0, 2, 4, 6, 8, 10, 12, and 20 h, 0.6 mL-samples were taken from the lagoon, measured discretely for luminescence, and centrifuged to remove cells. Supernatants were combined with an equal volume of 50% glycerol and stored in the freezer.
Relative phage ratios in supernatant samples were assayed using a PCR and analytical restriction digestion. 1 μL of each supernatant was added to a 20 μL qPCR reaction (iQ SYBR Green Mastermix, Biorad) with primers JC1163 (5′-GGAGTACGCTGCATCGAGATGCTCA-3′) and JC1488 (5′-GTAGAAATCAGCCAGTACATCACAAGACTC-3′) and amplified by PCR with SYBR Green fluorescence monitored following each amplification cycle. Samples were removed individually from the PCR block after exceeding a pre-determined fluorescence threshold, then placed on ice. Samples were quantitated again by SYBR Green fluorescence in a plate reader, and approximately equal amounts of DNA from the reactions (normalized based on the fluorescence readings) were added to restriction digestion reactions. The digestion reactions used AvaII in buffer NEB4 and were performed according to manufacturer instructions in 20-μL total volumes. Digestions were combined with 0.2 volumes of 5× loading dye containing Orange G and xylene cyanol, and analyzed by agarose gel electrophoresis (1% agarose, 0.5× TBE, 120V, 80 min). The electrophoresed gel was stained with SYBR Gold (Invitrogen) and imaged on a Typhoon laser scanner (excitation 488, emission 520/40 nm).
Continuous evolution of T3-selective RNA polymerases
A chemostat culture of S1030 cells carrying a DP encoding Ppsp-tet-gene III with a mutagenesis cassette (pJC184d5) and an AP encoding PT3-gene III (pJC174f) was prepared and equilibrated overnight at a flow rate of 400 mL/h in a chemostat volume of 250 mL. The next morning, two lagoons (50 mL each, flow rate 100 mL/h) were started, with their waste lines diverted through the in-line luminescence monitor.
Lagoons were seeded with 105 pfu of SP-T7WT. Lagoons received supplements delivered by syringe pump (flow rate 1 mL/h) consisting of either 20 μM ATc (in 500 mM L-arabinose) for lagoon 1 or vehicle alone (500 mM L-arabinose) for lagoon 2. At t = 12 h, lagoon 1's supplement was changed to 2 μM ATc (in 500 mM L-arabinose). At t = 28 h, the first chemostat was discontinued and both lagoons began receiving cells from a second chemostat containing S1030 cells harboring an MP (pJC184), a PT3-gene III AP (pJC174f), and a PT7-theophylline riboswitch-gene III-neg APneg (pJC173f-R5). Both lagoons received 500 mM L-arabinose. At t = 32 h, half of lagoon 1 was transferred to a new lagoon, lagoon 3. Lagoons 1 and 3 were brought to 40 mL total volumes with culture from the chemostat, and lagoons were equilibrated for 40 min. Lagoons 1 and 2 then received 100 mM theophylline (dissolved in 0.1 M NaOH, flow rate 1 mL/h), while lagoon 3 received vehicle only (0.1 M NaOH). At t = 52 h, the second chemostat was discontinued and all lagoons began receiving cells from a third chemostat containing S1030 cells harboring an MP (pJC184), a reduced RBS PT3-gene III (pJC174k) and an enhanced RBS/copy number PT7-gene III-neg APneg (pJC173g-SD8). All lagoon volumes were reduced to 40 mL each (flow rate maintained at 100 mL/h) and were supplemented with 500 mM L-arabinose. At t = 70.5 h, lagoon volumes were reduced to 30 mL each, while the flow rate was maintained at 100 mL/h.
Periodically, 0.6 mL-samples were taken from the lagoon, measured discretely for luminescence, and centrifuged to remove cells. Supernatants were combined with an equal volume of 50% glycerol and stored in the freezer.
In vivo gene expression measurements on evolved RNA polymerases
Cells for luminescence assays were S1030 cells described above containing APs encoding Ppsp-gene III (pJC184c-rrnB) and either PT7-luxAB (pJC173e4) or PT3-luxAB (pJC174e4). Cells were grown to mid-log phase and 20-μL aliquots were distributed into a deep-well plate. 10 μL of clonal phage aliquots were added to these wells and plates were incubated at 37 °C for 15 min. Wells were supplemented with 500 μL media, grown to mid-log phase, and cells were transferred to a 96-well plate for luminescence measurement.
In vivo gene expression measurements on RNA polymerase forward mutants
Cells for luminescence assays were S1030 cells described above containing an expression plasmid (EP) expressing a subcloned forward mutant T7 RNAP and APs encoding either PT7-luxAB (pJC173e4) or PT3-luxAB (pJC174e4). Cultures were grown overnight and used to seed 500-μL cultures in 96-well blocks that were grown to mid-log phase, after which cells were transferred to a 96-well plate for luminescence measurement.
Supplementary Material
Acknowledgments
The authors thank Kevin Esvelt, David Thompson, Brent Dorr, and Kevin Davis for helpful discussions. This research was supported by DARPA HR0011-11-2-0003, DARPA N66001-12-C-4207, and HHMI. J.C.C. was supported by the Harvard Chemical Biology Graduate Program. A.H.B. was supported by a National Science Foundation Graduate Research Fellowship. D.A.G.-N. was supported by the Harvard Biophysics Graduate Program.
Footnotes
Author Contributions: J.C.C. designed the research, prepared materials, and performed experiments. A.H.B. prepared materials and performed experiments. D.A.G.-N. designed and constructed the real-time luminescence monitor. D.R.L. designed and supervised the research. All authors contributed to the manuscript.
Competing Financial Interests: The authors have filed a provisional patent application on PACE and related improvements.
References
- 1.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson FK, Friedman SM, Smith GP. Filamentous phage DNA cloning vectors: a noninfective mutant with a nonpolar deletion in gene III. Virology. 1981;108:338–350. doi: 10.1016/0042-6822(81)90442-6. [DOI] [PubMed] [Google Scholar]
- 3.Calendar R. The bacteriophages. 1st. Oxford Univ. Press; 2006. pp. 376–455. [Google Scholar]
- 4.Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W. Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev. 2006;58:1622–1654. doi: 10.1016/j.addr.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan L, Kurek I, English J, Keenan R. Laboratory-directed protein evolution. Microbiol Mol Biol Rev. 2005;69:373–392. doi: 10.1128/MMBR.69.3.373-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leconte AM, et al. A population-based experimental model for protein evolution: effects of mutation rate and selection stringency on evolutionary outcomes. Biochemistry. 2013;52:1490–1499. doi: 10.1021/bi3016185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci U S A. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc. 2006;128:2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- 9.Tracewell CA, Arnold FH. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol. 2009;13:3–9. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boeke JD, Model P, Zinder ND. Effects of bacteriophage f1 gene III protein on the host cell membrane. Mol Gen Genet. 1982;186:185–192. doi: 10.1007/BF00331849. [DOI] [PubMed] [Google Scholar]
- 11.Brissette JL, Weiner L, Ripmaster TL, Model P. Characterization and sequence of the Escherichia coli stress-induced psp operon. J Mol Biol. 1991;220:35–48. doi: 10.1016/0022-2836(91)90379-k. [DOI] [PubMed] [Google Scholar]
- 12.Brissette JL, Russel M, Weiner L, Model P. Phage shock protein, a stress protein of Escherichia coli. Proc Natl Acad Sci U S A. 1990;87:862–866. doi: 10.1073/pnas.87.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakonjac J, Jovanovic G, Model P. Filamentous phage infection-mediated gene expression: construction and propagation of the gIII deletion mutant helper phage R408d3. Gene. 1997;198:99–103. doi: 10.1016/s0378-1119(97)00298-9. [DOI] [PubMed] [Google Scholar]
- 14.Bonner G, Lafer EM, Sousa R. Characterization of a set of T7 RNA polymerase active site mutants. J Biol Chem. 1994;269:25120–25128. [PubMed] [Google Scholar]
- 15.Rakonjac J, Feng J, Model P. Filamentous phage are released from the bacterial membrane by a two-step mechanism involving a short C-terminal fragment of pIII. J Mol Biol. 1999;289:1253–1265. doi: 10.1006/jmbi.1999.2851. [DOI] [PubMed] [Google Scholar]
- 16.Bennett NJ, Rakonjac J. Unlocking of the filamentous bacteriophage virion during infection is mediated by the C domain of pIII. J Mol Biol. 2006;356:266–273. doi: 10.1016/j.jmb.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 17.Grant RA, Lin TC, Konigsberg W, Webster RE. Structure of the filamentous bacteriophage fl. Location of the A, C, and D minor coat proteins. J Biol Chem. 1981;256:539–546. [PubMed] [Google Scholar]
- 18.Lopez J, Webster RE. Minor coat protein composition and location of the A protein in bacteriophage f1 spheroids and I-forms. J Virol. 1982;42:1099–1107. doi: 10.1128/jvi.42.3.1099-1107.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch SA, Gallivan JP. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009;37:184–192. doi: 10.1093/nar/gkn924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raskin CA, Diaz G, Joho K, McAllister WT. Substitution of a single bacteriophage T3 residue in bacteriophage T7 RNA polymerase at position 748 results in a switch in promoter specificity. J Mol Biol. 1992;228:506–515. doi: 10.1016/0022-2836(92)90838-b. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399:80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 22.Imburgio D, Rong M, Ma K, McAllister WT. Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry. 2000;39:10419–10430. doi: 10.1021/bi000365w. [DOI] [PubMed] [Google Scholar]
- 23.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dove SL, Hochschild A. Conversion of the omega subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 1998;12:745–754. doi: 10.1101/gad.12.5.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xi L, Cho KW, Tu SC. Cloning and nucleotide sequences of lux genes and characterization of luciferase of Xenorhabdus luminescens from a human wound. J Bacteriol. 1991;173:1399–1405. doi: 10.1128/jb.173.4.1399-1405.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammar M, Arnqvist A, Bian Z, Olsen A, Normark S. Expression of two csg operons is required for production of fibronectin- and congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol. 1995;18:661–670. doi: 10.1111/j.1365-2958.1995.mmi_18040661.x.. [DOI] [PubMed] [Google Scholar]
- 27.Danese PN, Pratt LA, Dove SL, Kolter R. The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol Microbiol. 2000;37:424–432. doi: 10.1046/j.1365-2958.2000.02008.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Preston JF, 3rd, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology. 2001;147:3241–3247. doi: 10.1099/00221287-147-12-3241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.