

Abstract
Epilepsy refers to a cluster of neurological disease characterized by seizures. While many forms of epilepsy have a well-defined immune etiology, in other forms of epilepsy an altered immune response is only suspected. In general, the hypothesis that inflammation contributes to seizures is supported by experimental results. Additionally, antiepileptic maneuvers may act as immunomodulators and anti-inflammatory therapies can treat seizures. Triggers of seizure include a bidirectional communication between the nervous system and organs of immunity. Thus, a crucial cellular interface protecting from immunological seizures is the blood-brain barrier. Here, we summarize recent advances in the understanding and treatment of epileptic seizures which derive from a non-neurocentric viewpoint and suggest key avenues for future research.
Keywords: inflammation, anti-epileptic drugs, blood-brain barrier, corticosteroids, immunomodulatory axis, vagus nerve stimulation, infection
Glossary
Hyperexcitable and hypersynchronous
Epileptic seizures are characterized by increased neuronal excitability and hypersynchronous activity in the cortical network. The term “hyperexcitable” refers to a neuron or to a neuronal network characterized by an increased probability of firing an action potential or a series of action potentials in response to a stimulus that normally elicits a sub-threshold response or a single spike. Neuronal networks can oscillate between a resting and firing state activity in response to either intrinsic (pacemaker) properties or as a result from activity of many neurons. In individual neurons, oscillations can appear either as oscillations in membrane potential or as rhythmic patterns of action potentials. Synchronized activity of large numbers of neurons occurs during epileptic seizures. The summation of electrical signals from this large assemblies of neurons is the basis of the EEG appearance during a seizure, which is characterized by large amplitude (voltage) signals.
Seizure threshold
This term is used to describe how susceptible one is to seizures at a given time. Both internal and external factors and stimuli contribute towards this threshold. As described in this review, ions, transmitters, inflammatory mediators and body temperature are examples of internal factors that alter the epileptogenic threshold. External stimuli may be sensory, electrical or chemical. These are often used to trigger experimental seizures (kainic acid, electrical stimulation of the amygdala). A complex interaction between external and internal factors explains why precipitating events of comparable potency may or not trigger seizures.
Cryptogenic, idiopathic and symptomatic epilepsy
A cryptogenic or idiopathic disease is a disease with unknown etiology. In the case of epilepsy, these terms refer to patients where no genetic or metabolic disorder is identified and imaging (MRI) of the cortex and hippocampus does not reveal detectable abnormalities. The term symptomatic epilepsy is, by contrast, used to define an epileptic disorder due to a structural or metabolic condition, genetic or acquired, that has been demonstrated to be associated with a substantially increased risk of developing epilepsy. Lesional epilepsy is the antonym of cryptogenic and refers to patients with a distinct abnormality visible in MRI scans.
Interictal, ictal EEG
The EEG associated with epileptic seizures (referred to as “ictal”, from ictus Latin for a stroke or blow) is characterized by an abrupt change of the signal. Focal seizures are typically characterized by the appearance of local low-voltage fast activities progressively replaced by slower quasi-rhythmic activities often spreading to the neighboring regions. Between seizures, the EEG may appear normal or feature interictal epileptic abnormalities (e.g. spikes, sharp waves, slow waves) isolated or in brief discharges.
Ictogenic process, epileptogenesis
Seizures are symptoms of epilepsy, a cluster of neurological diseases. Ictogenesis refers to the events leading to the development of a seizure, including the prodromic features named “auras” and EEG changes that predict seizure onset (“ripples”, “slowing”, etc.). Epileptogenesis refers to the events occurring during the often silent (no seizures) period between an insult (e.g., traumatic brain injury) and the development of a first seizure. The epileptogenic process may last days to years.
Status epilepticus and super-refractory status epilepticus
According to the “Glossary of Descriptive Terminology for Ictal Semiology” of the International League Against Epilepsy (ILAE), the term status epilepticus (SE) refers to a seizure which shows no clinical signs of arresting after a duration encompassing the great majority of seizures of that type in most patients or recurrent seizures without resumption of baseline central nervous system function interictally. The most common SE is generalized tonic-clonic status epilepticus, a potentially fatal condition associated with neuronal injury and respiratory and metabolic dysfunction. Although the ILAE does not define the minimum duration for a seizure to be defined as status, the operational definitions propose to start treatment when seizure activity continues beyond 5 minutes. Refractory SE is defined as SE that has not responded to first-line therapy with a benzodiazepine or second-line therapy, and which requires the application of general anesthesia. Super-refractory status epilepticus is defined as SE that has continued or recurred despite 24 hours of general anesthesia.
Inflammatory reflex
This term refers to the neuronal circuits responsible for the control of systemic inflammation. In analogy to the regulation of heart rate by adrenergic and cholinergic nerves, the inflammatory reflex has a “motor” component that either increases or decreases the activity of systemic inflammatory organs and cells. The inflammatory reflex is regulated by cytokines and other mediators of the immune response. The best understood inflammatory reflex consists of the anti-inflammatory effects of parasympathetic nicotinic synapses on organs such as the spleen. As in many cholinergic systems, opposing effects are achieved by muscarinic receptor activation.
Immunological synapses
In analogy to the chemical synapse in neurons, the immunological synapse refers to the microenvironment hosting the interface between an antigen-presenting cell and a lymphocyte such as an effector T cell or Natural Killer cell. These immune-immune cell interactions are modulated by the presence of closely associated adrenergic or cholinergic nerve terminals.
Seizures and epilepsy
A seizure is a paroxysmal event due to an excessive, hypersynchronous discharge from central nervous system (CNS) neurons or neuronal networks. This abnormal electrical activity causes a range of clinical/behavioral manifestations, ranging from dramatic convulsions often associated with loss of consciousness to experiential phenomena not readily discernible by an observer [1]. The term seizure should be carefully distinguished from epilepsy. Epilepsy is a syndrome of two or more unprovoked or recurrent seizures on more than one occasion. Epilepsy specifically refers to a condition in which a person has recurrent seizures due to a chronic or genetically predetermined underlying process, while seizures are symptoms of epilepsy or standalone manifestations of altered brain function also occurring in non-epileptics (due to drug overdose, alcohol withdrawal, etc.). Epileptic patients oscillate unpredictably between the “ictal” state (seizures present, grossly abnormal electroencephalogram (EEG)) and the “interictal” state (often no clinical symptoms, slight or no EEG changes).
The orthodox view of epilepsy centers on neurons as the main culprit of seizures; targeting of neuronal ion channels, GABA and glutamate receptors has been, for decades, the mainstream pharmacological approach to eradicate seizures. While the ultimate effectors of seizures are neurons, recent advances in experimental neurology have revealed that inflammation can precipitate seizures or sustain seizure activity [2, 3]. Two distinct inflammatory processes have been linked to seizures. Neuroinflammation is present in epileptic brain where it exacerbates seizures or increases their frequency [2, 4]. On the other hand, systemic inflammation can cause epileptiform neuronal discharge via loss of ionic (e.g., potassium [5-7]) and neurotransmitter (e.g., glutamate [7, 8]) homeostasis. While neuroinflammation directly affects neurovascular and glial function, the effects of systemic inflammation are mediated or facilitated by loss of blood-brain barrier (BBB) function [9]. BBB disruption (BBBD) can be triggered by a direct insult to the endothelium [10] or by systemic factors, including activation of circulating [11-15] leukocytes and release of molecular mediators that increase vascular permeability [16, 17].
The discovery of the unexpected role of inflammation in epilepsy has changed our view on what factors contribute to seizures and may help elucidate why, in an epileptic brain, seizures occur rather infrequently and are interspersed by long intervals of relatively normal, “interictal” neuronal activity [11, 12, 18-35]. In other words, an epileptic patient always has an “epileptic brain” but rarely does this brain produce symptoms (seizures). With this in mind, it is not surprising that the etiological mechanisms underlying the development of an epileptic brain are not the same which are involved in the generation of seizures. The epileptic brain phenotype is the consequence of developmental, genetic and molecular factors while the transition from interictal-to-ictal neuronal firing may be due to inflammation-driven changes in the neuronal environment and BBBD [36-38]. The pathophysiological rationale for this hypothesis is as follows: i) “Static” or persistent, inherited or acquired defects such as expression of abnormal ion channels [39] or malformations of cortical development [40] are unlikely triggers of seldom occurring seizures. These pathophysiological features are instead hallmarks of epilepsy and contribute to the epileptic pathology as a whole (i.e., mental retardation, psychiatric comorbidities, etc.); ii) The epileptic brain often displays loss- or gain-of-function mutations, such as loss-of-function mutations in the sodium channel NaV1.1 which cause Severe Myoclonic Epilepsy of Infancy (SMEI or Dravet syndrome [39]); iii) On an epileptic brain background the transition from interictal to ictal neuronal firing is influenced by changes in ionic homeostasis (e.g., diminished action potential repolarization due to elevated extracellular potassium concentration ([K+]out), acute or transient antibody-mediated “loss-of-function” (i.e., antibodies against voltage-gated potassium channels, glutamic acid decarboxylase and glutamate receptors [41]), or altered glutamate uptake by astrocytes [39, 42]. This hypothesis builds on several experimental reports linking BBBD, neuroinflammation and epilepsy but also suggests a systemic inflammatory explanation for the neurovascular changes that trigger seizures. Thus, here we argue that while abnormal neuronal excitability and synchronization “cause” seizures, the mechanisms responsible for abnormal neuronal firing may involve non-neuronal players such as the BBB.
Inflammatory mechanisms involved in blood-brain barrier disruption
If BBBD is responsible for loss of CNS homeostasis and abnormal neuronal firing, how and when do BBB cells lose their physiological function? Owing to its intravascular location, the BBB is prone to incursions by circulating inflammatory signals [9, 27, 43-46]. These attacks could be facilitated by increased expression of adhesion molecules on endothelial cells seen in epileptic brain [47]. In addition to leukocyte-endothelial interactions, BBBD may also result from other factors. These are summarized in Figure 1 and Table 1 and reviewed in the following paragraphs.
Figure 1. Schematic representation of the immunological players involved in seizure disorders.

Coexistence of central and peripheral inflammatory mechanisms which are potentially epileptogenic requires numerous checkpoints to ensure that infectious or other pro-inflammatory signals are fully activated only under extreme conditions. In this scenario, electrophysiological control of seizure threshold (e.g., endowment of neuronal ion channels) interacts with ictogenic alterations of the brain milieu (soluble inflammatory factors). The latter either directly (potassium ions) or indirectly (albumin) affect neuronal firing. CNS levels of these factors are ultimately controlled by the blood-brain barrier. The most commonly reported excitability changes occur when potassium or glutamate homeostasis is altered [7]. A typical downstream event of inflammation (increased vascular permeability – dotted arrow) will not only alter immediate gene expression or cause sudden excitability changes but also sustain gliosis and activation of microglia [3]. Both principal neurons and interneurons are prone to electrophysiological changes facilitated by pro-inflammatory signals (e.g., COX-2 and IL-1β) or by an abnormal angiogenesis (Ang-2, VEGF). Other brain cells (astrocyte and microglia) contribute to delay recovery from pathologic interstitial homeostasis (e.g., albumin and IgG extravasation). Please see Table 1 for a summary of the molecular players involved.
Table 1. Brain and peripheral immunological determinants of seizures.
A summary of concordant and divergent experimental and clinical data is provided. The occurrence of neuroglial, vascular and systemic immune modulation is consistent with a possible role of a brain-periphery pro-inflammatory cross talk in seizure disorders.
| Cell type | Subtype / localization | Molecular players | Animal Human |
Model | Stage | Ref |
|---|---|---|---|---|---|---|
| Neuron | Principal cell Interneurons | APOE4 | Rodent | KA | Chronic | [104] |
|
| ||||||
| TLR4 / IL-1β | Rodent | LPS, KA, Electr.Stim., etc. | Acute/Chronic | [2, 54, 105, 106] | ||
| Human | ||||||
|
| ||||||
| TLR4 / HMGB1 | Rodent | KA | Acute/Chronic | [54] | ||
|
| ||||||
| COX2 | Rodent | Pilo | Acute/Chronic | [52] | ||
|
| ||||||
| VEGF | Rodent | KA | Acute/Chronic | [58] | ||
|
| ||||||
| IgG | Human Autoimmune Epilepsy | [75, 107] | ||||
|
|
||||||
| BBB Neuro-Glia | Astrocytes | mTOR | Human | Tuberous Sclerosis Complex | Chronic | [108-110] |
| Rodent | ||||||
|
| ||||||
| TGF-α K+ and Glut. Uptake IL-1β | Rodent | Bile salt KA, Electr. Stim. | Acute/Chronic | [2, 36, 60, 106] | ||
|
| ||||||
| CXCL12, CXCR4, MCP-1, CCR2 | Rodent | Pilo | Acute/Chronic | [55, 56] | ||
| KA | ||||||
|
| ||||||
| Microglia | Activation (Iba-1) | Rodent | Rasmussen KA, electr. stim. | Chronic | [111, 112] | |
| Human | ||||||
|
| ||||||
| CCR5-ligand | Rodent | KA | Chronic | [113] | ||
|
| ||||||
| Selectins | Guinea pig | Acute | [47] | |||
|
| ||||||
| VEGF–R | Rodent | KA | Acute/Chronic | [58, 59] | ||
|
|
||||||
| BBB interface | Bone marrow | Leukocytes Brain and blood cytokines | Rodent | Pilo | Acute/Chronic | [20] |
|
| ||||||
| Leukocytes Circulating soluble factors | T-cells Blood cytokines | Human | Ictal / interictal | [12, 19, 25, 29, 33, 48, 49, 100] | ||
| Rodent | (Li)-Pilo | Acute – post SE | ||||
|
| ||||||
| T-cells Integrins | Rodent | Pilo / KA | Acute/Chronic | [11, 113] | ||
|
| ||||||
| CD3, CD4, CD8 Macrophages | Rodent | KA | Acute/Chronic | [15, 24] | ||
|
| ||||||
| B cells circulating | Kainate receptors | Human | KA | NA | [114] | |
Animal models of seizures typically rely on chemical or electric methodologies to induce an acute status epilepticus that may evolve into chronic seizures [14, 24, 48]. In these models, pro-inflammatory events leading to seizures have been shown to occur in the brain and peripherally (Figure 1 and Table 1). However, findings derived from experimental models have produced contrasting findings (e.g. [11, 13, 15, 24, 48, 49]).
Targeting seizure-induced brain inflammation reduces seizure severity and number [2, 50], while extravasation of pro-inflammatory molecules into brain across a leaky BBB or their ex novo expression by brain cells is ictogenic or can exacerbate abnormal ictal activity [44, 51]. The molecular players involved in seizure-related inflammation are often the same that participate in systemic inflammation. For example, altered brain expression of cyclooxygenase-2 (COX-2) and prostaglandin during seizures affects neuronal excitability [52] with a mechanism similar to inflammation-derived peripheral pain. Seizure-dependent neuronal COX-2 tilts the scale in favor of early neuroprotection but causes a delayed neurodegeneration of pyramidal cells (Table 1). The high-mobility group box (HMGB) proteins are immune activators that have multiple functions in the regulation of immunity and inflammation [53]. Blocking TLR-4 and HMGB-1 signaling decreases kainate-induced seizures [54].
Neurons are not the only brain cells to display an inflammatory phenotype in epileptic brain since other brain cells contribute to seizure-related immune response (Table 1) [37]. Adhesion molecules (P- and E- selectin) are up-regulated in response to electrographic seizures at the luminal side of the endothelium forming the BBB [47], chemokines and their receptors (CXCL12 and CXCR4; CCL2 and CCR2) are up-regulated in glia during seizures [17, 55, 56] and regions characterized by a “leaky” BBB have been consistently reported in epileptic brain [57]. An example of a synergism between BBBD and its downstream consequences is vascular endothelial growth factor (VEGF). VEGF is a potent modulator of vascular permeability; increased VEGF levels in the brain cause BBB leakage. VEGF is released by neurons in response to seizures, and after release binds to its receptor, VEGF-R2, on endothelial cells (Figure 1). This interaction triggers angiogenesis and vascular remodeling [58, 59]. The formation of new, “leaky” vessels may further promote seizures by the mechanisms described earlier. Together these studies suggest that numerous inflammatory processes and cell types are involved in the disruption of immune privilege and brain homeostasis that precedes ictal events mediated by BBBD. But how do all these events translate into abnormal neuronal function?
How does inflammation affect neuronal behavior?
One of the most remarkable features of the mammalian BBB is its ability to maintain ionic and osmotic gradients between brain and blood (recently reviewed in [37]). Consequences of BBBD all seem to conspire towards increased neuronal firing [36, 37]. While the following paragraphs focus on the role of potassium homeostasis, other mechanisms are also crucial to ictogenesis after BBBD [39, 41, 42]. Conclusive evidence that BBBD can cause seizures was derived from the experimental or clinical disruption of the BBB induced by an osmotic shock. Osmotic challenges delivered to the endothelial cells to disrupt tight junctions trigger seizures in human subjects and animal models [10, 60]. While most of the clinical data derive from the osmotic shock approach to BBBD to improve chemotherapy for brain tumors [10], experimental data suggest that the trigger used for BBBD is not a determining factor in seizure induction. In fact, systemic inflammation, aberrant angiogenesis, and reperfusion damage all decrease seizure threshold by a mechanism involving increased BBB permeability [36, 58, 59, 61].
An obvious candidate for BBBD-mediated changes in homeostasis is extracellular potassium. The effects of increased potassium in epileptogenesis are however not novel, since many laboratories have shown its powerful actions on neurons [62, 63]. Owing to its asymmetric concentrations across the endothelium, BBBD increases brain potassium concentrations [37, 64, 65]. A parsimonious explanation for how BBBD triggers seizures may therefore be ascribed to a sudden increase of extracellular potassium triggering broad neurophysiological changes. However, this appears to be an overly simplistic mechanism. An unexpected consequence of BBBD is albumin extravasation and its uptake by astrocytes via a TGF-β-dependent mechanism [42]. This leads to loss of inward rectifying potassium Kir 4.1 channels and reduced buffering of [K+]out. Loss of spatial buffering further increases [K+]out which in turn depolarizes neurons and causes increased firing. This leads to additional accumulation of potassium ions in the extracellular space owing to loss of neuronal potassium during action potential repolarization. Thus, BBBD creates synergistic sequelae that result in a dramatic increase of [K+]out.
In agreement with a role for potassium homeostasis in seizure disorders is the finding that loss of inward rectifying potassium channels is a hallmark of post-traumatic epilepsy [62], where BBB leakage is an early event [66]. An additional synergistic link between the electrophysiological consequences of inflammation and exacerbation of the inflammatory process itself has been recently proposed [67]. This study demonstrated that potassium ions can act as a signaling factor controlling one of the key pro-inflammatory cytokines, IL-1β. This finding is an example of a mechanism of seizure perpetuation, whereby BBB leakage, altered potassium homeostasis and pro-inflammatory mediators are linked.
Clinical evidence linking inflammation to seizures
The first description of epilepsy due to inflammation dates back to 1958 when Theodore Rasmussen from the Montreal Neurological Institute described a few children operated on for intractable focal seizures, and progressive hemiparesis, and in whom the pathology of the brain tissues demonstrated hemispheric inflammatory changes [68]. Neuropathology clarified that the brain inflammation in what is now termed Rasmussen encephalitis (RE) is dominated by T cells (granzyme B positive CD8+ cells), by microglial activation and microglial nodules, and is followed by neuronal loss and astro-gliosis restricted to the affected hemisphere [69]. Although the etiology of RE remains unclear, it is now acknowledged that a key role in the pathogenesis of RE is played by the cytotoxic effect of T-cells that causes apoptotic death of neurons and astrocytes in neocortex and white matter [68].
More recent clinical evidence links the immune system with seizure onset or maintenance in other forms of epilepsy. Anti-inflammatory drugs, such as steroids and intravenous immunoglobulins, are useful in selected drug-resistant epileptic syndromes, while fever, immunization and trivial infection can precipitate seizures, providing a solid link between inflammation and seizures [3, 70]. Seizures can also occur as a consequence of autoimmune disorders, such as lupus or celiac disease [71, 72]. In addition, inflammatory markers have been detected in surgical brain specimens from epileptic patients and markers of inflammation or CNS autoantibodies are associated with a number of epileptic disorders, further linking abnormal immune responses with seizures (e.g., [73]). More recently, the presence of anti-nuclear antibodies located within neuronal nuclei was reported [74]. In the same cohort, circulating autoantibodies were found in sera from patients affected by a broad range of epilepsies, including temporal lobe epilepsy due to cortical malformations.
Considering all epilepsies with an inflammatory etiology, two scenarios become apparent depending on the presence or absence of inflammatory markers and the response to treatment. On the one hand there are epilepsies where inflammation plays a pivotal and recognized role in the ictogenic process, while on the other are seizure disorders in which the role of immunity is advocated with varying levels of clinical evidence, but lacks definitive proof. The following are instances of epilepsies with suspected or recognized inflammatory disease. These examples are presented to underscore the fact that the experimental data presented in the previous sections have a clear cut counterpart in the clinical setting and that both clinical and pre-clinical evidence supports a link between inflammatory events and seizures.
Encephalitis with prominent epileptic seizures is an antibody-associated disease reported in patients with full-blown brain neuropathology and in association with cryptogenic epilepsy (e.g., normal brain MRI). These auto-antibodies are directed against neuronal surface proteins such as the NR1 subunit of NMDA receptor, the potassium channel complex protein LGI1, or against the intracellular protein glutamic acid decarboxylase (GAD) [75]. Consistent with an immunologic origin of the disease, in these patients, seizures are refractory to antiepileptic drugs but are controlled by steroids or other immunomodulatory treatments [68].
Encephalopathy associated with NMDAR antibodies, also reported in association with ovarian teratoma, is characterized by the abrupt onset of seizures, behavioral and movement disorders [76]; MRI in these patients is often normal. The presence of autoantibodies together with the response to traditional anti-inflammatory treatments underscores the systemic inflammatory component involved in these epilepsies. The absence of obvious abnormalities on brain MRI further support the hypothesis that epileptic seizures can occur as a consequence of altered systemic immunity even in absence of measurable brain pathology. In other words, these findings (normal MRI, circulating antibodies, and response to immunomodulators) show how a neurological disorder characterized by chronic epileptic seizures can result from systemic events (B cell activation and production of autoantibodies; BBB permeability increase allowing brain entry for these antibodies) leading to neuronal misfiring due to altered microenvironment (antibodies against ion channels; loss of potassium and glutamate homeostasis).
A common event involved in seizure generation is fever, a factor known to lower seizure threshold in children [77]. Beside simple febrile convulsions, which are a benign, most likely genetic, and self-limited condition, fever may precipitate seizures in chronic epilepsy. A susceptibility to fever- induced seizures is a hallmark of definite epileptic syndrome due to channelopathies (e.g., in SCN1A related epilepsies [78]), or to mutations in genes that are crucial for brain development and synaptic transmission, such as PCDH19 [79]. Why fever triggers seizures is not clear, although the role of inflammatory mechanisms in inducing hyperexcitability has been suggested [80].
At the extreme end of the spectrum of fever induced seizure are FIRES (febrile infection-related epilepsy) and IHHE (idiopathic hemiconvulsion-hemiplegia-epilepsy syndrome), two conditions collectively known as “acute encephalopathy with inflammation mediated status epilepticus” [81]. These are rare syndromes characterized by the occurrence of status epilepticus in a previously healthy child, during or closely after a febrile episode. In IHHS after the unilateral status epilepticus the child develops atrophy of one hemisphere, contralateral hemiparesis and epilepsy; in FIRES, the refractory status, which mainly involves the perisilvian areas and mesial temporal structures, is followed by drug resistant epilepsy and mental deterioration, and, in a subset of patients, by bilateral mesial temporal atrophy. In both conditions, seizures are refractory to antiepileptic drugs. The etiology of FIRES and IHHS is unknown and there is no evidence of brain infection. Clinical features and experimental models point to the hypothesis of a vicious circle involving inflammation and seizure activity facilitated by brain maturation [81].
Tuberous sclerosis, a highly epileptogenic disease is due to mutations of TSC1 or TSC2 tumor suppressor genes, which inhibit the mammalian target of rapamycin complex1 (mTOR1). The mTOR pathway is essential in controlling cell proliferation and metabolism, but also plays a role in immune cell homeostasis. Inflammation in brain tubers is revealed by the presence of extravasated macrophages, by altered expression of TNFα, NF-κB and cell adhesion molecules in astrocytes and dysplastic neurons [82]. Clinical trials with rapamycin may clarify if a targeted immunosuppressant treatment is effective in reducing seizures independent from its effects on tumor growth but experimental results have already shown that rapamycin may reduce seizure burden by restoring BBB function or by preventing its failure [83].
Taken together, clinical findings favor an immunologic etiology in many syndromes associated with seizures. These pathologies should constitute the translational bases for rational and clinically relevant scientific and experimental investigations.
Pharmacological evidence linking inflammation to seizures
Epilepsy is a complex disease and the pharmacology of anti-epileptic drugs (AED) is comparably multifaceted. Since the ultimate goal of AED is to prevent or abort the abnormal electrical firing of neurons, their mechanism of action has traditionally been ascribed to blockade of excitatory neurons and ion currents or to augmentation of inhibitory interneurons and ionic conductances. Thus, two broad categories of AED can be described, drugs reducing sodium, calcium or glutamate receptor-mediated ion currents or drugs increasing GABA-ergic inhibition. However, a review of mechanisms of action of anti-seizure maneuvers suggests and anti-inflammatory component (Table 2, [18, 84, 85] and [21, 86-93]).
Table 2. Pharmacological and therapeutic evidences supporting how immunological processes acting as mediators of seizures may be exploited to treat epilepsy.
Note that with a few exceptions (e.g., carbamazepine), most antiepileptic interventions encompass an immunomodulatory effect. Novel approaches such as vagal nerve stimulation are also modulators of the immune response.
| Drug or therapy (type) | Traditional mechanism(s) of action | Immunomodulatory effects – known or proposed | Ref. |
|---|---|---|---|
| Valproate (AED) | Na+ and Ca++ Currents-other unknown; | Inhibits NFkB | [85] |
| Phenytoin (AED) | Na+ currents | Decreased T cells IgA deficiency Immunosuppression | [94, 95] |
| Vigabatrin (AED) | GABA | No effects | [18] |
| Levetiracetam (AED) | Synaptic transmission (SV-2) | No effects | [18] |
| Diazepam (AED) | GABA-A | T cells and IFN-γ inhibition | [96] |
| Carbamazepine (AED) | Na+ currents | No consistent effects; pro-inflammatory | [18] |
| Corticosteroids (Immunomodulators) | Immunodepression | Similar to NFkB inhibition | [98] |
| Propofol (Anesthetic short-acting hypnotic agent) | GABA agonist | Inhibits NFkB | [86-88] |
| Thiopental (anesthetic short-acting hypnotic agent) | GABA agonist | Inhibits NFkB | [89] |
| Ketamine (Anesthetic) | NMDA antagonist | Inhibits NFkB IL-1β, TNF-α surge | [90] [91] |
| Magnesium (electrolyte) | Electrolyte; NMDA blocker | Restores NMDA receptor blockade after BBB disruption | [92] |
| Vagal nerve stimulator | Device | Nicotinic receptors; Protection of BBB | [21, 22, 45, 101, 102] |
| Ketogenic diet | Dietary regimen | Protection of BBB | |
| Hypothermia | Medical management | Inhibits NFkB; Protection of BBB | [93] |
Many AEDs can affect both humoral and cellular immunity, modifying T cell behavior and expression of inflammatory mediators [18]. The transcription factor NF-κB mediates the immune modulatory effects of some AEDs but whether this is relevant for AED efficacy is unknown (Table 2 and [18]). Phenytoin decreases suppressor T cell activity and causes reversible IgA deficiency in epileptics [18, 94, 95]. Diazepam inhibits T cell function via peripheral benzodiazepine receptors and by decreasing interferon (IFN)-γ production [96]. The immune modulatory effects of AED should be taken into account when these drugs are administered in combination with anti-inflammatory agents such as adrenocorticotropic hormone (ACTH) or glucocorticosteroids because of possible cumulative immunosuppressant actions. Drugs or interventions commonly used to tackle super-refractory status epilepticus (the most severe and life-threatening form of seizures) may also act by a mechanism encompassing an anti-inflammatory effect and repair of the cerebrovasculature [84]. Since many AED have, in addition to electrophysiological effects, an anti-inflammatory mechanism of action, one may expect that these drugs can be used to treat diseases characterized by an excessive immune response. In fact, AED are used for the treatment of chronic pain, a condition where inflammation is an accepted and recognized etiological mechanism [97].
Many of the molecular pathophysiological mechanisms described in human epileptic brain or in animal models are also found in diseases unrelated to epilepsy such as multiple sclerosis (MS). For example, animal models of MS or patients affected by MS display increased BBB permeability, presence of autoantibodies in serum and abnormal levels of pro-inflammatory cytokines. If similarities between MS and epilepsy are consistent with a comparable underlying inflammatory mechanism, then one may predict that drugs that are beneficial for MS patients may also be used to prevent epileptic seizures. Since MS is a neuroimmune disorders, a number of immunomodulators have been used to treats its symptoms and progression. The following are a few examples of drugs that were traditionally used for MS or other immunological disorders, drugs that today are also administered to epileptic patients. This further underscores the immunological component in seizure disorders.
Glucocorticosteroids (prednisone, dexamethasone, cortisone) can be used to treat MS but also effectively decrease seizure burden in multiple drug-resistant epileptics [98]. The IL-1β receptor antagonist IL1-RA (Kineret, Anakinra) exerts antiepileptic activity in experimental models [2], and a humanized monoclonal antibody against the cell adhesion molecule α4-integrin (natalizumab) has anecdotal anti-seizure efficacy [28, 99]. Clinical data have shown that immunomodulators are effective in many forms of epilepsy, but before their use becomes widely accepted additional data are needed to address the following: i) Given the anti-inflammatory effects of traditional AED, are there risks in administering a combination of immunomodulators and AED? ii) Are there particular forms of epilepsy (for example, pediatric vs. adult; focal vs. generalized) that more consistently respond to these anti-inflammatory therapy? and, iii) Are the side effects of immunomodulation acceptable in these chronic diseases? Future clinical trials will elucidate these points and eventually expand the use of anti-inflammatory drugs in epilepsy.
Communication between peripheral and neuroinflammation: possible role in seizure disorders
Seizures do not only impact the cerebral cortex but can spread to nuclei involved in autonomic regulation and neuroendocrine function. Conversely, peripherally generated nervous or chemical signals can impact brain physiology [43]. The integration of these signals may influence seizure threshold. A broad range of stress stimuli (e.g., social, physical, consequent to mood disorders) activate the hypothalamic–pituitary–adrenal (HPA) and the sympathetic–adrenal–medullary (SAM) axes. Both pathways target the adrenal glands but SAM can extend its reach to activate lymph node-resident immunocompetent cells. This is achieved by a multipronged mechanism including i) direct stimulation of the adrenal medulla to produce adrenaline and noradrenaline; ii) ‘hard-wiring’, through sympathetic-nervous-system innervating lymphoid organs; and, iii) stress hormones acting on lymphoid cells [27]. Whether stressors cause pro- versus anti-inflammatory effects probably depends on the nature of the stressor and its duration (acute vs. chronic) [27]. In addition to the previously mentioned results showing that resident (lymph node, spleen) immune cells are activated by stress associated with seizures, clinical data show that circulating natural killer (NK) and T-helper CD4+ cell immune levels also respond to the inter-ictal to ictal transition (seizure onset) or are sensitive to post-ictal silencing [12, 25, 30]. This suggests that activation of the peripheral immune system, and changes in cytokines, occurs at time of seizure or recovery from the same [19, 29, 33, 100]. The aforementioned involvement of the autonomic nervous system acting on leukocytes via immunological synapses supports the hypothesis of systemic determinants of seizures.
In addition to the HPA and SAM axes, the vagus and the inflammatory reflex contribute to the control of the innate immune response and inflammation [31]. The vagus is a parasympathetic cholinergic cranial nerve involved in a variety of functions spanning from the regulation of heart rate to control of systemic inflammation. As in many cholinergic nerves, both muscarinic and nicotinic receptors are present on target organs; these usually exert opposing functions and are under normal conditions activated to achieve equilibrium. The vagal inflammatory reflex includes a “pro-inflammatory” afferent and an “anti-inflammatory” efferent arm (Figure 2). The cholinergic nicotinic receptor alpha-7 acetylcholine subunit mediates vagal anti-inflammatory effects[46]. Noteworthy, vagus nerve stimulation (VNS) is used to reduce seizure burden in drug resistant epileptic subjects ([101]; see also Table 2). While anti-seizures effects of VNS have been attributed to a brain pacemaker effect, VNS also affects the immune system (Table 2). VNS treatment influences serum IL-6, IL-10 and cortisol levels in epileptic subjects [102]. In vitro, LPS-driven IL-8 secretion by leukocytes obtained from patients 6 months after VNS was reduced compared to baseline[21]. In an experimental model of seizures, serum corticosterone levels increased after VNS, suggesting an effect of VNS on the hypothalamic pituitary-adrenal axis [22]. A comprehensive study correlating seizure outcome and immunomodulation is required to fully elucidate whether VNS immune modulation contributes to the anti-seizures effect. In summary, the peripheral nervous system may influence seizure threshold by centrifugal and centripetal mechanisms involved in the control of inflammation.
Figure 2. Proposed routes of brain-periphery immune communication in seizure disorders.
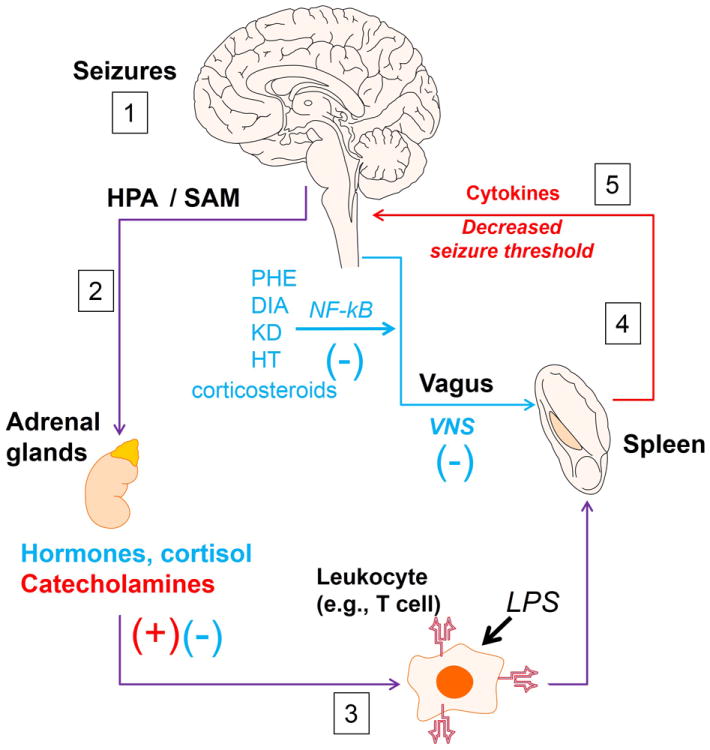
Pro-inflammatory events occurring peripherally (experimentally induced or infectious: e.g., LPS, clinically relevant: e.g., colitis) are transmitted to the brain (afferent vagus – red line, and circulating cytokines) possibly tilting the neurons toward a pro-seizure condition. Conversely, a brain-derived stress signal stimulates the adrenal glands (via hypothalamic-pituitary-adrenal gland (HPA) and sympathetic adrenal-medullary (SAM) axes); activation of these results in secretion of pro- (red) and anti-inflammatory (blue) factors. The brain-peripheral cross talk comprises peripheral organ of immune competency (e.g., spleen, lymph nodes, bone marrow, resident cells in gut [43]) and activation of circulating leukocytes. In the inflammatory reflex, a centripetal sensory input travels after infection or injury through the afferent vagus nerve to the brainstem; the efferent vagus carries centrifugal spleen-bound signals that modulates acetylcholine-release, transmitting neural signals to other immune cells by action on α-7 nicotinic receptors (see asterisk and [46]). Glucocorticosteroids can reduce seizures while commonly prescribed anti-epileptic drugs (e.g., phenytoin and diazepam, indicated in blue) have anti-inflammatory properties. Other anti-seizure therapeutic intervention (ketogenic diet, KD and hypothermia, HT) have anti-inflammatory effects. See also Table 2.
Concluding remarks, important caveats and future directions
Despite the increasing evidence supporting inflammatory processes triggering or sustaining seizures, a number of questions remain unresolved. For example, whether brain inflammation is the initiator or the consequence of systemic inflammatory processes is not a purely academic question. From the therapeutic point of view, if systemic inflammation is to be targeted, then issues of trans-BBB drug delivery are mute therapeutic implications [84]. Conversely, selective direct targeting of brain inflammation could have fewer side effects such as persistent immunosuppression.
It is also unclear whether the findings summarized herein warrant the use of anti-inflammatory drugs in clinical epilepsies where an infectious or febrile context is not suspected. A recent paper described the predictive value of white blood cells in the diagnosis of epilepsy [103]. The specificity of pro-inflammatory mechanisms related to epilepsy is not absolute since many neurological diseases have a recognized inflammatory etiology. This raises the question of whether pro-inflammatory processes may be an epiphenomenon with little significance to the true etiological mechanisms of seizures. However, the evidence against a role for inflammation in seizure disorders is not as compelling as the pro-inflammatory hypothesis.
As this field of research continues to grow, new inflammatory molecules, cellular players and organs are added to the mix. While fundamental questions remain to be answered, there is little doubt that inflammation has integrated clinical and pre-clinical epilepsy research into the field of neuroimmunology. Preventing and reducing the inflammatory burden taking place during seizures is likely to become a viable therapeutic option in drug resistant cases where AED alone are not sufficient to halt seizures.
Table 3.
Summary of clinical findings that constitute the backbone of research aimed at modeling and understanding of inflammatory mechanisms in epilepsy. This list is weighted towards epilepsy syndromes that may share an inflammatory component or ictogenic mechanism. However, many other epilepsy syndromes exist, and in these an inflammatory or infectious trigger has not been reported. Examples are genetic epilepsies with mutations of neuronal ion channels.
| Type of seizure disorders | Signs and symptoms | Mechanisms | ||
|---|---|---|---|---|
|
| ||||
| Epilepsies with an inflammatory component | Epilepsies with seizures directly linked to inflammation (demonstrated or highly suspected) | Rasmussen | MRI reveals encephalopathy or Elevated inflammatory markers in blood or brain or Neuropathology or Fever or Responds to immunomodulators or ketogenic diet (FIRES) |
T-cells |
| CD8+ | ||||
| Granzyme | ||||
| B cells | ||||
| Blood-brain barrier disruption | ||||
|
|
|
|||
| Autoimmune encephalitis | CNS autoantibodies (NMDA, GAD, NR1, LGI) Oligoclonal bands | |||
|
|
|
|||
| Acute encephalopathy with inflammation-mediated status epilepticus (FIRES HHE) | unknown | |||
|
| ||||
| Epilepsies with seizures possibly linked to (or facilitated by) inflammation | Infantile spasms | Markers not always present MRI findings not always present May Respond to immunomodulators (spasms, TS) |
unknown | |
| Tuberous sclerosis | mTOR | |||
| Fever related epilepsy | unknown | |||
TS=tuberous sclerosis.
Highlights.
Traditional and novel anti-epileptic treatments encompass immunomodulatory effects
Acute and chronic immunological triggers contribute to acute and chronic seizures
Etiological factors of seizure disorders include brain and peripheral pro-inflammatory changes and their interplay
The blood-brain barrier (BBB) is a dynamic interface between the CNS and periphery
The BBB reacts to pro-inflammatory stimuli from either the brain or periphery
Acknowledgments
Supported by R01NS078307 (NM and DJ), R01NS43284, R41MH093302, R21NS077236, R42MH093302, UH2TR000491 and R21HD057256 (DJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Panayiotopoulos CP. The new ILAE report on terminology and concepts for the organization of epilepsies: critical review and contribution. Epilepsia. 2012;53:399–404. doi: 10.1111/j.1528-1167.2011.03381.x. [DOI] [PubMed] [Google Scholar]
- 2.Vezzani A, et al. Inflammation and epilepsy. Handb Clin Neurol. 2012;107:163–175. doi: 10.1016/B978-0-444-52898-8.00010-0. [DOI] [PubMed] [Google Scholar]
- 3.Vezzani A, Janigro D. Inflammation. In: Engel G, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Second. Lippincott Williams and Wilkins; 2008. pp. 267–276. [Google Scholar]
- 4.Vezzani A, et al. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in rat hippocampal slices. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- 6.Amzica F, et al. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22:1042–1053. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.David Y, et al. Astrocytic Potassium and Glutamate Buffering Controls Synaptic Responses in A Frequency-Dependent Manner. Epilepsia. 2009;50:86–86. [Google Scholar]
- 8.Heinemann U, et al. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia. 2012;60:1251–1257. doi: 10.1002/glia.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivest S. Interactions between the immune and neuroendocrine systems. Prog Brain Res. 2010;181:43–53. doi: 10.1016/S0079-6123(08)81004-7. [DOI] [PubMed] [Google Scholar]
- 10.Marchi N, et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabene PF, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer S, et al. NK and CD4+ T cell changes in blood after seizures in temporal lobe epilepsy. Exp Neurol. 2008;211:370–377. doi: 10.1016/j.expneurol.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Marchi N, et al. Blood-brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ravizza T, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Zattoni M, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31:4037–4050. doi: 10.1523/JNEUROSCI.6210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Librizzi L, et al. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann Neurol. 2012;72:82–90. doi: 10.1002/ana.23567. [DOI] [PubMed] [Google Scholar]
- 17.Xu JH, et al. CCR3, CCR2A and macrophage inflammatory protein (MIP)-1a, monocyte chemotactic protein-1 (MCP-1) in the mouse hippocampus during and after pilocarpine-induced status epilepticus (PISE) Neuropathol Appl Neurobiol. 2009;35:496–514. doi: 10.1111/j.1365-2990.2009.01022.x. [DOI] [PubMed] [Google Scholar]
- 18.Beghi E, Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011;52(Suppl 3):40–44. doi: 10.1111/j.1528-1167.2011.03035.x. [DOI] [PubMed] [Google Scholar]
- 19.Choi J, et al. Increased levels of HMGB1 and pro-inflammatory cytokines in children with febrile seizures. Journal of neuroinflammation. 2011;8:135. doi: 10.1186/1742-2094-8-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa-Ferro ZS, et al. Transplantation of bone marrow mononuclear cells decreases seizure incidence, mitigates neuronal loss and modulates pro-inflammatory cytokine production in epileptic rats. Neurobiol Dis. 2012;46:302–313. doi: 10.1016/j.nbd.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 21.De HV, et al. Effects of vagus nerve stimulation on pro- and anti-inflammatory cytokine induction in patients with refractory epilepsy. J Neuroimmunol. 2009;214:104–108. doi: 10.1016/j.jneuroim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 22.De HV, et al. Increased rat serum corticosterone suggests immunomodulation by stimulation of the vagal nerve. J Neuroimmunol. 2009;212:102–105. doi: 10.1016/j.jneuroim.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 23.de Vries HE, et al. Inflammatory events at blood-brain barrier in neuroinflammatory and neurodegenerative disorders: Implications for clinical disease. Epilepsia. 2012;53:45–52. doi: 10.1111/j.1528-1167.2012.03702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deprez F, et al. Adoptive transfer of T lymphocytes in immunodeficient mice influences epileptogenesis and neurodegeneration in a model of temporal lobe epilepsy. Neurobiol Dis. 2011;44:174–184. doi: 10.1016/j.nbd.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 25.Eeg-Olofsson O, et al. Abnormalities of T-lymphocyte subsets in epileptic patients. Acta Neurol Scand. 1985;72:140–144. doi: 10.1111/j.1600-0404.1985.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 26.Galic MA, et al. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33:116–125. doi: 10.1016/j.yfrne.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 28.Granata T, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother. 2009;9:1791–1802. doi: 10.1586/ern.09.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao LY, et al. Interictal interleukin-17A levels are elevated and correlate with seizure severity of epilepsy patients. Epilepsia. 2013;54:e142–145. doi: 10.1111/epi.12337. [DOI] [PubMed] [Google Scholar]
- 30.Nowak M, et al. Interictal alterations of cytokines and leukocytes in patients with active epilepsy. Brain Behav Immun. 2011;25:423–428. doi: 10.1016/j.bbi.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex-linking immunity and metabolism. Nat Rev Endocrinol. 2012;8:743–754. doi: 10.1038/nrendo.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayyah M, et al. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–1080. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Uludag IF, et al. Interleukin-6, interleukin-1 beta and interleukin-1 receptor antagonist levels in epileptic seizures. Seizure : the journal of the British Epilepsy Association. 2013;22:457–461. doi: 10.1016/j.seizure.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Yamanaka G, et al. Increased level of serum interleukin-1 receptor antagonist subsequent to resolution of clinical symptoms in patients with West syndrome. J Neurol Sci. 2010;298:106–109. doi: 10.1016/j.jns.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 35.Eeg-Olofsson O, et al. Immunological studies in focal epilepsy. Acta neurologica Scandinavica. 1988;78:358–368. doi: 10.1111/j.1600-0404.1988.tb03671.x. [DOI] [PubMed] [Google Scholar]
- 36.Friedman A. Blood-brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia. 2011;52(Suppl 8):19–20. doi: 10.1111/j.1528-1167.2011.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Janigro D. Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia. 2012;53(Suppl 1):26–34. doi: 10.1111/j.1528-1167.2012.03472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchi N, et al. Blood-brain barrier dysfunction and epilepsy: Pathophysiologic role and therapeutic approaches. Epilepsia. 2012;53:1877–1886. doi: 10.1111/j.1528-1167.2012.03637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy) Handb Clin Neurol. 2013;111:627–633. doi: 10.1016/B978-0-444-52891-9.00065-8. [DOI] [PubMed] [Google Scholar]
- 40.Spreafico R, Tassi L. Cortical malformations. Handb Clin Neurol. 2012;108:535–557. doi: 10.1016/B978-0-444-52899-5.00047-2. [DOI] [PubMed] [Google Scholar]
- 41.Graus F, et al. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. 2010;257:509–517. doi: 10.1007/s00415-009-5431-9. [DOI] [PubMed] [Google Scholar]
- 42.Ivens S, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 43.Qureshi IA, Mehler MF. Towards a ’systems’-level understanding of the nervous system and its disorders. Trends in neurosciences. 2013 doi: 10.1016/j.tins.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riazi K, et al. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89:34–42. doi: 10.1016/j.eplepsyres.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 45.Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009;64:28–32. doi: 10.1016/j.neuron.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tracey KJ. Understanding immunity requires more than immunology. Nat Immunol. 2010;11:561–564. doi: 10.1038/ni0710-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Librizzi L, et al. Expression of adhesion factors induced by epileptiform activity in the endothelium of the isolated guinea pig brain in vitro. Epilepsia. 2007;48:743–751. doi: 10.1111/j.1528-1167.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- 48.Marchi N, et al. In vivo and in vitro effects of pilocarpine: Relevance to ictogenesis. Epilepsia. 2007;48:1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holtman L, et al. Blood plasma inflammation markers during epileptogenesis in post-status epilepticus rat model for temporal lobe epilepsy. Epilepsia. 2013;54:589–595. doi: 10.1111/epi.12112. [DOI] [PubMed] [Google Scholar]
- 50.Ravizza T, et al. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497:223–230. doi: 10.1016/j.neulet.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 51.Riazi K, et al. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U S A. 2008;105:17151–17156. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serrano GE, et al. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci. 2011;31:14850–14860. doi: 10.1523/JNEUROSCI.3922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanai H, et al. High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol. 2012;33:633–640. doi: 10.1016/j.it.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Maroso M, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- 55.Foresti ML, et al. Chemokine CCL2 and its receptor CCR2 are increased in the hippocampus following pilocarpine-induced status epilepticus. J Neuroinflammation. 2009;6:40. doi: 10.1186/1742-2094-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartman NW, et al. CXCL12-mediated guidance of migrating embryonic stem cell-derived neural progenitors transplanted into the hippocampus. PLoS One. 2010;5:e15856. doi: 10.1371/journal.pone.0015856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- 58.Morin-Brureau M, et al. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: role of VEGF signaling pathways. J Neurosci. 2011;31:10677–10688. doi: 10.1523/JNEUROSCI.5692-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rigau V, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130:1942–1956. doi: 10.1093/brain/awm118. [DOI] [PubMed] [Google Scholar]
- 60.Seiffert E, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marchi N, Lerner-Natoli M. Cerebrovascular Remodeling and Epilepsy. Neuroscientist. 2012 doi: 10.1177/1073858412462747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D’Ambrosio R, et al. Impaired K(+)homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. Journal of Neuroscience. 1999;19:8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Janigro D, et al. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. Journal of Neuroscience. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keep RF, et al. Blood-brain barrier mechanisms involved in brain calcium and potassium homeostasis. Brain Res. 1999;815:200–205. doi: 10.1016/s0006-8993(98)01155-x. [DOI] [PubMed] [Google Scholar]
- 65.Lapilover EG, et al. Peri-infarct blood-brain barrier dysfunction facilitates induction of spreading depolarization associated with epileptiform discharges. Neurobiol Dis. 2012;48:495–506. doi: 10.1016/j.nbd.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korn A, et al. Focal cortical dysfunction and blood-brain barrier disruption in patients with Postconcussion syndrome. J Clin Neurophysiol. 2005;22:1–9. doi: 10.1097/01.wnp.0000150973.24324.a7. [DOI] [PubMed] [Google Scholar]
- 67.Arlehamn CS, et al. The role of potassium in inflammasome activation by bacteria. J Biol Chem. 2010;285:10508–10518. doi: 10.1074/jbc.M109.067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bien CG, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis - A European consensus statement. Brain. 2005;128:454–471. doi: 10.1093/brain/awh415. [DOI] [PubMed] [Google Scholar]
- 69.Hart YM, et al. Double pathology in Rasmussen’s syndrome - A window on the etiology? Neurology. 1998;50:731–735. doi: 10.1212/wnl.50.3.731. [DOI] [PubMed] [Google Scholar]
- 70.Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 71.Ludvigsson JF, et al. Increased risk of epilepsy in biopsy-verified celiac disease: a population-based cohort study. Neurology. 2012;78:1401–1407. doi: 10.1212/WNL.0b013e3182544728. [DOI] [PubMed] [Google Scholar]
- 72.Lang B, et al. New autoantibody mediated disorders of the central nervous system. Curr Opin Neurol. 2003;16:351–357. doi: 10.1097/01.wco.0000073937.19076.d5. [DOI] [PubMed] [Google Scholar]
- 73.Iyer A, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia. 2010;51:1763–1773. doi: 10.1111/j.1528-1167.2010.02547.x. [DOI] [PubMed] [Google Scholar]
- 74.Iffland PH, et al. Intracellular and circulating neuronal antinuclear antibodies in human epilepsy. Neurobiol Dis. 2013;59:206–219. doi: 10.1016/j.nbd.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vincent A, et al. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10:759–772. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- 76.Armangue T, et al. Pediatric Anti-N-methyl-D-Aspartate Receptor Encephalitis-Clinical Analysis and Novel Findings in a Series of 20 Patients. J Pediatr. 2012 doi: 10.1016/j.jpeds.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cross JH. Fever and fever-related epilepsies. Epilepsia. 2012;53(Suppl 4):3–8. doi: 10.1111/j.1528-1167.2012.03608.x. [DOI] [PubMed] [Google Scholar]
- 78.Gambardella A, Marini C. Clinical spectrum of SCN1A mutations. Epilepsia. 2009;50(Suppl 5):20–23. doi: 10.1111/j.1528-1167.2009.02115.x. [DOI] [PubMed] [Google Scholar]
- 79.Nabbout R, et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology. 2011;76:1193–1194. doi: 10.1212/WNL.0b013e31820a9642. [DOI] [PubMed] [Google Scholar]
- 80.McClelland S, et al. Epileptogenesis after prolonged febrile seizures: mechanisms, biomarkers and therapeutic opportunities. Neuroscience letters. 2011;497:155–162. doi: 10.1016/j.neulet.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nabbout R, et al. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol. 2011;10:99–108. doi: 10.1016/S1474-4422(10)70214-3. [DOI] [PubMed] [Google Scholar]
- 82.Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia. 2011;52(Suppl 3):26–32. doi: 10.1111/j.1528-1167.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- 83.van Vliet EA, et al. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–1263. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- 84.Marchi N, et al. The blood-brain barrier hypothesis in drug resistant epilepsy. Brain. 2012;135:e211. doi: 10.1093/brain/awr343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ichiyama T, et al. Sodium valproate inhibits production of TNF-alpha and IL-6 and activation of NF-kappaB. Brain Res. 2000;857:246–251. doi: 10.1016/s0006-8993(99)02439-7. [DOI] [PubMed] [Google Scholar]
- 86.Sanchez-Conde P, et al. The comparative abilities of propofol and sevoflurane to modulate inflammation and oxidative stress in the kidney after aortic cross-clamping. Anesth Analg. 2008;106:371–378. doi: 10.1213/ane.0b013e318160580b. table. [DOI] [PubMed] [Google Scholar]
- 87.Schneemilch CE, et al. Effects of different anaesthetic agents on immune cell function in vitro. Eur J Anaesthesiol. 2005;22:616–623. doi: 10.1017/s0265021505001031. [DOI] [PubMed] [Google Scholar]
- 88.Schneemilch CE, et al. Effect of 2 anesthetic techniques on the postoperative proinflammatory and anti-inflammatory cytokine response and cellular immune function to minor surgery. J Clin Anesth. 2005;17:517–527. doi: 10.1016/j.jclinane.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 89.Roesslein M, et al. Thiopental protects human T lymphocytes from apoptosis in vitro via the expression of heat shock protein 70. J Pharmacol Exp Ther. 2008;325:217–225. doi: 10.1124/jpet.107.133108. [DOI] [PubMed] [Google Scholar]
- 90.Beilin B, et al. Low-dose ketamine affects immune responses in humans during the early postoperative period. Br J Anaesth. 2007;99:522–527. doi: 10.1093/bja/aem218. [DOI] [PubMed] [Google Scholar]
- 91.Welters ID, et al. Continuous S-(+)-ketamine administration during elective coronary artery bypass graft surgery attenuates pro-inflammatory cytokine response during and after cardiopulmonary bypass. Br J Anaesth. 2011;106:172–179. doi: 10.1093/bja/aeq341. [DOI] [PubMed] [Google Scholar]
- 92.Amtorp O, Sorensen SC. The ontogenetic development of concentration differences for protein and ions between plasma and cerebrospinal fluid in rabbits and rats. J Physiol. 1974;243:387–400. doi: 10.1113/jphysiol.1974.sp010759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–S202. doi: 10.1097/CCM.0b013e3181aa5241. [DOI] [PubMed] [Google Scholar]
- 94.Basaran N, et al. Serum immunoglobulins, complement levels and lymphocyte subpopulations in phenytoin-treated epileptic patients. Immunopharmacol Immunotoxicol. 1989;11:335–346. doi: 10.3109/08923978909005374. [DOI] [PubMed] [Google Scholar]
- 95.Hashiba N, et al. Phenytoin at optimum doses ameliorates experimental autoimmune encephalomyelitis via modulation of immunoregulatory cells. J Neuroimmunol. 2011;233:112–119. doi: 10.1016/j.jneuroim.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 96.Wei M, et al. Suppressive effect of diazepam on IFN-gamma production by human T cells. Int Immunopharmacol. 2010;10:267–271. doi: 10.1016/j.intimp.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 97.Galletti F, et al. Pathophysiological basis of migraine prophylaxis. Prog Neurobiol. 2009;89:176–192. doi: 10.1016/j.pneurobio.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 98.Marchi N, et al. Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoS One. 2011;6:e18200. doi: 10.1371/journal.pone.0018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sotgiu S, et al. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report BMC Neurol. 2010;10:84. doi: 10.1186/1471-2377-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lehtimaki KA, et al. Levels of IL-1beta and IL-1ra in cerebrospinal fluid of human patients after single and prolonged seizures. Neuroimmunomodulation. 2010;17:19–22. doi: 10.1159/000243081. [DOI] [PubMed] [Google Scholar]
- 101.Englot DJ, et al. Vagus nerve stimulation for epilepsy: a meta-analysis of efficacy and predictors of response. J Neurosurg. 2011;115:1248–1255. doi: 10.3171/2011.7.JNS11977. [DOI] [PubMed] [Google Scholar]
- 102.Majoie HJ, et al. Vagus nerve stimulation in refractory epilepsy: effects on pro- and anti-inflammatory cytokines in peripheral blood. Neuroimmunomodulation. 2011;18:52–56. doi: 10.1159/000315530. [DOI] [PubMed] [Google Scholar]
- 103.Sarkis RA, et al. Patients with generalised epilepsy have a higher white blood cell count than patients with focal epilepsy. Epileptic Disord. 2012;14:57–63. doi: 10.1684/epd.2012.0493. [DOI] [PubMed] [Google Scholar]
- 104.Zhang XM, et al. Overexpression of apolipoprotein E4 increases kainic-acid-induced hippocampal neurodegeneration. Exp Neurol. 2012;233:323–332. doi: 10.1016/j.expneurol.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 105.Rodgers KM, et al. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–2486. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vezzani A, et al. Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information; Bethesda, MD: 2012. [PubMed] [Google Scholar]
- 107.Vincent A, Crino PB. Systemic and neurologic autoimmune disorders associated with seizures or epilepsy. Epilepsia. 2011;52(Suppl 3):12–17. doi: 10.1111/j.1528-1167.2011.03030.x. [DOI] [PubMed] [Google Scholar]
- 108.Marcotte L, et al. Cytoarchitectural alterations are widespread in cerebral cortex in tuberous sclerosis complex. Acta Neuropathol. 2012;123:685–693. doi: 10.1007/s00401-012-0950-3. [DOI] [PubMed] [Google Scholar]
- 109.Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends in molecular medicine. 2011;17:734–742. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 110.Wong M, Crino PB. Tuberous sclerosis and epilepsy: role of astrocytes. Glia. 2012;60:1244–1250. doi: 10.1002/glia.22326. [DOI] [PubMed] [Google Scholar]
- 111.Wirenfeldt M, et al. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol Dis. 2009;34:432–440. doi: 10.1016/j.nbd.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wirenfeldt M, et al. Microglia - insights into immune system structure, function, and reactivity in the central nervous system. Histol Histopathol. 2011;26:519–530. doi: 10.14670/HH-26.519. [DOI] [PubMed] [Google Scholar]
- 113.Louboutin JP, et al. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J. 2011;25:737–753. doi: 10.1096/fj.10-161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sturgill JL, et al. Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J Neuroimmunol. 2011;233:80–89. doi: 10.1016/j.jneuroim.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]