

Abstract
Bcl-2 family proteins act as essential regulators and mediators of intrinsic apoptosis. Several lines of evidence suggest that the anti-apoptotic members of the family, including Bcl-2, Bcl-xL and Mcl-1, exhibit functional redundancy. However, the current evidence is largely indirect, and based mainly on pharmacological data using small-molecule inhibitors. In order to study compensation and redundancy of anti-apoptotic Bcl-2 proteins at the molecular level, we used a combined knockdown/overexpression strategy to essentially replace the function of one member with another. The results show that HeLa cells are strictly dependent on Mcl-1 for survival and correspondingly refractory to the Bcl-2/Bcl-xL inhibitor ABT-263, and remain resistant to ABT-263 in the context of Bcl-xL overexpression because endogenous Mcl-1 continues to provide the primary guardian role. However, if Mcl-1 is knocked down in the context of Bcl-xL overexpression, the cells become Bcl-xL-dependent and sensitive to ABT-263. We also show that Bcl-xL compensates for loss of Mcl-1 by sequestration of two key pro-apoptotic Bcl-2 family members, Bak and Bim, normally bound to Mcl-1, and that Bim is essential for cell death induced by Mcl-1 knockdown. To our knowledge, this is the first example where cell death induced by loss of Mcl-1 was rescued by the silencing of a single BH3-only Bcl-2 family member. In colon carcinoma cell lines, Bcl-xL and Mcl-1 also play compensatory roles, and Mcl-1 knockdown sensitizes cells to ABT-263. The results, obtained employing a novel strategy of combining knockdown and overexpression, provide unique molecular insight into the mechanisms of compensation by pro-survival Bcl-2 family proteins.
Keywords: Bcl-2, Bcl-xL, Mcl-1, apoptosis, ABT-263
Introduction
Bcl-2 family proteins act as essential mediators of intrinsic apoptosis by regulating the permeability of the outer mitochondrial membrane and the release of cytochrome c and other apoptogenic factors into the cytosol. Bcl-2 proteins can be categorized into three main groups based on their function and sequence homology within their α-helical Bcl-2 homology (BH)1 domains [1]; multi-domain pro-apoptotic Bax and Bak, anti-apoptotic (e.g. Bcl-2, Bcl-xL, and Mcl-1), and pro-apoptotic BH3-only proteins (Bad, Bid, Bim, Puma, Noxa, and others) [2]. Several models have been proposed on how interactions of Bcl-2 family members determine whether or not a cell undergoes apoptosis [3-8]. In the direct activation model, a sub-group of the BH3-only proteins, known as activators and including Bim, Bid and perhaps Puma, directly bind and activate Bax and Bak resulting in mitochondrial outer membrane permeabilization and apoptosis [3-5]. Anti-apoptotic Bcl-2 family proteins block cell death by sequestering the direct activators and also by binding monomeric Bax and Bak before they oligomerize [6]. A second group of sensitizer BH3-only proteins, such as Bad, Noxa and Bik, act by displacing activator BH3-only proteins from the anti-apoptotic proteins. In the indirect activation model, BH3-only proteins displace anti-apoptotic proteins from binding activated forms of Bax and Bak [7]. While a recent study using mathematical modeling and robustness analysis favored the direct activation model [9], it is important to note that the two models are not mutually exclusive. The embedded-together model [8] has recently been proposed to take into account the fact that binding to membranes is essential for key Bcl-2 protein interactions to occur.
Anti-apoptotic members of the Bcl-2 protein family are commonly misregulated in cancer cells. In addition to being elevated in follicular lymphoma [10], Bcl-2 has also been demonstrated to provide a survival advantage as well as resistance to cancer treatments for many cancers including prostate, melanoma, breast and non-small-cell lung carcinoma [11-14]. Additionally, genes encoding Mcl-1 and Bcl-xL are two of the most commonly amplified genes across a variety of human cancers, and elevated expression of Mcl-1 and Bcl-xL are required to maintain cancer cell survival [15]. Based on these observations, drugs have been developed to mimic their natural antagonist BH3-only proteins [16]. The first of these agents to show promise, ABT-737 or its orally available analog ABT-263, is a BH3 mimetic related to the BH3-only protein Bad that was demonstrated to have high affinity for Bcl-2, Bcl-xL and Bcl-w but low affinity for Mcl-1 and A1 [17]. Notably, cells which express high levels of Mcl-1 were found to be resistant to ABT-737 [17, 18], and evidence that Mcl-1 expression is the key feature for ABT-737 resistance has since been demonstrated in many cell lines as well as primary patient samples [19-22]. Furthermore, ABT-737 resistance is circumvented through down regulation of Mcl-1 [5, 23-25]. To date, a specific Mcl-1 inhibitor has yet to be developed. However, due to the very short half-life of Mcl-1 (~ 30 min) [26], agents which act as general transcriptional repressors preferentially decrease Mcl-1 expression level [27]. Interestingly, cells which express high levels of Bcl-xL were found to be resistant to the loss of Mcl-1 [27]. Similarly, a recent study showed that Mcl-1 and Bcl-xL are capable of compensating for the loss of one another in the cellular response to oxidative stress [28]. These findings highlight the functional redundancy of anti-apoptotic Bcl-2 family proteins and emphasizes that for certain cancers inhibition of all family members may be required to overcome blocks to apoptosis.
The current evidence for redundancy among anti-apoptotic Bcl-2 proteins is largely indirect, and based mainly on pharmacological data where small-molecule inhibition of one member fails to elicit cell death if another member is expressed at a sufficient level to provide compensation. Furthermore, it is widely assumed that redundancy is mediated by the ability of one anti-apoptotic Bcl-2 member to bind and sequester pro-apoptotic Bcl-2 proteins normally bound by another. However, only limited molecular evidence in direct support of functional compensation and the underlying mechanisms is available. In neuroblastoma cell lines selected for resistance to ABT-263, increased Mcl-1 expression was observed which co-precipitated with Bim, suggesting that ABT-263 resistance resided in Bim sequestration by overexpressed Mcl-1 [29]. In non-small cell lung cancer cell lines, overexpression of Bcl-xL reduced the amount of Bim bound by Mcl-1, but evidence that Bcl-xL bound Bim was lacking [27].
In order to investigate compensation mechanisms among anti-apoptotic Bcl-2 proteins, we used a combination of knockdown and overexpression strategies to probe their functional redundancy in HeLa and colon carcinoma cell lines. We show that HeLa cells are strictly dependent on Mcl-1 for survival and correspondingly refractory to ABT-263, and remain resistant to ABT-263 in the context of Bcl-xL overexpression because endogenous Mcl-1 continues to provide the primary guardian role. However, if Mcl-1 is knocked down in the context of Bcl-xL overexpression, the cells become Bcl-xL-dependent and sensitive to ABT-263. Importantly, Bcl-xL compensates for loss of Mcl-1 by sequestration of two key pro-apoptotic Bcl-2 family members, Bak and Bim, which to our knowledge is the first direct demonstration of pro-apoptotic protein swapping mediated by anti-apoptoticBcl-2 proteins. We also show that Bim is essential for cell death induced by Mcl-1 knockdown. Thus by employing a novel strategy of combining knockdown and overexpression, cells with replacement of one pro-survival Bcl-2 protein with another were obtained, which enabled unique molecular insight into the mechanisms of compensation exhibited by these key proteins.
Materials and methods
Materials
Antibodies against Bcl-xL (2762), Bim (2933), Bak (6947) and GAPDH (14C10) were purchased from Cell Signaling (Beverly, MA); antibodies against Bcl-2 (sc-509), Mcl-1 (sc-819) and Protein A/G PLUS-agarose (sc-2003) beads were purchased from Santa Cruz (Santa Cruz, CA); antibody against active NT-Bak (06536) was purchased from Millipore (Temecula, CA); antibody for Poly(ADP-ribose) polymerase (PARP) was purchased from BD Biosciences (San Jose CA); Ambion Silencer Select siRNA against Bcl-2 (s1916), Bcl-xL (s224530), Bim (s195012), Mcl-1 (120644), and negative control siRNA (4390844) were purchased from Invitrogen (Carlsbad, CA); vinblastine was purchased from Sigma Aldrich (St. Louis, MO) and thymidine was purchased from EMD Biosciences (Gibbstown, NJ). Plasmids encoding Bcl-2 (Plasmid 8768, Stanley Korsmeyer, Yamamoto 1999) and Bcl-xL (Plasmid 8749, Stanley Korsmeyer, Chao 1995) were purchased from Addgene (Cambridge, MA).
Cell culture, preparation of whole cell extracts, and immunoblotting
HeLa human cervical carcinoma cell line was maintained in monolayer culture at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine growth serum, 2 mM L-glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin. Whole cell extracts for immunoblot analysis were prepared as described previously [30, 31] and protein concentration was determined using the Bradford method. Apparent molecular weights derived from SDS-PAGE/immunoblotting of the proteins examined are as follows: Bak, 25 kDa; Bcl-2, 24 kDa; Bcl-xL, 30 kDa; Bim-EL,- L, -S; 23, 15, 12 kDa, respectively; GAPDH, 37 kDa; Mcl-1, 40 kDa, PARP, 112 and 85 kDa for uncleaved and cleaved, respectively.
Immunoprecipitation
Immunoprecipitation was performed by lysing cells in 0.3 ml of 40 mM HEPES (pH 7.5), 0.12 M NaCl, 1% CHAPS, 1 mM EDTA, containing phosphatase and protease inhibitors for 45 min on ice, centrifuged at 13000 × g for 15 min. The extract (1.0 mg) was precleared with Protein A/G PLUS-agarose beads according to the manufacturer's instructions (Santa Cruz), and the supernatant was collected. 5 μg of antibody was added to the supernatant and mixed overnight at 4°C followed by incubation with Protein A/G PLUS-agarose beads for 4h. After centrifugation, the supernatant was recovered, and the immunoprecipitate was washed and resuspended in Laemmli sample buffer. Equivalent portions of supernatant and precipitate were subjected to 12.5% acrylamide SDS-PAGE and immunoblotting.
Transfections
Depletion of anti-apoptotic Bcl-2 family proteins was performed using siRNA at a final concentration of 50 nM with a non-coding siRNA as a negative control. Transfections were performed using Lipofectamine 2000 (Invitrogen) in Opti-MEM according to manufacturer’s instructions. Transfection media was replaced with fresh DMEM containing serum 6 h post-transfection. Knockdown efficiency was monitored through immunoblotting.
Generation of cell lines stably expressing Bcl-2 proteins
Cells stably overexpressing human untagged Bcl-2, and Bcl-xL, were generated by transfecting HeLa cells at 70% confluence using Lipofectamine 2000 in serum-free Opti-MEM (Invitrogen). After 6 h, the transfection medium was replaced with DMEM containing 10% fetal bovine serum. After an additional 24 h, G418 was added to the medium to a final concentration of 1 mg/ml, and the cells were maintained for 2 weeks. Drug-resistant colonies were selected, expanded, and maintained in growth medium containing 0.4 mg/ml G418. Clones were screened for overexpression of the transfected Bcl-2 family protein by immunoblotting using specific antibodies.
Cell viability assays
Cell viability was determined using (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) reagent as described. Cells (2000/well) were seeded in 96-well plates and indicated drug concentrations were added the following day in a fixed final concentration of 0.1% DMSO. Controls received vehicle (0.1% DMSO) alone. After 72 h, MTT reagent (50 μg/well) was added for 4 h at 37°C followed by solubilization with DMSO until color became homogeneous. Cell viability for RS4;11 suspension cells was determined using a Cell Proliferation Kit 1 (MTT) purchased from Roche Applied Science (Indianapolis, IN) according to manufactures instructions. Absorbance readings were taken at 540 nm. Data are expressed relative to vehicle-treated controls (100% viability).
Caspase-3 assay
Caspase-3 activity was measured using a Caspase-3 Colorimetric Assay Kit (Millipore) according to manufacturer’s instructions. Briefly, 100 μg whole cell extract was incubated with caspase-3 substrate (5 μg) at 37°C for 3 h followed by quantification of the cleaved substrate by measuring absorbance at 405 nm. Values were corrected for background absorbance obtained in assays conducted in the absence of sample.
Statistical analysis
Data were analyzed by Student’s t-test with p < 0.05 being considered significantly different. All experiments were conducted with replicates of at least 3 and data reported as mean ± S.D. All key experiments have been repeated at least twice with essentially identical data.
Results
HeLa cells are dependent on Mcl-1 for survival and refractory to ABT-263
In order to test the hypothesis that HeLa cells are dependent on anti-apoptotic Bcl-2 family proteins for survival, and if so whether a specific member was dominant, a series of knockdown experiments using siRNA targeting Bcl-2, Bcl-xL, or Mcl-1 was performed followed by monitoring for cell death and cell viability. Extensive knockdown of Bcl-2, Bcl-xL, or Mcl-1 was achieved by 48 h and levels remained diminished through at least 72 h (Fig. 1A). Knockdown of Bcl-2 or Bcl-xL or both was without significant effect on PARP, a marker for caspase activation which remained intact, whereas, in contrast, knockdown of Mcl-1 resulted in complete PARP cleavage by 72 h (Fig. 1A). These results were further validated using a cell viability assay in which the percentage of viable cells was determined following knockdown of each anti-apoptotic protein for 72 h (Fig. 1B). As before, knockdown of Bcl-2 or Bcl-xL had no discernible effect on cell viability whereas knockdown of Mcl-1 led to a significant decrease in cell viability, when compared to cells transfected with a non-coding negative control siRNA. In order to more specifically address whether Mcl-1 knockdown induced apoptotic cell death, caspase-3 assays were conducted (Fig. 1C). Knockdown of Mcl-1 led to a robust increase in caspase-3 activity whereas knockdown of Bcl-2 or Bcl-xL resulted in minimal caspase-3 activation similar to that of non-coding control siRNA. The results of Fig. 1 demonstrate that loss of Mcl-1 is sufficient to induce apoptosis in HeLa cells and strikingly does so without any additional death stimulus. HeLa cells are therefore strictly dependent on Mcl-1 for survival.
Fig. 1.
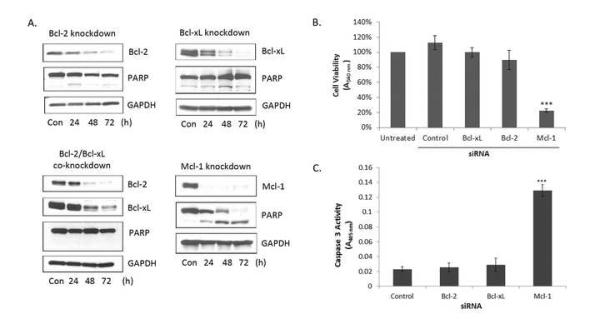
HeLa cells are dependent on Mcl-1 for survival. Cells were transfected with siRNA (50 nM) targeting Bcl-2, Bcl-xL, Bcl-2 + Bcl-xL, Mcl-1 or with a negative control siRNA. A. Cells were harvested 24-72 h post-transfection and extracts subjected to immunoblot analysis for the indicated proteins. GAPDH was used as a loading control. B. Relative cell viability was determined by MTT assay 72 h post transfection following siRNA treatment targeting Bcl-2, Bcl-xL or Mcl-1. Non-coding control siRNA was used as a negative control. Results given are mean ± standard deviation (n = 6). *** = p < 0.001. C. Caspase-3 activity was measured 48 h post transfection following siRNA treatment targeting Bcl-2, Bcl-xL or Mcl-1, as described in Materials and Methods. Non-coding control siRNA was used as a negative control. Results given are mean ± standard deviation (n = 3). *** = p < 0.001.
Recently, the development of small molecule Bcl-2/Bcl-xL inhibitors, such as ABT-263, has shown great promise in the treatment of a variety of cancers either alone or when combined with traditional chemotherapeutic agents [16, 17, 20, 32, 33]. Resistance to ABT-263 is frequently and most often observed in cells which express high levels of Mcl-1 [18-20], which is hypothesized to compensate for the inhibition of Bcl-2 and Bcl-xL. We therefore tested whether ABT-263 would be effective in HeLa cells when tested alone or in combination with the microtubule inhibitor vinblastine (Fig. 2). HeLa cells were found to be completely refractory to ABT-263 at concentrations as high as 10 μM, consistent with their dependence on Mcl-1 rather than Bcl-2 or Bcl-xL for survival. In contrast, RS4;11 leukemia cells, which have previously been shown to be Bcl-2 dependent [21], were sensitive to ABT-263 with an IC50 of 225±20 nM (Fig. 2A, left). Immunoblotting showed that RS4;11 cells express much higher levels of Bcl-2 and lower levels of Mcl-1 compared to HeLa cells (Fig. 2A, right). Additionally, combining ABT-263 and vinblastine showed no significant difference from treatment with vinblastine alone (Fig. 2B). Thus ABT-263 failed to sensitize HeLa cells to a conventional cytotoxic agent. Taken together, these results highlight the dependence of HeLa cells on Mcl-1 for survival and support previous evidence that expression of Mcl-1 is a marker of ABT-263 resistance [16, 18-20].
Fig. 2.
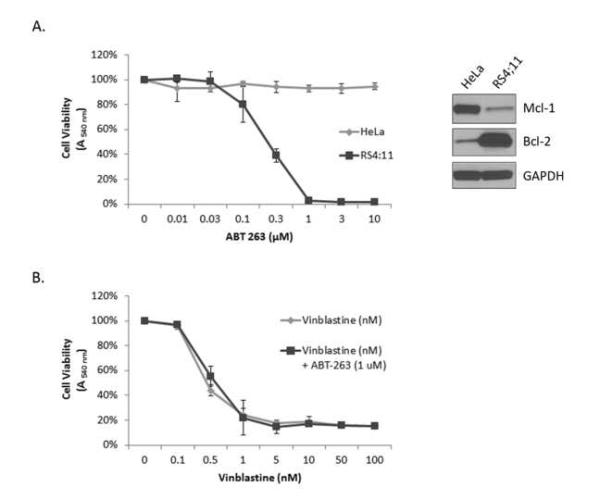
HeLa cells are refractory to the Bcl-2/Bcl-xL inhibitor ABT-737. A. HeLa cells or RS4;11 cells were treated with vehicle (0.1% DMSO) (100% viability) or treated with ABT-263 (0.01-10 μM) for 72 h followed by assesment for cell viability by MTT assay. Results given are mean ± standard deviation (n = 6). The right panel shows immunoblot analysis of indicated proteins in HeLa versus RS4;11 cells. B. HeLa cells were treated with vehicle (0.1% DMSO) (100% viability) or treated with vinblastine (0.1-100 nM) alone or with 1 μM ABT-263 for 72 h followed by assesment for cell viability by MTT assay. Results given are mean ± standard deviation (n = 6).
Redundancy and compensation in the anti-apoptotic Bcl-2 protein sub-family
Anti-apoptotic Bcl-2 sub-family proteins share significant homology and it is hypothesized that loss of one member could be compensated for by another anti-apoptotic Bcl-2 family member. As was demonstrated in Fig. 1 and Fig. 2, expression of Mcl-1 is sufficient to maintain survival of HeLa cells in the context of severely reduced levels or inhibition of both Bcl-2 and Bcl-xL. We next sought to determine whether the reverse was true, that is, whether Bcl-2 or Bcl-xL could compensate for loss of Mcl-1. To test this hypothesis, Mcl-1 was knocked down in HeLa cells generated to stably overexpress full length untagged Bcl-2 or Bcl-xL, as we have found that HA-tagged Bcl-2 proteins localize aberrantly [34]. Following knockdown of Mcl-1 for 72 h, cell viability was assessed in the Bcl-2/Bcl-xL overexpressing cells compared with control HeLa cells (Fig. 3A). Both Bcl-2 and Bcl-xL overexpressing cells showed significant protection from Mcl-1 knockdown, with Bcl-xL overexpression providing complete protection. Fig. 3B shows an immunoblot verifying Mcl-1 knockdown in the presence of Mcl-1 siRNA and demonstrating Bcl-2/Bcl-xL overexpression in the respective cell lines. Note that the level of Bcl-2 overexpression is such that endogenous Bcl-2 is not detected under these exposure conditions.
Fig. 3.

Bcl-2 and Bcl-xL overexpression compensates for the loss of Mcl-1 and confers sensitivity to ABT-263. A. Control HeLa cells or HeLa cells stably overexpressing Bcl-2 or Bcl-xL were transfected with siRNA targeting either Mcl-1 or a negative control siRNA (100% viability). Cell viability was assessed 72 h post-transfection using an MTT assay. Results given are mean ± standard deviation (n = 6). B. Immunoblot analysis of indicated proteins in control HeLa cells or cells stably overexpressing Bcl-2 or Bcl-xL before and after Mcl-1 knockdown. C. HeLa cells stably overexpressing Bcl-xL were co-treated with siRNA targeting either Mcl-1 or a negative control siRNA along with ABT-263 (10 μM) for 72 h and cell viability determined. Results given are mean ± standard deviation (n = 6). *** = p < 0.001.
We next tested the effect of ABT-263 in the context of Bcl-xL overexpression. In control HeLa cells, knockdown of Mcl-1 reduced cell viability (Fig. 1) and ABT-263 had no effect (Fig. 2). As shown in Fig. 3C, ABT-263 alone was not effective in killing Bcl-xL overexpressing cells, as presumably endogenous levels of Mcl-1 are able to compensate for inhibition of Bcl-xL. To test this directly, Mcl-1 was knocked down in Bcl-xL overexpressing which were subsequently treated with ABT-263 (Fig. 3C). In this case, cell viability was greatly reduced. Thus through the use of a combined knockdown/overexpression strategy, which essentially replaced Mcl-1 function with Bcl-xL, ABT-263 sensitivity was conferred to otherwise ABT-263 resistant cells. Taken together, these results indicate that the major anti-apoptotic Bcl-2 family proteins are functionally redundant and are able to compensate for one another.
Bcl-2 protein family dependency, redundancy, and compensation were next investigated in two colon carcinoma cell lines (DLD-1 and HT29). We focused on Bcl-xL and Mcl-1 as these cell lines have virtually undetectable levels of Bcl-2 [35]. Additionally, phosphorylation and inactivation of Bcl-xL and Mcl-1 in these cell lines has previously been shown to be critical for their response to microtubule inhibitors [35]. DLD-1 and HT29 cells were transfected with siRNA targeting Bcl-xL, Mcl-1, both Bcl-xL and Mcl-1, or a non-coding negative control for 72 h followed by assessment of cell viability (Fig. 4). Knockdown of Bcl-xL or Mcl-1 alone in DLD-1 cells led to only a modest but significant decrease in cell viability. Strikingly, co-knockdown of both Bcl-xL and Mcl-1 led to a major decrease in cell viability (Fig. 4A). Thus in DLD-1 cells, Bcl-xL and Mcl-1 play compensatory roles. In HT29 cells (Fig. 4B), a modest decrease in cell viability was observed after Bcl-xL knockdown, and knockdown of Mcl-1 led to an apparent increase in cell viability which may be attributable to increased proliferation of the cells over 72 h, as Mcl-1 has been shown to be a cell cycle inhibitor [36]. Co-knockdown of both Bcl-xL and Mcl-1 in HT29 cells led to only a slight decrease in cell viability when compared to knockdown of Bcl-xL alone (Fig 4B). When protein expression levels were examined, knockdown of each protein was not as efficient when both siRNAs were utilized in co-treatment conditions versus when individual siRNAs were used independently (Figs. 4A, 4B, right panels). Thus residual levels of both proteins, especially in the case of HT29 cells, may be sufficient to sustain cell viability under co-treatment conditions. Alternately, other anti-apoptotic Bcl-2 proteins such as A1 or Bcl-w may play a role. Also evident from the immunoblots of Figs. 4A and 4B was the striking observation that both DLD-1 and HT29 cells compensated for knockdown of Bcl-xL by increasing expression of Mcl-1, though the reverse was not true, with Bcl-xL levels similar before and after Mcl-1 knockdown. Mcl-1 expression has previously been shown to be transiently increased in cells which are resistant to ABT-263 [19], which together with the results presented here, indicates that upregulation of Mcl-1 expression is a key compensatory mechanism in response to inhibition or removal of Bcl-xL function.
Fig. 4.

Effect of Mcl-1, Bcl-xL knockdown on viability of DLD-1 and HT29 cells. DLD-1 (A) or HT29 (B) cells were transfected with 100 nM control siRNA (-) or siRNA to Bcl-xL or Mcl-1 for 24 h, trypsinized and seeded (2000 cells/well) in a 96-well culture dish. After 48 h, cell viability was determined. Results given are mean ± standard deviation (n = 6). ** = p < 0.01, *** = p < 0.001. Immunoblot analysis for determination of knockdown efficiency is shown on the right of each panel. GAPDH was used as a loading control. (C) Mcl-1 knockdown sensitizes HT-29 cells to ABT-263. HT29 cells were transfected with 100 nM control siRNA or siRNA to Mcl-1 for 24 h as indicated, trypsinized, and seeded (2000 cells/well) in a 96-well culture dish, and treated with increasing concentrations of ABT-263 (0-10 μM). After 48 h, cell viability was determined. Results given are mean ± standard deviation (n = 6). Immunoblot analysis for determination of knockdown efficiency is shown on the right with GAPDH as a loading control.
Because it was not possible to efficiently knockdown both Bcl-xL and Mcl-1 in HT-29 cells, a combination of Mcl-1 knockdown and treatment with ABT-263 was employed. Thus cells were treated with control siRNA or siRNA against Mcl-1 for 24 h, and then treated with increasing concentrations of ABT-263 for an additional 48 h, and cell viability assessed (Fig. 4C). After pre-treatment with control siRNA, HT29 cells were sensitive to ABT-263, with an IC50 value of 753 ± 104 nM (Fig. 4C), although they were less sensitive than RS4;11 cells (IC50 value of 225 ± 20 nM; Fig. 2A). However, after Mcl-1 knockdown, HT29 cells were srongly sensitized to ABT-263, with an IC50 value of 165 ± 7 nM (Fig. 4C). These results confirm that Bcl-xL and Mcl-1 play compensatory roles in HT-29 cells, and cell viability is most strongly diminished when both proteins are functionally compromised.
Bcl-xL compensates for loss of Mcl-1 through sequestration of Bak and Bim
We next sought to test the hypothesis that Bcl-xL is able to compensate for the loss of Mcl-1 by sequestering pro-apoptotic proteins normally sequestered by Mcl-1. Several BH3-only family members, including Bim, Bid, Bmf and Puma, as well as multi-domain Bak, bind to both Mcl-1 and Bcl-xL [37]. In order to identify pro-apoptotic Bcl-2 proteins bound to Mcl-1 in HeLa cells, co-immunoprecipitation was performed (Fig. 5A). Mcl-1 was efficiently immunoprecipitated and specificity was demonstrated as Mcl-1 remained in the supernatant in the absence of antibody (Fig. 5A, lanes 9-14). Immunoblotting of the immunoprecipitated material revealed the presence of Bak and several forms of Bim (Bim-EL, Bim-L, and Bim-S) (Fig. 5A, lane 13) but not Bax, Bid, or Puma (results not shown). Levels of immunoprecipitated Mcl-1 were comparable between control HeLa cells and HeLa overexpressing Bcl-xL, but levels of Bak and Bim were clearly reduced (Fig. 5A, compare lanes 13 and 14). To directly test whether overexpressed Bcl-xL assumed a role in binding Bak and Bim, Bcl-xL was immunoprecipitated (Fig. 5A, lanes 3-8). Immunoblotting confirmed the presence of increased levels of Bcl-xL in the overexpressing cells versus control HeLa cells and moreover clearly showed Bak and Bim to be bound (Fig. 5A, lane 8). While endogenous Bcl-xL may bind to some degree to these pro-apoptotic proteins, it is evident that overexpressed Bcl-xL has taken over the primary role from Mcl-1. These results show a direct switch in the sequestering of pro-apoptotic Bcl-2 family proteins by anti-apoptotic family members, and emphasize the critical role anti-apoptotic Bcl-2 protein redundancy and compensation plays.
Fig. 5.

Overexpressed Bcl-xL sequesters pro-apoptotic Bak and Bim. Extracts from HeLa cells (H) or HeLa cells stably overexpressing Bcl-xL (B) were subjected to immunoprecipitation using antibodies specific for Bcl-xL (lanes 7 and 8) or Mcl-1 (lanes 13 and 14). Precipitations conducted in the absence of antibody (N) are shown in lanes 6 and 12, and the corresponding supernatants for Bcl-xL (lanes 3-5) or Mcl-1 (lanes 9-11) are also indicated, with whole cell extracts shown in lanes 1 and 2. Immunoblot analysis was performed for the indicated proteins and the three isoforms of Bim, extra long (EL), long (L), and short (S), are indicated. GAPDH was used as a loading control. Note that for the Bak blot, a longer exposure time was used to visualize the bands in lanes 1-5 versus the bands in lanes 6-14. B. Loss of Mcl-1 leads to the activation of Bak. Cell extracts (WCE) were prepared from HeLa cells 48 h post-transfection with siRNA targeting Mcl-1 or a negative control siRNA. Immunoprecipitations (IP) were performed using an antibody (Ab) specific to active Bak (NT-Bak) or in the absence of active Bak antibody as indicated. The corresponding supernatants (Sup) were also analyzed. Immunoblotting was performed using an antibody recognizing total Bak. GAPDH was used as a loading control.
Loss of Mcl-1 leads to activation of Bak
Because a primary role for Mcl-1 in HeLa cells is the sequestration of Bak and Bim, we reasoned that knockdown of Mcl-1 would lead to Bak activation. To test this hypothesis, immunoprecipitation using an antibody that recognizes the active form of Bak (NT-Bak) was performed on HeLa cells following knockdown of Mcl-1. As hypothesized, loss of Mcl-1 led to the activation of Bak, while total levels of Bak remained unchanged, and active Bak was not detectable in cells transfected with control siRNA (Fig. 5B). These results confirm the hypothesis that loss of Mcl-1 leads to the activation of Bak, which in turn initiates apoptosis.
Bim is essential for cell death induced by Mcl-1 knockdown
The immunoprecipitation results of Fig. 5A indicated that Mcl-1 sequesters the activator Bim as well as the effector Bak. Because knockdown of Mcl-1 leads to loss of sequestration of Bim and Bak and subsequent Bak activation, a key question is whether the release of Bak is sufficient, or whether release of the activator Bim is also required to initiate apoptosis. To answer this question, siRNA targeting Bim was used, either alone or together with siRNA targeting Mcl-1. As shown in Fig. 6A, efficient knockdown of all three forms of Bim (Bim-EL, Bim-L, and Bim-S) was observed, and efficient knockdown of both Bim and Mcl-1 was achieved when cells were co-treated with siRNAs targeting both proteins. Importantly, while knockdown of Mcl-1 alone resulted in PARP cleavage (Fig. 6A) and loss of cell viability (Fig. 6B), as expected, under conditions of co-knockdown of both Mcl-1 and Bim, cells were essentially completely protected, with no evidence of PARP cleavage and retention of cell viability. These results demonstrate conclusively that cell death in HeLa cells induced by Mcl-1 knockdown requires release of the activator Bim.
Fig. 6.
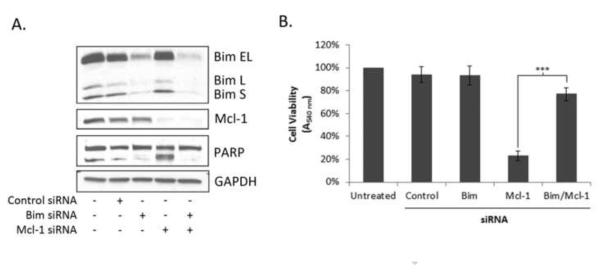
Depletion of Bim protects cells from loss of Mcl-1. Cells were transfected with siRNA (50 nM) targeting Bim, Mcl-1, Bim + Mcl-1, or with a negative control siRNA. A. Cells were harvested 48 h post-transfection and extracts subjected to immunoblot analysis for the indicated proteins. GAPDH was used as a loading control. B. Relative cell viability was determined by MTT assay 72 h post transfection. Results given are mean ± standard deviation (n = 6). *** = p< 0.001.
Discussion
It has become increasingly evident that dysregulation of cell death pathways, especially intrinsic apoptosis, plays a key role in cancer cell survival and chemoresistance [38]. Indeed, many cancer cells exhibit abnormal phenotypes, such as chromosomal instability and oncogene activation that would normally promote apoptotic signaling but fail to do so due to specific blocks in downstream death pathways [39]. Recently, the innovative technique of BH3 profiling has been introduced and developed to distinguish these selective blocks in apoptosis [39]. Broadly, cancer cells may exhibit defects upstream at the level of activation of BH3-only proteins, may be defective at the level of the effectors Bax and Bak, or may exhibit an apoptotic block due to overexpression of pro-survival Bcl-2 proteins. Cancers in this latter group are considered primed for death because the pro-survival Bcl-2 proteins are typically and largely occupied by activator BH3-only proteins [39]. Interestingly, dependence on a particular pro-survival Bcl-2 protein is dictated not so much by its overall expression level but more by its level of occupancy by activator BH3-only proteins [39]. Genetic alterations, including chromosomal translocations [10, 40] and gene amplification [15], as well as changes in micro-RNAs [41, 42], appear to be in part responsible for different cancers becoming dependent on different members of the anti-apoptotic Bcl-2 family for survival.
In this study we combined knockdown with overexpression to investigate compensation and redundancy in this critically important Bcl-2 subfamily. A key initial finding was that HeLa cells are strictly dependent on Mcl-1 for survival, despite the presence of other anti-apoptotic Bcl-2 proteins, and highlights the critical role played by members of the pro-survival Bcl-2 subfamily in guarding against apoptotic death. Precisely why HeLa cells exhibit dependence on Mcl-1 and not other anti-apoptotic Bcl-2 proteins is not clear, but presumably genetic alterations leading to Mcl-1 overexpression provided a survival advantage at some point during genesis and progression of such cervical carcinomas. Importantly, while endogenous levels of Bcl-2 and Bcl-xL were not sufficient to compensate for loss of Mcl-1, HeLa cells were unaffected by Mcl-1 knockdown if either Bcl-2 or Bcl-xL was overexpressed (Fig. 3A). This result suggests that endogenous levels of Bcl-2 and Bcl-xL do not provide a sufficient reservoir for sequestration of pro-apoptotic proteins freed through the loss of their normal guardian, Mcl-1. Unlike HeLa cells, the two colon carcinoma cell lines tested were not strictly dependent on Mcl-1 for survival. Instead, the protective role of anti-apoptotic Bcl-2 family proteins appears to be shared by multiple family members. Thus, knockdown of Mcl-1 and knockdown or inhibition of Bcl-xL were necessary to maximally decrease cell viability in DLD-1 and HT-29 cells (Fig. 4). These results highlight the need for the development of anti-apoptotic Bcl-2 family protein inhibitors that target multiple family members.
A strategy combining knockdown of Mcl-1 with overexpression of Bcl-xL provided a novel and convenient means to essentially replace the function of one with the other. Under these conditions, HeLa cells became Bcl-xL-dependent and sensitivite to ABT-263 (Fig. 3C). The ability of Bcl-xL to assume primary guardianship from Mcl-1 resided in its ability to bind to Bak and Bim, which were the major pro-apoptotic proteins found in complex with Mcl-1, and which become available for binding to Bcl-xL upon loss of Mcl-1 via knockdown (Fig. 5A). To our knowledge, the data of Fig. 5A represent one of the most striking and illustrative examples of pro-apoptotic Bcl-2 protein swapping yet reported. These results further emphasize another important feature of the pro-survival Bcl-2 proteins, namely their shared ability to bind to activator BH3-only proteins and effector pro-apoptotic Bcl-2 proteins. Thus Mcl-1 and Bcl-xL have in common the ability to bind with high affinity to Bim, and both have the ability to bind to the BH3 domain of Bak [43]. These results provide molecular evidence for the mechanism by which anti-apoptotic Bcl-2 proteins function redundantly and are able to compensate for the loss of one family member through expression of another.
Upon Mcl-1 knockdown, Bak became activated which typically requires interaction with an activator BH3-only protein [44], and Mcl-1 was found to bind to both Bak and Bim. This raised the interesting question of whether cell death in response to Mcl-1 knockdown was dependent on Bim. This was tested by co-knockdown of both Mcl-1 and Bim and it was conclusively demonstrated that Bim is essential for cell death induced by Mcl-1 knockdown. To our knowledge, this is the first example where apoptosis induced by loss of Mcl-1 was rescued by the silencing of a single BH3-only Bcl-2 family member. Interestingly, in several other studies, loss of Bim was not sufficient to reverse cell death mediated by Mcl-1 deletion [45-47]. For example, deletion of Bim and Puma failed to reverse cell death mediated by Mcl-1 deletion in myeloid progenitor cells, resulting in neutropenia and loss of mature neutrophils [45]. However, Mcl-1 deficient macrophages exhibited increased sensitivity to death during bacterial phagocytosis that was abolished by co-deletion of Bim [45]. Tripathi et al. showed that loss of Bim did not prevent Mcl-1 deficient T cells from undergoing death in response to virus infection [46]. These results, taken together, emphasize that anti-apoptotic Mcl-1 may play selective roles in cellular differentiation and setting an apoptotic threshold in mature cells which may be cell line-specific. Intriguingly, in HeLa cells, we observed that a single pro-survival Bcl-2 family member, Mcl-1, sequestered both a critical activator, Bim, and an effector, Bak, in the pro-apoptotic Bcl-2 subfamily. It will be of interest to determine whether other cancer cell lines dependent on a particular pro-survival Bcl-2 family member also display similar Bcl-2 binding characteristics, that is, the co-binding of an activator and effector to a single pro-survival member. Conversely, in cancer cells not strictly dependent on one pro-survival Bcl-2 protein, it will be of interest to determine whether such responsibility is divided between different pro-survival members. Emerging biophysical techniques which allow the assessment of the extent and type of pro-apoptotic Bcl-2 protein occupancy by pro-survival Bcl-2 proteins will be enlightening in this regard [48].
Highlights.
HeLa cells were found to be strictly dependent on Mcl-1 for survival and refractory to the Bcl-2 antagonist ABT-263
In the absence of Mcl-1 and with Bcl-xL overexpressed, the cells became Bcl-xL-dependent and ABT-263-sensitive
Bcl-xL compensated for Mcl-1 loss by binding Bak and Bim normally bound to Mcl-1
Bim was essential for cell death induced by Mcl-1 loss
Bcl-xL and Mcl-1 also play compensatory roles in colon carcinoma cells
Acknowledgement
This work was supported by the National Institutes of Health Grant CA-109821 from the National Cancer Institute to TCC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
- BH
- Bcl-2 homology
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- DMEM
- Dulbecco’s modified Eagle’s medium
- PARP
- poly (ADP-ribose) polymerase
REFERENCES
- 1.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 16442004:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 1162004:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 172005:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 4.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes & development. 192005:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer research. 672007:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 6.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer cell. 92006:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 7.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 3092005:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 8.Leber B, Lin J, Andrews DW. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene. 292010:5221–5230. doi: 10.1038/onc.2010.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Cui J, Zhang W, Shen P. Robustness analysis identifies the plausible model of the Bcl-2 apoptotic switch. FEBS letters. 5812007:5143–5150. doi: 10.1016/j.febslet.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 10.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 3351988:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 11.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 521992:6940–6944. [PubMed] [Google Scholar]
- 12.Jansen B, Schlagbauer-Wadl H, Brown BD, Bryan RN, van Elsas A, Muller M, Wolff K, Eichler HG, Pehamberger H. bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nat Med. 41998:232–234. doi: 10.1038/nm0298-232. [DOI] [PubMed] [Google Scholar]
- 13.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 551995:3902–3907. [PubMed] [Google Scholar]
- 14.Pezzella F, Turley H, Kuzu I, Tungekar MF, Dunnill MS, Pierce CB, Harris A, Gatter KC, Mason DY. bcl-2 protein in non-small-cell lung carcinoma. N Engl J Med. 3291993:690–694. doi: 10.1056/NEJM199309023291003. [DOI] [PubMed] [Google Scholar]
- 15.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 4632010:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer cell. 102006:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 4352005:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 18.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, Fesik SW, Shen Y. 'Seed' analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 262007:3972–3979. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 19.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 1152010:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer cell. 102006:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 1112008:2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boiani M, Daniel C, Liu X, Hogarty MD, Marnett LJ. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. The Journal of biological chemistry. 2882013:6980–6990. doi: 10.1074/jbc.M112.414177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Li G, Ma X, Wang Y, Liu G, Feng L, Zhao Y, Zhang G, Wu Y, Ye X, Qin B, Lu J. Norcantharidin enhances ABT-737-induced apoptosis in hepatocellular carcinoma cells by transcriptional repression of Mcl-1. Cellular signalling. 2012 doi: 10.1016/j.cellsig.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Russo M, Spagnuolo C, Volpe S, Tedesco I, Bilotto S, Russo GL. ABT-737 resistance in B-cells isolated from chronic lymphocytic leukemia patients and leukemia cell lines is overcome by the pleiotropic kinase inhibitor quercetin through Mcl-1 down-regulation. Biochemical pharmacology. 852013:927–936. doi: 10.1016/j.bcp.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Hikita H, Takehara T, Shimizu S, Kodama T, Shigekawa M, Iwase K, Hosui A, Miyagi T, Tatsumi T, Ishida H, Li W, Kanto T, Hiramatsu N, Hayashi N. The Bcl-xL inhibitor. ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib, Hepatology. 522010:1310–1321. doi: 10.1002/hep.23836. [DOI] [PubMed] [Google Scholar]
- 26.Adams KW, Cooper GM. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. The Journal of biological chemistry. 2822007:6192–6200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R, Golub TR. Chemical Genomics Identifies Small-Molecule MCL1 Repressors and BCL-xL as a Predictor of MCL1 Dependency. Cancer cell. 212012:547–562. doi: 10.1016/j.ccr.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eno CO, Zhao G, Olberding KE, Li C. The Bcl-2 proteins Noxa and Bcl-xL co-ordinately regulate oxidative stress-induced apoptosis. The Biochemical journal. 4442012:69–78. doi: 10.1042/BJ20112023. [DOI] [PubMed] [Google Scholar]
- 29.Goldsmith KC, Gross M, Peirce S, Luyindula D, Liu X, Vu A, Sliozberg M, Guo R, Zhao H, Reynolds CP, Hogarty MD. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 722012:2565–2577. doi: 10.1158/0008-5472.CAN-11-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. The Journal of biological chemistry. 2752000:29980–29985. doi: 10.1074/jbc.M003776200. [DOI] [PubMed] [Google Scholar]
- 31.Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Molecular and cellular biology. 302010:640–656. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell death and differentiation. 162009:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 33.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer research. 672007:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 34.JM Eichhorn, S Sakurikar, SE Alforde, R Chu, Chambers TC. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death and Disease. doi: 10.1038/cddis.2013.360. manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakurikar N, Eichhorn JM, Chambers TC. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. The Journal of biological chemistry. 2872012:39193–39204. doi: 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. The Journal of biological chemistry. 2752000:39458–39465. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]
- 37.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. Journal of cell science. 1222009:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 1442011:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nature reviews. Cancer. 82008:121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 40.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 2261984:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 41.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 1022005:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 3532005:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 43.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 372010:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Stepwise activation of BAX and BAK by tBID. BIM, and PUMA initiates mitochondrial apoptosis, Mol Cell. 362009:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood. 1132009:2805–2815. doi: 10.1182/blood-2008-05-159145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripathi P, Koss B, Opferman JT, Hildeman DA. Mcl-1 antagonizes Bax/Bak to promote effector CD4(+) and CD8(+) T-cell responses. Cell death and differentiation. 202013:998–1007. doi: 10.1038/cdd.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koss B, Morrison J, Perciavalle RM, Singh H, Rehg JE, Williams RT, Opferman JT. Requirement for antiapoptotic MCL-1 in the survival of BCR-ABL B-lineage acute lymphoblastic leukemia. Blood. 1222013:1587–1598. doi: 10.1182/blood-2012-06-440230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Saez AJ. The secrets of the Bcl-2 family. Cell death and differentiation. 192012:1733–1740. doi: 10.1038/cdd.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]