

Abstract
5′-Methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs) are bacterial enzymes that catalyze hydrolysis of the N-ribosidic bonds of 5′-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to form adenine and 5-thioribosyl groups. MTANs are involved in AI-1 and AI-2 bacterial quorum sensing and the unusual futalosine-based menaquinone synthetic pathway in Streptomyces,Helicobacter, and Campylobacter species. Crystal structures show MTANs to be homodimers with two catalytic sites near the dimer interface. Here, we explore the cooperative ligand interactions in the homodimer of Staphylococcus aureus MTAN (SaMTAN). Kinetic analysis indicated negative catalytic cooperativity. Titration of SaMTAN with the transition-state analogue MT-DADMe-ImmA gave unequal catalytic site binding, consistent with negative binding cooperativity. Thermodynamics of MT-DADMe-ImmA binding also gave negative cooperativity, where the first site had different enthalpic and entropic properties than the second site. Cysteine reactivity in a single-cysteine catalytic site loop construct of SaMTAN is reactive in native enzyme, less reactive when inhibitor is bound to one subunit, and nonreactive upon saturation with inhibitor. A fusion peptide heterodimer construct with one inactive subunit (E173Q) and one native subunit gave 25% of native SaMTAN activity, similar to native SaMTAN with MT-DADMe-ImmA at one catalytic site. Pre-steady-state kinetics showed fast chemistry at one catalytic site, consistent with slow adenine release before catalysis occurs at the second catalytic site. The results support the two catalytic sites acting sequentially, with negative cooperativity and product release being linked to motion of a catalytic site loop contributed by the neighboring subunit.
Bacterial 5′-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) are formed in reactions of the polyamine pathway and in methyl-transfer reactions where S-adenosylmethionine is the methyl donor. One fate of these products is to undergo N-ribosyl bond hydrolysis from the action of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs). The product adenine can be recycled to the adenine nucleotide pool via adenine phosphoribosyltransferase, whereas 5-methylthioribose can be converted to methionine for recycling to S-adenosylmethionine.1 MTA and SAH are product inhibitors of the enzymes that produce them, and MTAN plays a role in reducing their concentration and processing them for recycling. A low concentration of MTA and SAH is achieved in vivo by the catalytic efficiency of the MTANs, with typical values of approximately 107 M–1 s–1 for kcat/Km.2 In addition to the role in S-adenosylmethionine-dependent methyl-transfer reactions and polyamine synthesis, MTANs are also involved in bacterial quorum sensing (QS), which is an important strategy for gene-expression regulation in bacterial populations.
In QS, small molecules called autoinducers (AI) are synthesized and released as cell-to-cell signaling molecules.3 AI-1 are acylated homoserine lactones (AHL) that serve as signaling molecules between bacteria of the same species. A byproduct of AI-1 synthesis is MTA, which is also a product of the polyamine synthesis pathway. Through product inhibition, MTA is an inhibitor of AHL synthase and polyamine synthase. AI-2 is a signaling molecule for QS and is thought to play a role in bacterial communication between different species.1,3S-Ribosylhomocysteine (SRH) is formed by the action of MTANs on SAH and is the precursor for AI-2 synthesis. Inhibition of MTAN causes MTA and SAH accumulation and is proposed to inhibit both AI-1 and AI-2 production (Figure 1). In MTAN knockout strains or in the presence of tight-binding inhibitors of MTAN, production of AI-2 was dramatically reduced.4 Recently, MTAN has also been recognized as essential for QS-independent virulent factors in Neisseria meningitides and Staphylococcus aureus.5,6 In other bacterial species, MTAN is essential in an alternative menaquinone synthetic pathway involving futolosine.7−9 Because MTAN is not expressed in humans, it provides a potential target for antibacterial drug design both in QS signaling and in atypical menaquinone synthesis.
Figure 1.

MTAN in quorum sensing (top) and menaquinone synthesis (bottom).
The subunit protein fold of MTAN belongs to the same family as human purine nucleoside phosphorylase (PNP; 6-oxypurine nucleoside + phosphate → 6-oxypurine + α-d-ribose 1-phosphate).10,11 PNP is a trimeric enzyme, and filling one of its three monomer catalytic sites with a tight-binding transition-state analogue is sufficient to cause complete inhibition.12 This result is interpreted as the three sites acting sequentially, and interruption of the catalytic site cycling by a tight-binding inhibitor at one site causes full inhibition. However, in the crystal structure, all three sites can be saturated with inhibitor to give human PNP as a symmetrical trimer with the three active sites located at the interfaces of adjacent subunits.13 Another member of this family is human 5′-methylthioadenosine phosphorylase (MTAP; 5′-methylthioadenosine + phosphate → adenine +5-methylthio-α-d-ribose 1-phosphate). This trimeric structure is similar to PNP, but the three catalytic sites are independent such that filling one of the catalytic sites with a tight-binding inhibitor leaves the other two fully active. Thus, filling one or two of the three sites with inhibitor causes 1/3 or 2/3 inhibition of full catalytic activity.14
In the structurally related trimeric PNP and MTAN enzymes, the patterns of catalytic site cooperativity differ substantially. A similar analysis has not been reported for the related MTAN dimeric enzyme family. Here, we establish cooperativity and investigate the possibility of oligomerization changes by covalent coupling of the subunits. We find no evidence for subunit dissociation or aggregation. A functional dimer is consistent with the crystal structures of MTANs. Subunit interfaces reveal substantial interactions in the apoenzyme and increased interactions when catalytic site ligands are bound.4,15,16 Here, we characterize the interactions of SaMTAN dimer catalytic site interactions using kinetics, chemical reactivity, and transition-state analogue inhibitors as probes.
Approximately 30 crystal structures of MTANs from several bacterial and plant species are available in the PDB. All are homodimers with the active sites near the dimer interfaces.4,15,16 In the closed complexes with the catalytic sites filled, the catalytic site cavities are covered by a loop from the adjacent monomer. The center of these loops contains a phenylalanine that is located over the filled active site on the adjacent subunit (Phe104 in SaMTAN). The loop has been proposed to isolate the catalytic site from bulk solvent, and this loop also interacts with the 5′-methylthio group or homocysteine group of reactants. Finally, it serves as a potential link for cooperative communication between subunits.
On the basis of kinetic isotope effects and computational chemistry, MTAN transition-state structures are ribocationic at C1′ with a neutral, N7-protonated adenine as the leaving group.17,18 MTANs also stabilize a specific water molecule adjacent to C1′ as the incipient nucleophilic group.19 The chemical reactivity of ribocations is extremely high, requiring critical positioning of C1′ and the water molecule to prevent reaction of the ribocation with nearby protein nucleophiles. The loop is likely to play a role in stabilizing the reactants in a geometric position that permits them to reach the transition state with the appropriate water–ribocation geometry.
Isothermal titration calorimetry (ITC) studies suggested that dimeric MTANs from Escherichia coli, Salmonella enterica, and Vibrio cholerae all demonstrate negative cooperativity for inhibitor binding at the first and second sites.20 However, the role of catalytic site cooperativity was not extended to the catalytic function with normal substrates. In this study, we show that the monomers of S. aureus MTAN (SaMTAN) have negative cooperativity upon catalytic site filling and this cooperativity extends to the catalytic function of SaMTAN.
Materials and Methods
Chemicals
MT-DADMe-ImmA was synthesized as previously described and generously provided by Dr. Gary B. Evans of the Carbohydrate Chemistry Team, Ferrier Institute, Victoria University of Wellington, Lower Hutt, New Zealand. Xanthine oxidase (Grade III) and 5′-deoxy-5′-methylthioadenosine (MTA) were purchased from Sigma-Aldrich (Saint Louis, MO). 5,5′-Dithiobis-2-nitrobenzoic acid (DTNB) was purchased from Sigma-Aldrich (Milwaukee, WI). All other chemicals were purchased at the highest purity commercially available and used without further purification.
Enzyme Purification
Native and mutant SaMTANs were purified as described earlier.2,19,20 In brief, a plasmid containing SaMTAN and encoding an N-terminal His6 tag was transformed into BL21 (DE3) E. coli cells. Bacterial cells were grown to an OD of 0.6 at 600 nm before induction by with 1 mM IPTG. Cells were harvested by centrifugation after 20 h of additional growth at 20 °C. Cells from 6 L of culture were broken by sonication, and the soluble portion was harvested after centrifugation. The sample was applied to a 20 mL Ni-NTA column that was pre-equilibrated with 50 mM HEPES (pH 7.6). The column was washed with 200 mL of buffer containing 50 mM HEPES (pH 7.6) and 60 mM imidazole. SaMTAN was eluted from the Ni-NTA column by gravity flow with 50 mL of buffer containing 50 mM HEPES (pH 7.6) and 250 mM imidazole. The protein was concentrated to ∼10 mL and dialyzed against 100 mL of buffer containing 50 mM HEPES (pH 7.2), 100 mM NaCl, and 1% (w/v) charcoal with three changes. The protein co-purifies with tightly bound adenine, and the charcoal step is required to remove this adenine. The protein was further purified using a Superdex 200 (26/60) gel-filtration column. The purified SaMTAN reaches >95% purity by this method, and the protein has no bound adenine. The concentration of SaMTAN was determined by adsorption at 280 nm, and the extinction coefficient was determined by the ProtParam program (corrected using the Pace method).
Kinetic Assays and Inhibitor Titration
Kinetic assays for inhibitor interactions were performed at 100 μM MTA in 50 mM HEPES, pH 7.2, and the formation of adenine was monitored by the absorption decrease at 274 nm. The MTAN catalytic activity was also detected in continuous assays at 25 °C by monitoring the conversion of adenine to ATP and detection by firefly luciferase (PerkinElmer) in 50 μL reaction mixtures using an assay based on published methods.21,22 Differences included addition of PPDK (pyruvate phosphate dikinase) and APRTase (adenine phosphoribosytransferase) to 780 and 310 μU per well, respectively. MTAN was added to a final concentration of 0.02 to 0.3 nM. MTA concentrations ranged from 0.013 to 4.0 μM. Direct comparisons of catalytic rates from the 274 nm direct kinetic assay and the luciferase assay gave relative kcat values of 10.2 and 1.8, respectively, because of the buffer and coupling reagents used in the luciferase assay. All kinetic results are normalized to the rates for the direct kinetic assay.
The concentration of MT-DADMe-ImmA was determined by the absorption at 275 nm with an extinction coefficient of 8500 M–1 cm–1. SaMTAN was incubated with MT-DADMe-ImmA for 30 min with an enzyme concentration of 50 μM prior to activity assay. Assays were initiated by adding 3.5 μL of the enzyme–inhibitor solution to 1 mL assay solutions. The rate progression curves were linear, indicating no inhibitor loss from the SaMTAN complex during the assay. Data were fitted using KaleidaGraph.
Isothermal Titration Calorimetry Studies
The binding of MT-DADMe-ImmA (600 μM) to SaMTAN (40 μM) was measured with a VP-ITC microcalorimeter (MicroCal) at 25 °C. The inhibitor and enzyme were in the same buffer containing 30 mM sodium phosphate (pH 7.2) and 100 mM NaCl. Other experimental details were as described previously.20 Titration of MT-DADMe-ImmA into enzyme-free buffer was measured and used as the control. No significant heat signal was observed. Data were fitted to the two sets of sites model according to the user’s manual for the VP-ITC microcalorimeter, where a Kd value of 1.4 nM was fixed for the first site.
Mutagenesis and Cloning
Covalently linked heterodimer or covalently linked native dimers of SaMTAN were generated by PCR amplification of the N-terminal encoding region using primer pair 1 (forward: TTTCTAGAAGGAGGTAAAACATATG and reverse: CACCTTCGGATCCACCTCCCTCGGAACCTCCAGCGCCCGAGGCCAATTGGCTAACCAGAGCCTCG) and PCR amplification of the C-terminal encoding region using primer pair 2 (forward: TTCCGAGGGAGGTGGATCCGAAGGTGGAACCTCGGGAGCTACAATGATCGGCATTATTGGTGCTAT and reverse: GGCTCGAGTTATCACAATTGGC). The linear DNAs from both PCR reactions were mixed and joined by ligation. The resulting DNA was digested using XbaI and XhoI and incorporated into expression vector pj214 from DNA2.0.
DTNB Labeling
MT-DADMe-ImmA and F104C/C181S SaMTAN were incubated for 30 min at stoichiometries of 0, 0.2, 0.5, 0.8, and 1.5 per enzyme subunit (catalytic site), respectively. Reaction mixtures were equilibrated at 25 °C, and reactions were initiated by adding 2 mM DTNB in 50 mM HEPES, pH 7.2, to cuvettes to a final concentration of 300 μM DTNB and 23 μM F104C/C181S SaMTAN. Reactions were monitored continuously in a CARY 300 spectrophotometer with cell changers to follow the absorbance increase at 412 nm.
Rapid Chemical-Quench Experiments
Rapid chemical-quench experiments were carried out using an apparatus from KinTek (Model RQF-3) whose temperature was maintained by circulating ice water (∼ 0 °C). The reaction was initiated by rapid mixing of SaMTAN (0.1 mM) and MTA (1 mM). After appropriate delay times, the SaMTAN reaction was quenched using 5 M HCl from a third syringe, and product (adenine) formation was quantitated by UPLC calibrated to a standard curve. Using KaleidaGraph (Synergy Software), the rapid-burst phase for formation of adenine was fitted to single exponential (kburst), and the linear portion of the curve was fitted to a straight line (klinear), yielding values of 52 ± 18 and 1.2 ± 0.2 s–1, respectively.
Solvent Viscosity Studies
Solvent viscosity effects were performed using glycerol or sucrose as the viscogens. To achieve relative viscosity values of 1, 2, 3 and 4, the concentrations of glycerol were 0, 24, 35, and 42% (w/w), and for sucrose, the concentrations were 0, 21, 29, and 34% (w/w).
Results and Discussion
Steady-State Kinetic Analysis of SaMTAN
Kinetic analysis of SaMTAN saturation with MTA was unusual in giving a negative cooperativity (Hill plot = 0.9, not shown) and high apparent affinity (nanomolar) for substrate saturation (Figure 2). The high affinity for MTA required use of a sensitive luciferase assay for adenine quantitation.21,22 Substrate saturation curves did not conform to the Michaelis–Menten equation (v = kcatA/(Km + A)) or to the equation for two catalytic sites acting independently with distinct kinetic constants (v = (kcat1A/(Km1 + A)) + (kcat2A/(Km2 + A))). The data conformed to a kinetic mechanism where the first site to fill is active but when both sites are filled, distinct kinetic parameters are observed according to the expression first named by W. W. Cleland as a “2/1” inhibitor function23 and later adapted to cooperative multiple catalytic sites in the form v = kcat2((A2 + kcat1Km2A/kcat2))/(A2 + Km2A + Km1Km2).24,25 In this equation, v is the initial reaction rate, A is substrate concentration, kcat2 is the rate of product formation from saturated enzyme (EA2), kcat1 is the rate of product formation from the enzyme with one site filled (EA1), Km2 is the Michaelis constant for the filling the second substrate site, and Km1 is the Michaelis constant for filling the first binding site. Least-squares fits of the data to the 2/1 equation gave kcat2/kcat1 = 4.3, Km1 = 0.1 μM, and Km2 = 0.9 μM. The rate of product formation from the EA2 complex (kcat2) is 10.2 s–1, and from the EA1 complex (kcat1), is 2.4 s–1 (1.8 and 0.42 s–1 in the luciferase assay, Figure 2, see Materials and Methods). We sought to validate the steady-state kinetic analysis experimentally by direct physical chemistry measurements, including residual catalytic site activity as one site is filled with a transition-state analogue.
Figure 2.

Catalytic activity of SaMTAN dimer as a function of MTA using the coupled assay for detection of adenine. The line is the best fit to the experimental points for the 2/1 equation described in the text. There are 21 experimental rate measurements in the fitted curve, partially obscured by overlap. The wide substrate range is needed to define the saturation of both catalytic sites. The kinetic constants from the fit are indicated in the inset.
Titration of SaMTAN with Tight-Binding Inhibitor
Purified apo-SaMTAN was titrated with MT-DADMe-ImmA, a transition-state analogue with a dissociation constant of 1.4 nM (Figure 3).2 MT-DADMe-ImmA is a close analogue of the transition state for hydrolysis of MTA by MTANs with a 5′-methylthioribocationic center and a N7-protonated adenine leaving group. The enzyme (at a concentration of 50 μM) was incubated with the inhibitor for >10 min, and samples were taken to initiate steady-state assays. The assay mixtures contained relatively high enzyme concentrations (100–500 nM) to prevent fractional dissociation of the inhibitor during assays. With a Ki value of 1.4 nM, less than 1% of the MT-DADMe-ImmA is expected to dissociate from SaMTAN during the 0.5 to 2 min assay time. This assumption was supported by linear initial reaction rates in a direct recording spectroscopic assay. MTAN catalytic activity decreased as a linear function of inhibitor concentration until saturation of the first catalytic site was achieved. With one site filled, 80% of the total catalytic activity was lost (Figure 3). Titration of the second site led to complete inhibition of catalytic activity at a stoichiometry of 1:1 of inhibitor to catalytic sites. Although both subunits have high affinity for the inhibitor relative to enzyme and inhibitor concentrations in the titration, binding to the second site has lower affinity than to the first. In the crystal structure, SaMTAN is a symmetric dimer. Thus, the decreased inhibitor affinity of the second site as the first catalytic site is filled can be attributed to negative cooperativity and can be correlated with catalytic site activity. When one site only is filled with substrate (EA1), catalysis is 1/4.3 of the rate from EA2. When one site is filled with inhibitor, the IEA1 complex gives rise to product at 1/5 of the rate from EA2. Another test of negative cooperativity is direct analysis of the thermodynamic parameters for catalytic sites in ITC titration experiments.
Figure 3.

Residual catalytic activity (v/vo) as a function of MT-DADMe-ImmA titration of SaMTAN. Symbols v and v0 are reaction rates in the presence and absence of inhibitor, respectively. Approximately 80% inhibition occurs when the first site is filled, and complete inhibition occurs as the second site is filled. The abscissa is the ratio of MT-DADMe-ImmA to SaMTAN dimer. The line is a linear fit to the experimental points as the MT-DADMe-ImmA/SaMTAN ratio increases to 1.0.
Site-Specific Thermodynamics of Tight-Binding Inhibitor
Thermodynamic features of the two catalytic sites were resolved by isothermal titration calorimeter (ITC) experiments for SaMTAN binding to MT-DADMe-ImmA at 25 °C (Figure 4). ITC technology has a lower limit of ∼10 nM for Kd values to permit full thermodynamic analysis by titration.26 The kinetically determined Ki value is 1.4 nM for MT-DADMe-ImmA binding; therefore, active-site titration is near-stoichiometric with little accumulation of free ligand. The ITC data used the kinetically determined Kd value (1.4 nM) as a fixed constant into the fitting algorithm for binding to the first site of the SaMTAN dimer. The Kd value of the second site was a variable and was fitted from the titration data to give a value of 9 nM. Thus, affinity of MT-DADMe-ImmA to the second subunit decreased 6-fold relative to the first site. Binding of MT-DADMe-ImmA to the first site yielded a ΔH of −10.1 kcal/mol and a −TΔS of −1.9 kcal/mol (ΔG = −12 kcal/mol), and binding to the second subunit yielded a ΔH of −5.3 kcal/mol and a −TΔS of −5.7 kcal/mol (ΔG = −11.0 kcal/mol). The first catalytic site has a more favorable enthalpy by 4.8 kcal/mol and less favorable entropic change by 3.8 kcal/mol than the second site. Enthalpic contributions are usually attributed to the formation of hydrogen bond or ionic interactions from the ligand binding, and the contribution of entropy are attributed to dynamic components, water exclusion, or hydrophobic differences. Thus, the second-site thermodynamic differences can be viewed as structural, dynamic, and hydrophobic rearrangements around the unbound second subunit when the first subunit is occupied. Although the affinity of SaMTAN for MTA and MT-DADMe-ImmA are very different, it is significant that the ratio of Km2/Km1 = 9 for substrate interaction is similar to the Kd2/Kd1 value of 6.4 found for MT-DADMe-ImmA binding. With these differences in catalytic site reactivity and transition-state analogue binding, we investigated the chemical reactivity of a probe placed near the catalytic sites.
Figure 4.

Isothermal titration of MT-DADMe-ImmA to SaMTAN. The abscissa is the ratio of MT-DADMe-ImmA to SaMTAN dimer.
Chemical Reactivity of Catalytic Site Loops
Phe104 is located on a flexible loop in SaMTAN and functions to cover the entrance of the active site in closed complexes of SaMTAN (Figure 5). The Phe104 position was selected to serve as a reporter of active-site loop accessibility as the catalytic sites are filled. A mutant of SaMTAN containing a single cysteine (F104C/C186S SaMTAN) was created and gave an enzyme with steady-state kinetic properties similar to the native enzyme (Table 1). The kinetic rates of Cys reactivity in F104C/C181S SaMTAN were measured with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) at 25 °C as a function of MT-DADMe-ImmA titration (Figure 5).
Figure 5.

DTNB labeling of C181S/F104C SaMTAN. Top: the active-site entrance is covered by F104, which was replaced with a cysteine for DTNB-labeling study. Bottom: Reaction of C181S/F104C SaMTAN complexes with DTNB. The reaction rates are for the reaction of DTNB with MT-DADMe-ImmA and SaMTAN dimer complexes at ratios of no inhibitor (magenta), 0.4:2.0 (green), 1.0:2.0 (yellow), 1.6:2.0 (blue), and 2.0:2.0 (black). All traces are corrected for the background rate of hydrolysis of DTNB.
Table 1. Kinetic Constants of Native and Engineered Constructs of SaMTAN.
| SaMTAN construct | kcat (s–1) | Km (μM) | kcat/Km (M–1 s–1) |
|---|---|---|---|
| native homodimer | 10.2 ± 0.1 | 0.9 ± 0.3 | 1.13 × 107 |
| F104C/C186S homodimer | 7.2 ± 0.4 | 2.9 ± 1.2 | 2.48 × 106 |
| native fusion homodimer | 8.3 ± 0.2 | 1.3 ± 0.1 | 6.38 × 106 |
| E173Q + native fusion heterodimer | 2.0 ± 0.2 | 0.5 ± 0.2 | 4.0 × 106 |
| E173Q homodimer | <0.1 | not measured | not measured |
With empty catalytic sites, F104C/C181S SaMTAN reacted with DTNB to give a single exponential rate constant of 0.095 ± 0.001 min–1 (Figure 5) and reacted completely in 30 min. The single exponential rate indicated that the catalytic site loops from both subunits share a common chemical environment. When MT-DADMe-ImmA filled both sites, F104C/C181S SaMTAN gave no significant reaction with DTNB, indicating inaccessible C104 loops (Figure 5). The crystal structures of apo and inhibitor-bound E. coli and S. enterica MTANs show that the catalytic site loops can be open when catalytic sites are empty, but binding of transition-state analogues caused highly organized catalytic site loops in inhibited complexes. In S. enterica MTAN, a tyrosine (Tyr107) hydroxyl is in hydrogen bond distance of 5′ extensions of enzyme-bound inhibitors containing 5′ substituent groups capable of hydrogen bonding.27 Thus, the reduced labeling rate constant in F104C/C181S SaMTAN by DTNB is due to the altered conformation and reduced dynamics of the C104 loop opening to expose the reactive cysteine to solvent and DTNB. When one subunit of the dimer was bound to MT-DADMe-ImmA, the labeling rate was slowed to 0.021 ± 0.001 min–1, 22% of that for the apo enzyme. Despite the slowed reaction rate, the extent of Cys reaction is comparable to enzyme without inhibitor bound. Chemical modification of both catalytic site C104 groups is described by a single exponential rate. If the inhibitor remained in one subunit during the Cys labeling experiment, then the C104 loops for both filled and empty sites would react at different rates. The same reaction rate for both catalytic site Cys groups is readily explained by MT-DADMe-ImmA inhibitor release and catalytic site recapture at equilibrium on the time scale of the DTNB reaction (2 h). This shuffling of inhibitor between sites supports the equivalent reaction rates of C104 loops at both catalytic sites. This explanation is consistent with the observed C104 reaction kinetics and is also consistent with the nanomolar dissociation constant for MT-DADMe-ImmA. For a near-diffusion controlled binding on-rate of 107 M–1 s–1 and a dissociation constant of 1.4 nM, the off-rate is 0.014 s–1, 40 times faster than the C104 chemical reactivity rate of 0.021 min–1. Thus, the inhibitor equilibrates between sites on the time scale of a few minutes, whereas the DTNB reaction occurs on the time scale of 2 hours for half-saturated enzyme (Figure 5).
Pre-Steady-State Burst Kinetics for SaMTAN
The kcat2 steady-state rate of MTA hydrolysis by SaMTAN is 10.2 s–1, and we tested if this rate is limited by chemistry and/or site–site interactions. We hypothesized that adenine product release might limit steady-state kinetic rates because the enzyme is isolated with tightly bound adenine (see below). Likewise, the negative cooperativity between sites questions if both sites can react at the same rate in the presteady state. Rapid mixing of SaMTAN with near-saturating MTA followed by chemical quench at 25 °C gave conversion of one catalytic site equivalent of MTA to adenine (half the sites reactivity) within the 3 ms time resolution of the experiment. At 0 °C, it was possible to measure the rapid formation of adenine at one of the two catalytic sites with an observed rate of 52 ± 18 s–1 followed by a steady-state rate of product formation of 1.2 ± 0.2 s–1 (Figure 6). The catalytic burst for the single-site on-enzyme chemistry is 43 times faster than the steady-state rate at 0 °C. Assuming a similar temperature coefficient for steady-state and pre-steady-state rates for extrapolation to 25 °C, the pre-steady-state burst is estimated to be 442 s–1 followed by the steady state rate of 10.2 s–1. The slow steady-state rate is consistent with slow release of product from the first catalytic site being required for catalytic function of the second site. SaMTAN binds adenine tightly and copurifies with 0.4–0.6 mol tightly bound adenine per enzyme dimer. This is removed before experiments (see Materials and Methods). We propose that the slow release of adenine from the first catalytic site is the rate-limiting step in steady-state catalysis. Because only one site reacts in the rapid-burst phase, the result demonstrates sequential catalytic site function. Binding at both sites of MTANs is known to occur from the steady-state kinetic model and the two-site binding with MT-DADMe-ImmA (Figures 2 and 3) and substrate analogues.15 However, only one site is active at a time, and product release from the first site (C104 loop opening), or something with the same rate constant, is required to permit catalysis at the second site. The steady-state cycle suggests that adenine release gives rise to the observed 10.2 s–1 steady-state rate with the sites acting sequentially. If product release is rate limiting, then the catalytic rate would be expected to be sensitive to solvent viscosity.
Figure 6.

Rate-limiting step of SaMTAN. Chemical quench at 0 °C indicated a single subunit burst of 52 s–1 followed by a steady-state rate of 1.2 s–1 (upper panel). The kcat for SaMTAN is decreased as a linear function of viscosity in glycerol (η/ηo) (lower panel).
Viscosity Effects Support Adenine Product Release as the Slow Step
In enzyme systems where rate is governed by diffusion, including product release, microviscogens like glycerol or sucrose are expected to alter the diffusion rate and change the kinetic rate as a linear function of viscosity.28 The results support adenine release as a diffusion-controlled rate-limiting step during steady-state catalysis. Addition of glycerol to increase the viscosity of assay solutions slowed the kcat for SaMTAN as a linear function of viscosity in glycerol (Figure 6). A similar experiment used sucrose as a second microviscogen and gave similar results (not shown).
Chimeric Linked SaMTAN with One Functional Catalytic Site
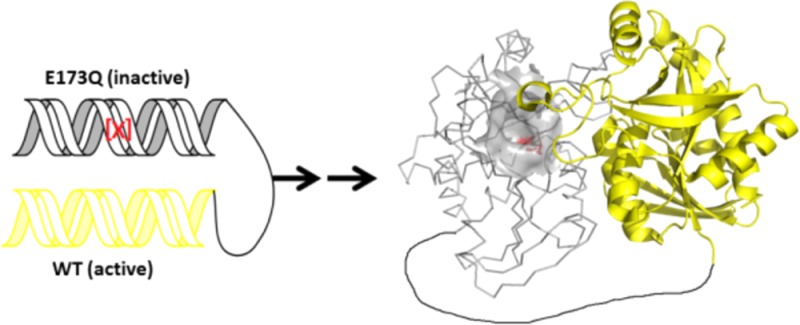
Pre-steady-state kinetics of MT-DADMe-ImmA inhibitor binding at one site and C104 chemical reactivity revealed aspects of negative cooperativity between subunits. We explored the properties of a SaMTAN in which one subunit is rendered catalytically inactive, the second is active, and the subunits are covalently linked. This approach explores one-site reactivity while preventing subunit-dissociation effects (Figure 7).
Figure 7.
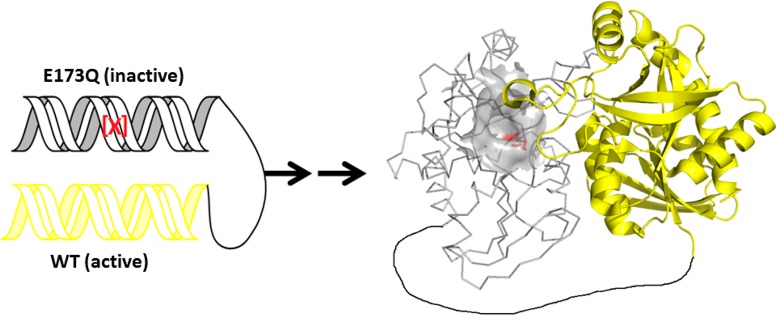
Creation of a covalently linked heterodimer. Gray subunit and yellow subunit are equally active before mutation. After mutation (indicated as red), the gray subunit is catalytically inactive. In the tertiary structure model (right), the active site of the gray subunit is in surface view, and residue 173 and its interacting site are in red. The linker (L20) is in black.
The inactive monomer contained an E173Q mutation, the dimer of which reduced kcat by more than 2 orders of magnitude (Table 1). To ensure active/inactive subunit stoichiometry and to decrease the possibility of subunit dissociation, the subunits were covalently linked by a 20 amino acid linker rich in serine and glycine to maximize linker flexibility (sequence: ASGAGGSEGGGSEGGTSGAT, named L20). The construct was expressed as a fusion protein with the E173Q inactive mutant in the N-terminal peptide and the native subunit in the C-terminal peptide. In the crystal structure of native MTANs, the distance between the N-terminus of one subunit to the C-terminus of the second subunit is 50 Å, and the fully extended length of the linker L20 is 70 Å. As a control, native MTAN monomers linked by L20 were also expressed and characterized. Its catalytic activity was similar to native MTAN (Table 1). Compared with native cross-linked SaMTAN, heterocross-linked-SaMTAN had a comparable Km value, but the value of kcat was 24% of the control enzyme. During catalysis, only the native subunit of the hetero-cross-linked-SaMTAN is active. It lacks the opportunity to alternate catalytic site action because of the inactive E173Q site. Chimeric SaMTAN does not benefit from the cooperative catalytic interaction of the neighboring subunit. Fused-dimer native SaMTAN has a kcat of 8.3 s–1, whereas the hetero-cross-linked-SaMTAN has a kcat of 2.0 s–1 for its functional catalytic site. This is the same value determined for kcat1 in the kinetic analysis (Figure 2). Because of limited expression and stability of this construct, we were unable to complete ITC and pre-steady-state kinetics with this construct.
Correlation of Structures with Function
Sui et al. reported the crystal structure of SaMTAN and compared it to E. coli MTAN, concluding that the proteins exhibited identical modes of inhibitor binding.16 A recent structure of S. enterica MTAN, which shares 53% sequence identity with SaMTAN, shows dramatic structural changes of several regions upon the binding of either substrate or the MT-DADMe-ImmA inhibitor.27 The crystal structure geometry of empty catalytic sites (apoenzyme) in EcMTAN and SeMTAN is open and similar if empty or if adenine-only is bound.15,16 Thus, the resting enzyme with one adenine bound is in a similar open catalytic site geometry to apoenzyme, and the empty second subunit is prepared to bind substrate, to catalyze the reaction, to facilitate adenine departure from the first substrate, and to proceed with substrate binding and chemistry at the first subunit while the second subunit is cleared by the motion of the 104 loop.
In the earlier structure–function analysis of SaMTAN, Sui et al.16 reported a kcat of 0.00973 s–1 for the enzyme using MTA as substrate in an assay oxidizing adenine with xanthine oxidase and coupling the reaction to the reduction of 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyltetrazolium chloride to form formazan with detection at 470 nm. As this chemical rate is 1048-fold slower than we measured for the direct observation of adenine formation by SaMTAN, this widely used formazan assay is perhaps not ideal for kinetic investigation of MTANs. The major findings from the crystal structure of SaMTAN are not affected by this difference,16 but the catalytic differences need to be considered in evaluating catalytic efficiency.
Sequential Mechanism for SaMTAN
Kinetic, binding, thermodynamic, and mutational analysis indicate that both subunits of SaMTAN can function alone or when its neighbor is filled. When both are filled, catalysis occurs sequentially, facilitated by slow product release, presumably by the rate-limiting 104 loop motion to open the catalytic site for adenine release. When only one catalytic site of SaMTAN is filled with substrate, the rate is slower. The high affinity of the first site (0.1 μM) permits the enzyme to scrub the organism of MTA and SAH but at a reduced turnover rate. At higher substrate concentrations (above 1 μM), where the second site is also filled, the enzyme can remove substrates at a substantially higher rate (Figure 8). For the first catalytic turnover with saturated enzyme, only one site reacts at 442 s–1, and catalysis does not occur at the second site until product release occurs from the first site, a slow 10.2 s–1 process. Thus, product release at the first site governs chemistry at the second site.
Figure 8.

Catalytic site chemistry and cooperativity for SaMTAN. Monomers of the dimer are shown in gray and yellow, where S represents the MTA substrate, P represents the products, and In represents the transition-state analogue. In the top reaction sequence, the burst kinetic rates at 25 °C are shown. The enzyme dimers are symmetric; thus, the first ligand to bind can bind to either catalytic site, and in substrate-saturated enzyme, either site can react first, leading to branched pathways. For simplicity of illustration, these statistical paths are not shown. Product release of one subunit is necessary before chemistry can occur at the second subunit.
Conclusions
Structural similarity exists at the level of tertiary protein folds between the dimeric MTANs and the mechanistically related purine nucleoside phosphorylases (PNPs) and methylthioadenosine phosphorylases (MTAPs).10,11 Human PNP and MTAP are trimers with distinct subunit communication. PNP steady-state kinetics are Michaelis–Menten; however, the complete inhibition of the trimer with one site filled with tight-binding inhibitor establishes an obligatory sequential catalytic site function.12 Human MTAP is a closely related structural trimer of the human PNP but has catalytic sites acting independently.14SaMTAN follows neither of the catalytic site interaction patterns of its structural relatives. Instead, dimeric SaMTAN has cooperative sites such that filling one site gives rise to physical and kinetic properties that are different at the catalytic sites when both catalytic sites are filled. SaMTAN shows rapid on-enzyme chemistry at 442 s–1 when exposed to saturating substrate. The rapid reaction is unusual in that it occurs at only one of the two catalytic sites. The second site forms product at 10.2 s–1, proposed to be the rate of product release. Thus, chemistry occurs at the saturated second site only as product is released from the first. This necessarily results in a sequential reaction sequence that alternates between the two sites.
Tight substrate binding and slow product release for dimeric SaMTAN results in an enzyme of high catalytic efficiency with catalytic site cooperativity both for substrate binding and catalytic efficiency. These catalytic site differences are structural, as evidenced by distinct entropic and enthapic components and similar properties in chimeric enzyme and enzyme with one site blocked by tight-binding inhibitors. The kinetic cooperativity described here for SaMTAN may extend to other MTANs, as ultrasensitive kinetic approaches are required to quantitate these catalytic site properties.
Acknowledgments
This work was supported by NIH research grant GM041916.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Parveen N.; Cornell K. A. (2011) Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez J. A.; Luo M.; Singh V.; Li L.; Brown R. L.; Norris G. E.; Evans G. B.; Furneaux R. H.; Tyler P. C.; Painter G. F.; Lenz D. H.; Schramm V. L. (2007) Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem. Biol. 2, 725–734. [DOI] [PubMed] [Google Scholar]
- Bassler B. L.; Losick R. (2006) Bacterially speaking. Cell 125, 237–246. [DOI] [PubMed] [Google Scholar]
- Gutierrez J. A.; Crowder T.; Rinaldo-Matthis A.; Ho M. C.; Almo S. C.; Schramm V. L. (2009) Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 5, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurlier K.; Vendeville A.; Halliday N.; Green A.; Winzer K.; Tang C. M.; Hardie K. R. (2009) Growth deficiencies of Neisseria meningitidis pfs and luxS mutants are not due to inactivation of quorum sensing. J. Bacteriol. 191, 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T.; Zhao L.; Sun B. (2013) LuxS/AI-2 system is involved in antibiotic susceptibility and autolysis in Staphylococcus aureus NCTC 8325. Int. J. Antimicrob. Agents 41, 85–89. [DOI] [PubMed] [Google Scholar]
- Dairi T. (2009) An alternative menaquinone biosynthetic pathway operating in microorganisms: An attractive target for drug discovery to pathogenic Helicobacter and Chlamydia strains. J. Antibiot. 62, 347–352. [DOI] [PubMed] [Google Scholar]
- Li X.; Apel D.; Gaynor E. C.; Tanner M. E. (2011) 5′-Methylthioadenosine nucleosidase is implicated in playing a key role in a modified futalosine pathway for menaquinone biosynthesis in Campylobacter jejuni. J. Biol. Chem. 286, 19392–19398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Haapalainen A. M.; Yan F.; Du Q.; Tyler P. C.; Evans G. B.; Rinaldo-Matthis A.; Brown R. L.; Norris G. E.; Almo S. C.; Schramm V. L. (2012) A picomolar transition state analogue inhibitor of MTAN as a specific antibiotic for Helicobacter pylori. Biochemistry 51, 6892–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Cornell K. A.; Riscoe M. K.; Howell P. L. (2001) Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure 9, 941–953. [DOI] [PubMed] [Google Scholar]
- Pugmire M. J.; Ealick S. E. (2002) Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem. J. 361, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R. W.; Tyler P. C.; Furneaux R. H.; Bagdassarian C. K.; Schramm V. L. (1997) One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 37, 8615–8621. [DOI] [PubMed] [Google Scholar]
- Fedorov A.; Shi W.; Kicska G.; Fedorov E.; Tyler P. C.; Furneaux R. H.; Hanson J. C.; Gainsford G. J.; Larese J. Z.; Schramm V. L.; Almo S. C. (2001) Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry 40, 853–860. [DOI] [PubMed] [Google Scholar]
- Guan R.; Ho M. C.; Brenowitz M.; Tyler P. C.; Evans G. B.; Almo S. C.; Schramm V. L. (2011) Entropy-driven binding of picomolar transition state analogue inhibitors to human 5′-methylthioadenosine phosphorylase. Biochemistry 50, 10408–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Singh V.; Evans G. B.; Tyler P. C.; Furneaux R. H.; Cornell K. A.; Riscoe M. K.; Schramm V. L.; Howell P. L. (2005) Structural rationale for the affinity of pico- and femtomolar transition state analogues of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Biol. Chem. 280, 18274–18282. [DOI] [PubMed] [Google Scholar]
- Siu K. K.; Lee J. E.; Smith G. D.; Horvatin-Mrakovcic C.; Howell P. L. (2008) Structure of Staphylococcus aureus 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 64, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Schramm V. L. (2007) Transition-state analysis of S. pneumoniae 5′-methylthioadenosine nucleosidase. J. Am. Chem. Soc. 129, 2783–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Lee J. E.; Nunez S.; Howell P. L.; Schramm V. L. (2005) Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry 44, 11647–11659. [DOI] [PubMed] [Google Scholar]
- Wang S.; Lim J.; Thomas K.; Yan F.; Angeletti R. H.; Schramm V. L. (2012) A complex of methylthioadenosine/S-adenosylhomocysteine nucleosidase, transition state analogue, and nucleophilic water identified by mass spectrometry. J. Am. Chem. Soc. 134, 1468–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas K.; Haapalainen A. M.; Burgos E. S.; Evans G. B.; Tyler P. C.; Gulab S.; Guan R.; Schramm V. L. (2012) Femtomolar inhibitors bind to 5′-methylthioadenosine nucleosidases with favorable enthalpy and entropy. Biochemistry 51, 7541–7550. [DOI] [PubMed] [Google Scholar]
- Sturm M. B.; Schramm V. L. (2009) Detecting ricin: Sensitive luminescent assay for ricin A-chain ribosome depurination kinetics. Anal. Chem. 81, 2847–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E. S.; Gulab S. A.; Cassera M. B.; Schramm V. L. (2012) Luciferase-based assay for adenosine: Application to S-adenosyl-l-homocysteine hydrolase. Anal. Chem. 84, 3593–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W. W. (1963) The kinetics of enzyme-catalyzed reactions with two or more substrates and products. Biochim. Biophys. Acta 67, 173–187. [DOI] [PubMed] [Google Scholar]
- Kowalik J.; Morrison J. F. (1968) Analysis of kinetic data for allosteric enzyme reactions as a nonlinear regression problem. Math. Biosci. 2, 57–66. [Google Scholar]
- Schramm V. L.; Morrison J. F. (1969) Kinetic studies of the mechanism and allosteric activation of the reaction catalyzed by nucleoside diphosphatase. Biochemistry 8, 3821–3828. [DOI] [PubMed] [Google Scholar]
- Freire E.; Schön A.; Velazquez-Campoy A. (2009) Isothermal titration calorimetry: general formalism using binding polynomials. Methods Enzymol. 455, 127–155. [DOI] [PubMed] [Google Scholar]
- Haapalainen A. M.; Thomas K.; Tyler P. C.; Evans G. B.; Almo S. C.; Schramm V. L. (2013) Salmonella enterica MTAN at 1.36 Å resolution: A structure-based design of tailored transition state analogs. Structure 21, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, pp 251–253, Wiley, New York. [Google Scholar]