

Abstract
Agrobacterium tumefaciens strain C58 can utilize d-galacturonate as a sole source of carbon via a pathway in which the first step is oxidation of d-galacturonate to d-galactaro-1,5-lactone. We have identified a novel enzyme, d-galactarolactone isomerase (GLI), that catalyzes the isomerizaton of d-galactaro-1,5-lactone to d-galactaro-1,4-lactone. GLI, a member of the functionally diverse amidohydrolase superfamily, is a homologue of LigI that catalyzes the hydrolysis of 2-pyrone-4,6-dicarboxylate in lignin degradation. The ability of GLI to catalyze lactone isomerization instead of hydrolysis can be explained by the absence of the general basic catalysis used by 2-pyrone-4,6-dicarboxylate lactonase.
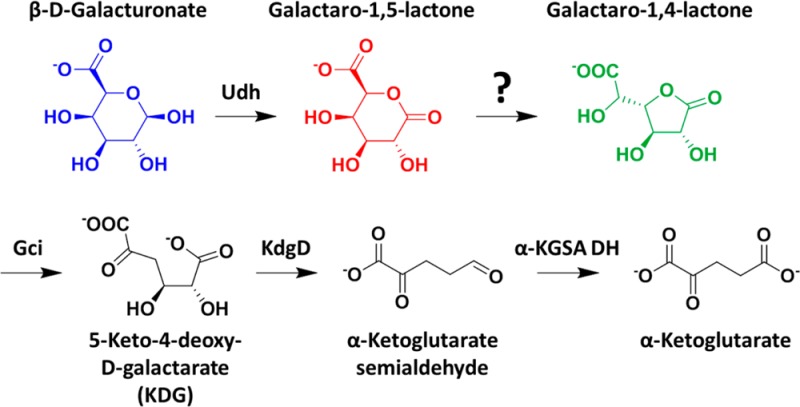
d-Galacturonate, the primary constituent of pectin, is an abundant polymer found in plant cell walls. Following hydrolysis by pectinases, d-galacturonate can be utilized as a sole carbon source by many soil bacteria. Several pathways are known for catabolism of d-galacturonate, including the oxidative pathway shown in Figure 1A.1 Our attention was drawn to this pathway in Agrobacterium tumefaciens strain C58 because it includes a reaction catalyzed by a member of the functionally diverse enolase superfamily.2 This enzyme, designated d-galactarolactone cycloisomerase (Gci, Atu3139), catalyzes the ring opening of d-galactaro-1,4-lactone to yield 2-keto-3-deoxy-l-threo-hexarate [hereafter designated 5-keto-4-deoxy-d-galactarate or 5-keto-4-deoxy-d-glucarate (KDG)] via a β-elimination reaction that is initiated by abstraction of the proton adjacent to the carboxylate group.2
Figure 1.
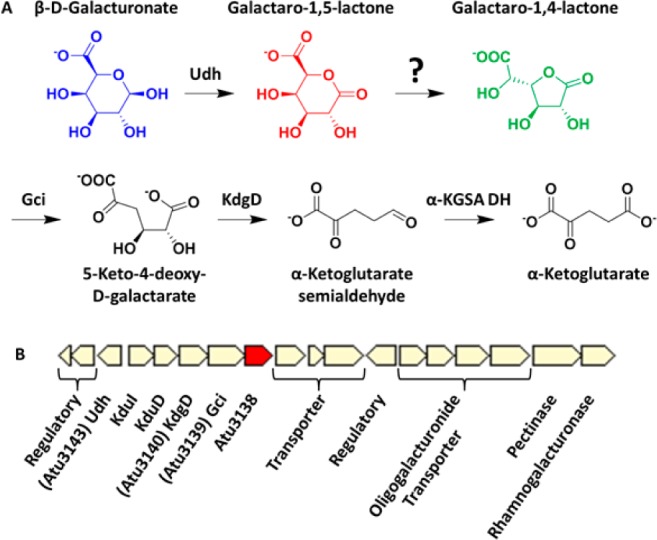
(A) d-Galacturonate oxidative catabolic pathway. (B) Gene cluster.
In this pathway, an NAD+-dependent uronate dehydrogenase (Udh, Atu3143) oxidizes the β-pyranose form of d-galacturonate to d-galactaro-1,5-lactone (δ-lactone). The assumption has been that the δ-lactone nonenzymatically isomerizes to d-galactaro-1,4-lactone (γ-lactone). Following this isomerization, Gci catalyzes the ring opening reaction of the γ-lactone to produce KDG that undergoes dehydration and decarboxylation to form α-ketoglutarate semialdehyde, which is oxidized to α-ketoglutarate, an intermediate in the citric acid cycle.1−4
Atu3139 is located in a gene cluster that includes Atu3138 and Atu3140 (Figure 1B); the transcription of the genes in this cluster is upregulated when d-galacturonate is the carbon source (unpublished results). This gene cluster also encodes orthologues of KduI and KduD that are involved in an alternate d-galacturonate utilization pathway that involves isomerization and reduction to produce 2-keto-3-deoxy-d-gluconate.5 On the basis of the sequence similarity, the closest functionally characterized homologue of the protein encoded by Atu3140 is a KDG dehydratase/decarboxylase (KdgD). Because KDG is the product of the reaction catalyzed by Gci, the proximity of these genes is not surprising. However, the function of the protein encoded by Atu3138 was unknown.
The protein encoded by Atu3138 (UniProt accession number A9CEQ7) is a member of the amidohydrolase superfamily (AHS), a very large functionally diverse enzyme superfamily.6 A9CEQ7 is a member of Pfam PF04909 (6287 sequences in release 27) and InterPro IPR006992 (11876 sequences in release 45.0). 2-Pyrone-4,6-dicarboxylate lactonase (PDC lactonase or LigI) is a structurally and mechanistically characterized member of these families.7
PDC, the substrate for LigI, is structurally similar to d-galactaro-δ-lactone: both are carboxy-substituted δ-lactones. In the pathway shown in Figure 1A, d-galactaro-δ-lactone has been proposed to isomerize to d-galactaro-γ-lactone nonenzymatically. However, on the basis of the proximity of the genes encoding A9CEQ7 and Gci and the structural similarity between PDC and d-galactaro-δ-lactone, we hypothesized that A9CEQ7 catalyzes the δ- to γ-lactone isomerization and therefore is a “missing” enzyme in this pathway. At neutral pH, the nonenzymatic isomerization is fast, so previous investigators may have assumed there would be no need for a catalyst. Also, because Gci catalyzes the ring opening reaction of d-galactaro-γ-lactone to KDG, no need for a lactonase is apparent, i.e., a reaction similar to that catalyzed by LigI. Here we report that A9CEQ7 is a galactaro δ-lactone isomerase (GLI), a novel activity for the AHS.
d-Galactaro-δ-lactone was prepared in D2O via bromine oxidation of d-galacturonate.8 This material could not be distinguished by 1H nuclear magnetic resonance (NMR) spectroscopy from the product obtained by the NAD+-dependent Udh (Atu3143). The synthetic δ-lactone is stable indefinitely at pD 4.8 and −80 °C.
The progress of the A9CEQ7-catalyzed reaction was monitored by 1H NMR spectroscopy (Figure 2). The 1H NMR spectrum of the synthetic δ-lactone at pD 6.4 is shown in Figure 2A; the red bars indicate the δ-lactone, the green bars the γ-lactone, and the blue bars the residual d-galacturonate from the synthesis. The spectra in Figure 2B (recorded at 2 min intervals) show the progress of the reaction. As the reaction proceeds, the intensities of the resonances associated with the δ-lactone decrease as the intensities of the resonances associated with the γ-lactone increase. After 1 h (Figure 2C), the only resonances (in addition to those associated with the residual d-galacturonate) are those associated with the γ-lactone. No meso-galactarate is detected by hydrolysis of either lactone.
Figure 2.
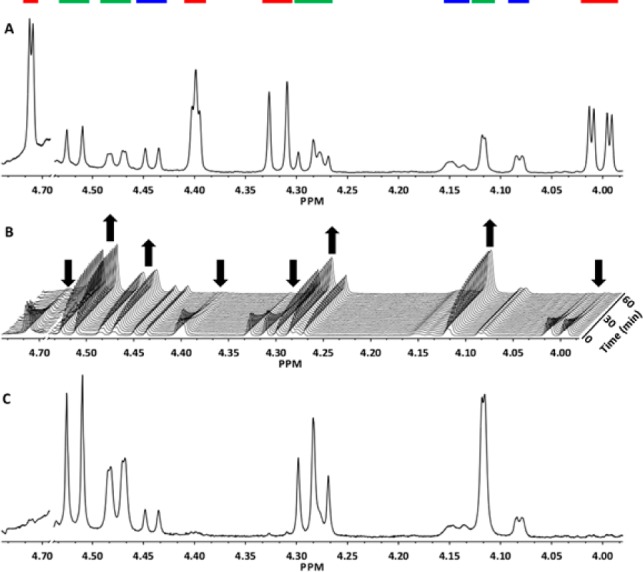
1H NMR spectra of the isomerase substrate and product. (A) Synthetic δ-lactone immediately after the pD had been adjusted from 4.8 to 6.4. (B) Time course of the reaction. (C) Reaction after 1 h. Resonances are color-coded to match the peaks associated with the structures in Figure 1A.
The reaction progress was monitored with a polarimeter. In Figure 3, the black line shows the change in the optical rotation during the nonenzymatic isomerization at pD 6.4; the red line shows the change in the optical rotation during the reaction in the presence of A9CEQ7. The kinetic parameters were measured for the A9CEQ7-catalyzed reaction: kcat = 440 s–1, and kcat/Km = 8.3 × 104 M–1 s–1. The rate enhancement is 6.8 × 105. The rate of the nonenzymatic reaction is dependent on pH, with a pD of 6.4 providing sufficient kinetic stability of the δ-lactone to allow confident rate measurements.
Figure 3.
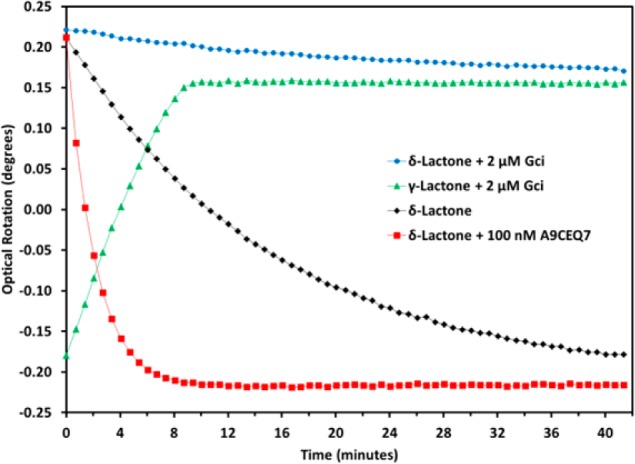
Polarimetric profiles of 5.5 mM d-galactaro-δ-lactone without enzyme (black diamonds), with A9CEQ7 (red squares), and with Gci (blue circles). Profile of 5.5 mM d-galactaro-γ-lactone with Gci (green triangles).
We also investigated the specificity of Gci for the γ- and δ-lactones. In Figure 3, the green line shows the progress of the reaction when Gci is added to γ-lactone produced in the absence of A9CEQ7; the blue line shows the progress of the reaction when Gci is added to a reaction mixture containing the δ-lactone. These reactions reach the same final optical rotation. The rate constants for the nonenzymatic isomerization of the δ-lactone to the γ-lactone (black line) and the Gci-catalyzed production of KDG in the absence of A9CEQ7 (blue line) are the same, establishing that Gci does not catalyze the ring opening of the δ-lactone.
On the basis of these results, we assign the d-galactarolactone isomerase (GLI) function to A9CEQ7. Although several lactonases have been identified in the AHS, this is the first lactone isomerase reaction.
The structure of GLI was determined in the absence of a ligand to 1.6 Å by molecular replacement using an ensemble model of structural homologues from IPR006992. GLI has a typical amidohydrolase fold with a distorted (β/α)8-TIM barrel. The closest structural homologue as determined with PDBeFold9 is LigI from Sphingomonas paucimobilius (Protein Data Bank entry 4DI8) with a root-mean-square deviation (rmsd) of 2.10 Å and a sequence identity of 27% over 253 Cα atoms. Despite the low level of sequence identity and the high rmsd values, LigI and GLI have very similar core TIM-barrel structures (Figure 4A). As expected by the differences in their substrates, LigI is a flat planar dicarboxylate lactone and GLI is a monocarboxylate sugar lactone; most of their differences are localized to the loops at the N-terminal end of the barrel that play a role in substrate recognition. For example, Arg 130 in LigI, which coordinates the distal carboxylate of its substrate, is a proline in GLI (Pro 123).
Figure 4.
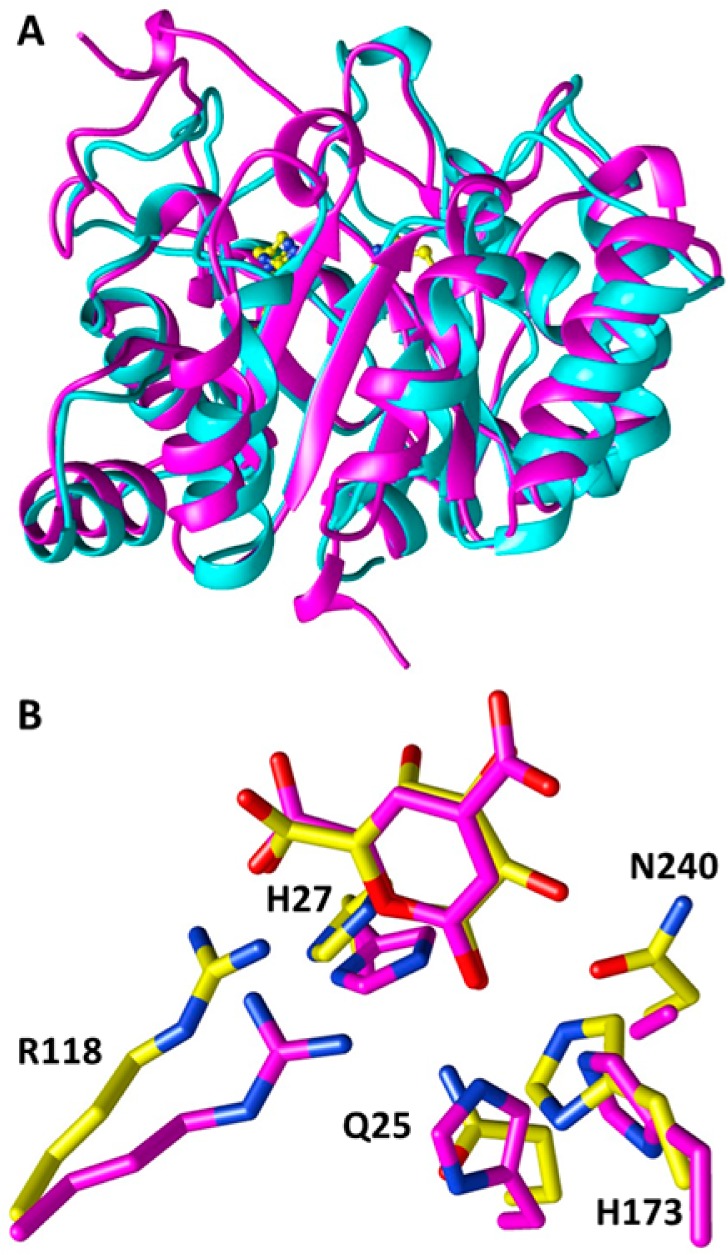
Structure of A9CEQ7. (A) Distorted (β/α)8-TIM barrel. A9CEQ7 (cyan) with supposed catalytic resides (yellow). LigI (4d8L) is colored magenta. (B) Superimposition of catalytic residues with those of LigI and the δ-lactone with PDC. Note an asparagine (N240) has replaced an aspartate involved in activating a water at the end of β-strand 8.
Unlike many members of the amidohydrolase superfamily, LigI does not require a divalent metal for its lactonase activity. The presence of Ca2+, Co2+, Mg2+, Mn2+, Ni2+, or Zn2+ does not increase the rate of the GLI reaction. These results suggest GLI also does not require a metal for catalytic activity.
In LigI, Asp 248 is the general base that activates a water molecule for nucleophilic attack on the lactone carbonyl group.7 A superposition of the active sites of GLI and the PDC-liganded D248A mutant of LigI at pH 8.5 is shown in Figure 4B. The active sites are essentially identical, with the notable exception that in GLI Asn 240 is the structural homologue of Asp 248 in LigI. The N240D substitution produced a 28-fold decrease in activity (kcat = 16 s–1, and kcat/Km = 3.6 × 103 M–1 s–1); no hydrolysis of the δ- or γ-lactone was observed in the presence of the D240N mutant.
Thus, the lactone isomerization reaction catalyzed by GLI can be explained by the lack of a general base for lactone hydrolysis. Inspection of the active site of GLI does not reveal either a general base that would activate the 4-OH group of the δ-lactone for intramolecular attack on the lactone carbonyl group or a nucleophile that would allow the formation of an acyl–enzyme intermediate that would partition between the δ- and γ-lactones. Therefore, in analogy to the proposed mechanism for the nonenzymatic reaction, we assume the modest rate enhancement results from preferential binding of the boat conformer of the δ-lactone to enforce the proximity of the 4-OH group and the lactone carbonyl group (Figure 5).
Figure 5.
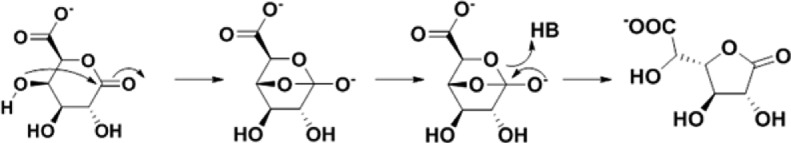
Proposed mechanism for A9CEQ7.
Supporting Information Available
A description of the experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors for GLI have been deposited as Protein Data Bank entry 4MUP.
Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the DOE under Contract DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Co., which operates the facility. This research was supported by National Institutes of Health Grants P01GM071790 and U54GM093342 to J.A.G.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Richard P.; Hilditch S. (2009) Appl. Microbiol. Biotechnol. 82, 597–604. [DOI] [PubMed] [Google Scholar]
- Andberg M.; Maaheimo H.; Boer H.; Penttilä M.; Koivula A.; Richard P. (2012) J. Biol. Chem. 287, 17662–17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer H.; Maaheimo H.; Koivula A.; Penttilä M.; Richard P. (2010) Appl. Microbiol. Biotechnol. 86, 901–909. [DOI] [PubMed] [Google Scholar]
- Parkkinen T.; Boer H.; Jänis J.; Andberg M.; Penttilä M.; Koivula A.; Rouvinen J. (2011) J. Biol. Chem. 286, 27294–27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condemine G.; Robert-Baudouy J. (1991) Mol. Microbiol. 5, 2191–2202. [DOI] [PubMed] [Google Scholar]
- Seibert C. M.; Raushel F. M. (2005) Biochemistry 44, 6383–6391. [DOI] [PubMed] [Google Scholar]
- Hobbs M. E.; Malashkevich V.; Williams H. J.; Xu C.; Sauder J. M.; Burley S. K.; Almo S. C.; Raushel F. M. (2012) Biochemistry 51, 3497–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbell H. S.; Frush H. L. (1943) J. Res. NBS 31, 33–44. [Google Scholar]
- Krissinel E.; Henrick K. (2004) Acta Crystallogr. D60, 2256–2268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.