

Abstract
The tumor microenvironment including glial cells and their inflammatory products regulates brain tumor development and progression. We have previously established that human glioma cells are exquisitely sensitive to IL-1 stimulation leading us to undertake a comparative analysis of the secretome of unstimulated and cytokine (IL-1)-stimulated glioblastoma cells. We performed label-free quantitative proteomic analysis and detected 190 proteins which included cytokines, chemokines, growth factors, proteases, cell adhesion molecules, extracellular matrix (ECM) and related proteins. Measuring area under the curve (AUC) of peptides for quantitation, the IL-1-induced secretome contained 13 upregulated and 5 downregulated extracellular proteins (p<0.05) compared to controls. Of these, IL-8, CCL2, TNC, Gal-1 and PTX3 were validated as upregulated and SERPINE1, STC2, CTGF and COL4A2 were validated as downregulated factors by immunochemical methods. A major representation of the ECM and related proteins in the glioblastoma secretome and their modulation by IL-1 suggested that IL-1 induces its effect in part by altering TGFβ expression, activity and signaling. These findings enhance our understanding of IL-1-induced modulation of glioma microenvironment, with implications for increased tumor invasion, migration and angiogenesis. They further provide novel targets for the glioblastoma intervention.
Keywords: interleukin-1, glioblastoma, secretome, label free quantitation, TGFβ, extracellular matrix, innate immunity
1. Introduction
Glioblastoma multiforme (GBM) is the most common brain tumor with very poor prognosis despite recent progress in chemotherapy and immunotherapy [1-3]. The poor prognosis of malignant gliomas is related to the ability of tumor cells to infiltrate the surrounding tissue, making the tumor non-resectable, as well as their high degree of neo-angiogenesis and tumor necrosis [4]. During tumorigenesis/progression, the normal balance between critical proteins such as proteases, their inhibitors, and adhesion molecules is dysregulated. It is also expected that other not yet identified proteins are involved in this process. In addition to the cell-associated proteins, glioma-produced secreted proteins also play a major role in cell-cell communication with other glioma cells, tumor vessels, inflammatory cells, and endogenous brain cells [3]. Therefore, analysis of proteins secreted by glioblastoma cells in an unbiased manner (secretome) is an important approach to understand these mechanisms.
Gliomas in vivo grow in a highly complex microenvironment. Microglia and astrocytes are among the brain cells implicated in facilitating glioma growth, invasion, angiogenesis and anti-tumor surveillance [3;5]. Tumor-associated microglia and astrocytes show an activated phenotype with a global change in their transcriptional, metabolic and secretory profiles, but the details of these changes and their impact on glioma progression are not known. Of the soluble mediators that are produced by activated microglia, IL-1 plays a particularly important role in the activation of glioma cells and initiating the neuroinflammatory cascade in the central nervous system (CNS) [6-9]. Human glioblastoma cells are exquisitely sensitive to IL-1 stimulation [10] (and this report). Importantly, evidence also supports that glioblastoma cells in vivo and in vitro also produce IL-1 [11;12] (Tarassishin and Lee, unpublished) and that IL-1 itself is also a potent inducer of IL-1 production in myeloid cells and in human glioma cells [13;14](manuscript in preparation). These results together indicate that IL-1-mediated cellular interactions involving GBM cells, tumor-associated microglia, neuroglial cells, and blood vessels can set off a potent proinflammatory, pro-tumor and neurotoxic cascade in the CNS [15-17].
Given the importance of inflammation in tumorigenesis and progression [18;19], and the central importance of IL-1 in the establishment of glioma microenvironment, in the present study we performed quantitative proteomic analysis of the glioblastoma secretome, comparing unstimulated and IL-1-stimulated glioblastoma cells (U251). This approach provides a platform of non-targeted and unbiased discovery for large number of differentially secreted proteins. We found 190 proteins including cytokines, growth factors, ECM and related proteins. Their altered abundance upon IL-1β stimulation imitates the inflammatory response and provides insight into the extracellular events resulting in pro-tumor environment.
2. Materials and methods
2.1 Cells, reagents, and sample preparation
Glioblastoma cell line U251 was cultivated in DMEM supplemented with 5% FBS and antibiotics (“Anti-anti” from Invitrogen/Life Technologies, Grand Island, NY). Recombinant human IL-1β was purchased from Peprotech (Rocky Hill, NJ). For cytokine stimulation, the cells were grown in 25 cm dishes until ~90% confluence, then incubated with IL-1β (10 ng/ml) for 15 min, which we previously determined to be sufficient to activate human astrocytes [20]. Cultures were then washed extensively with PBS to remove carry over cytokines and were further incubated with serum-free DMEM for an additional 24 h. Control medium was produced similarly except that the cytokine treatment was omitted.
Conditioned medium (CM) was collected by gentle aspiration and then centrifuged at 2,000 rpm for 10 min to remove cell debris. CM was concentrated using polyethersulfonate membrane > 5,000 MWCO (Sartorius Stedim Biotech GmBH, Gethingen, Germany), and washed twice with sterile ddH20 (x2 volume) in order to decrease the salt loading at MS. Samples were then denatured by boiling for 5 min and centrifuged at 10,000 rpm for 5 min to remove possible protein aggregates. Protein concentration was determined with Bio-Rad Protein Assay reagents (Bio-Rad, Hercules, CA). This process was repeated three times to collect three sets of controls and stimulated media.
2.2 SDS-PAGE and Western blotting
Ten (10) μg of samples were incubated with equal volume of 2x dissociation buffer for 5 min at 95 °C and applied to the Criterion 4-20% gradient gel or 10% gel as indicated. After electrophoresis, the gels were stained with Bio-Safe Coomassie Blue. All reagents were from Bio-Rad.
For Western blotting, the proteins were transferred to polyvinylidene difluoride membrane. The membrane was blocked in PBS containing 5% nonfat milk and then incubated with antibodies at 4°C for 16 h. Primary antibodies were: anti-Tenascin-C, anti-Galectin-1, and anti-Pentraxin 3 (1:1,000, R&D Systems, Minneapolis, MN), anti-Stanniocalcin-2 (1:100, ThermoScientific, Franklin, MA), anti-SERPINE1 (1:400, Sigma-Aldrich, St Louis, MO), anti-MMP2 (1:250, Cell Signaling, Beverly, MA), anti-CTGF (1:200, Santa Cruz Biotech, Santa Cruz, CA), and anti-COL4A2 (1:200, Santa Cruz Biotech). Secondary antibodies were: horseradish peroxidase (HRP) - conjugated anti-goat IgG (1:5,000, Rockland Immunochemicals, Gilbertville, PA), anti-rabbit IgG (1:500) and anti-mouse IgG (1:500, ThermoScientific) and incubated for 1 h at room temperature. Signals were developed using enhanced chemiluminescence (Super Signal West or Pico Chemiluminescent Substrate, ThermoScientific). After developing, the X-ray films were scanned and densitometry analyses were performed with NIH Image J software. Statistical analyses of western blot data were performed by Student’s t-test (Ctr vs. IL-1treated samples, n =3) using GraphPad Prism 5.0.
2.3 ELISA
ELISAs for IL-8, CCL2 and TGFβ1 were performed using R&D Systems DuoSet reagents following the manufacturer’s protocols. The sensitivity ranges for ELISA were 32.2 - 2,000 pg/ml for IL-8, 15.6 - 1,000 pg/ml for CCL2, and 31.2 - 2000 pg/ml for TGFβ1. The samples were diluted until the values fell within the linear range of detection. Statistical analyses of ELISA data were performed by Student’s t-test (Ctr vs. IL-1treated samples, n =3) using GraphPad Prism 5.0.
2.4 Mass Spectrometry Analysis of conditioned media
Concentrated and washed CM retentates from the membrane filters were loaded on a PAGE gel (12%) and run to ~6 mm to remove the dye from cell medium. Complete lanes were excised using disposable grid cutters (The Gel company, San Francisco, CA) to produce 6 bands with the size of 10X1X1mm3 from each samples. Two band slices were placed in each well of 96 well plates and in-gel digested using an Ettan Digester (GEHealthcare, Piscataway, NJ). Gel pieces were reduced with DTT and alkylated with iodoacetamide as is described previously [21].
Digested peptides from all 6 bands were pooled before MS analysis. An Agilent 1100 series Nano HPLC interfaced to a QStar XL mass spectrometer (AB Sciex, Ontario, Canada) was used for analysis. Extracted peptides were loaded onto a Zorbax 300SB-C18 trap column (5 μm, 300 Å, 0.3 mm) at a flow rate of 8 μl/min with 2% CH3CN/0.1% trifluoroacetic acid and delivered to an Acclaim 300 nanocolumn (C18, 3 μm, 300 Å, 75 μm i.d. × 15 cm, Dionex, Sunnyvale, CA, USA) by a switching mechanism. Peptides were eluted from the nanocolumn at a flow rate of 250 nl/min with 2% CH3CN/0.1% formic acid (solvent A) and 90% CH3CN/0.1% formic acid (solvent B). The gradients used were as follows: 0 to 30 min, 5% B (desalting); 30 to 80 min, 5 to 25% B; 80 to 95 min, 25 to 90% B; 95 to 110 min, 90% B; 110 to 120 min, 90 to 5% B; 120 to 130 min, 5% B. A nanospray voltage in the range of 2000 to 2400 V was optimized daily. All nanoLC-MS/MS data were acquired in the data-dependent acquisition mode in Analyst QS 1.1 (AB Sciex). TOF (time-of-flight) MS survey scans had a range of 300 to 1600 m/z for 1 s, followed by a product ion scan with a range of 50 to 1600 m/z for 2 s each. Collision energy was automatically controlled by the IDA CE Parameters script.
2.5 Protein identification
Once obtained, peak lists were generated from MS/MS spectra using AB SCIEX MS Data Converter ver 1.2 and searched against the IPI-Human database (version 3.73) concatenated with a reverse decoy using Mascot (version 2.3, Matrix Science). Fixed modification of cysteines to S-carbamidomethyl derivatives and variable oxidation (M) were defined for the database search. One missed cleavage was allowed with trypsin, and mass tolerance was set to 100 ppm for precursor ions and 0.2 Da for fragment ions. Searched results were exported as DTA files and grouped to protein matches using ProteoIQ (Ver.2.6.03, Nusep). Protein hits were filtered to include only those that were identified with less than 1% false discovery rate and 2 peptides detection.
2.6 Protein quantitation and Statistics
Wiff files from LC-MS/MS analysis were converted to mzXML with Trans Proteomic Pipeline ver 4.4 [22]. Intensity of peaks was extracted from mzXML using deisotoped accurate mass retention time (RT) pairs which were replicated throughout the entire experiments. Windows of the mass tolerance and RT were 100ppm and 15 seconds each. Then, those intensity measurements were matched to the redundant, tryptic peptides identified from each protein to determine an average, relative protein fold-change. Nonproteotypic peptides with significantly different ratios from proteotypic peptides of the same protein were excluded from quantitation. Systematic bias was corrected using intensity normalization using ProteoIQ. Proteins from 139 protein groups that demonstrated the median expression from the IL-1 simulated group were selected as “control” proteins for normalization. Normalization factors for each replicate were calculated for each control protein and then averaged across all proteins to determine normalization factors for each replicate. These factors were then applied to replicate intensities for all peptides in the dataset for quantitation. Presence of an interfering precursor which overlaps with the distribution of interest were removed from quantitation using the correlation coefficient measured between the theoretical and experimental isotopic distributions for the quantitative precursors.
Biological replicates were analyzed from different cultures and their analytical reproducibility was assessed by comparing protein expressions across biological replicates using average spectral counts and number of peptides. Average and replicate values from three measurements in each group were compared using Pearson correlation coefficient.
Statistical significance was measured using both Student’s t-test and Mann-Whitney U test. Student’s t-test was performed as implemented in ProteoIQ. For each protein, relative abundances for all peptides from all biological replicates were combined for the calculation, i.e. measurements from biological replicates were not treated separately. Thus, replicate sample size for each protein varied with number of peptide identifications. The p-value is calculated using the FDistribution function from the apache commons math library (http://commons.apache.org/proper/commons-math/download_math.cgi).
The Mann-Whitney U test was performed using a custom Java program as follows. For each peptide of each protein, the number of biological replicates that produced a measureable signal was determined for each sample, and from those counts we determined the minimum number of replicates from both samples. This value represented the maximum number of measurements to be used from each sample for the statistic. A reference value (normalized intensity) was then randomly chosen from the control sample replicates, and the relative abundance was calculated for each IL-1 sample (up to the maximum allowed) and placed in a list for IL-1.
Similarly, a list was created for the control sample by randomly choosing an IL-1 reference value and proceeding accordingly. The p-value was calculated from the two resulting lists using the Mann-Whitney function from the apache commons math3 library. Finally, in order to ensure that the calculation was not biased from any random choices, the calculation was repeated 1000 times, and the mean p-value was reported. Systematic bias was corrected with intensity normalization using TIC. Grouped peptides intensities were compared between control and IL-1 stimulated groups. T-test was performed to identify discriminating peptides between two groups.
2.7 Functional classification of secreted proteins
Acquired protein groups were annotated with GO using the Panther classification system [23] to determine the subcellular location and molecular function of them. FASTA files contains the sequences of all identified proteins were generated and uploaded to Secretome P 2.0 server [24] which produces ab initio predictions of non-classical protein secretion. Lastly, proteins and their ratios in table 1 were analyzed using Ingenuity Pathway® to identify the upstream transcriptional regulators in the dataset.
Table 1.
| Sequence Id | Sequence Name | P value# |
log2(ILβ/control) | ILβ/control ratio |
|---|---|---|---|---|
| IPI00215621* | IL8 Isoform 2 of Interleukin-8 | 0.0019 | 2.65 | 6.3 |
| IPI00412264 | PTN Pleiotrophin | 0.122 | 2 | 4.0 |
| IPI00029568* | PTX3 Pentraxin-related protein PTX3 | 0.0001 | 1.85 | 3.6 |
| IPI00009308 | CCL2 C-C motif chemokine 2 | 0.0001 | 1.7 | 3.2 |
| IPI00031008* | TNC Isoform 1 of Tenascin | 0.0015 | 1.07 | 2.1 |
| IPI00002714 | DKK3 Dickkopf-related protein 3 | 0.1213 | 0.9 | 1.9 |
| IPI00550991 | SERPINA3 highly similar to ALPHA-1- ANTICHYMOTRYPSIN |
0.0003 | 0.87 | 1.8 |
| IPI00470607 | FAM20C dentin matrix protein 4 | 0.1239 | 0.84 | 1.8 |
| IPI00009720 | LIF Leukemia inhibitory factor | <0.0001 | 0.81 | 1.8 |
| IPI00007221 | SERPINA5 Plasma serine protease inhibitor | 0.0003 | 0.62 | 1.5 |
| IPI00027497 | GPI Glucose-6-phosphate isomerase | 0.0926 | 0.56 | 1.5 |
| IPI00914848 | SERPINE2 Isoform 2 of Glia-derived nexin | 0.0044 | 0.56 | 1.5 |
| IPI00922213 | FN1 highly similar to Homo sapiens fibronectin 1 (FN1) |
0.0511 | 0.55 | 1.5 |
| IPI00016112 | PXDN Isoform 1 of Peroxidasin homolog | 0.0004 | 0.48 | 1.4 |
| IPI00008894 | CPA4 Carboxypeptidase A4 | 0.4189 | 0.45 | 1.4 |
| IPI00022822 | COL18A1 Isoform 2 of Collagen alpha-1(XVIII) chain |
0.1591 | 0.43 | 1.3 |
| IPI00009362 | SCG2 Secretogranin-2 | 0.1572 | 0.38 | 1.3 |
| IPI00219219 | LGALS1 Galectin-1 | 0.0049 | 0.35 | 1.3 |
| IPI00073454 | COL6A2 Isoform 2C2A' of Collagen alpha-2(VI) chain |
0.2688 | 0.3 | 1.2 |
| IPI00006273 | CYR61 CYR61 protein | 0.2598 | 0.22 | 1.2 |
| IPI00027166 | TIMP2 Metalloproteinase inhibitor 2 | 0.3788 | 0.22 | 1.2 |
| IPI00022418 | FN1 Isoform 1 of Fibronectin | 0.2783 | 0.19 | 1.1 |
| IPI00073196 | LTBP3 Isoform 1 of Latent-transforming growth factor beta-binding protein 3 |
0.4223 | 0.18 | 1.1 |
| IPI00296180 | PLAU Isoform 1 of Urokinase-type plasminogen activator |
0.1473 | 0.17 | 1.1 |
| IPI00002802 | LOX Protein-lysine 6-oxidase | 0.3572 | 0.16 | 1.1 |
| IPI00013976 | LAMB1 Laminin subunit beta-1 | 0.1865 | 0.15 | 1.1 |
| IPI00301579 | NPC2 cDNA FLJ59142, highly similar to Epididymal secretory protein E1 |
0.3973 | 0.13 | 1.1 |
| IPI00298281 | LAMC1 Laminin subunit gamma-1 | 0.2957 | 0.09 | 1.1 |
| IPI00291136 | COL6A1 Collagen alpha-1(VI) chain | 0.5165 | 0 | 1.0 |
| IPI00005686 | LIPG Isoform 1 of Endothelial lipase | 0.3959 | 0 | 1.0 |
| IPI00298971 | VTN Vitronectin | 0.4164 | 0 | 1.0 |
| IPI00003351 | ECM1 Isoform 1 of Extracellular matrix protein 1 | 0.4395 | −0.01 | 1.0 |
| IPI00027780 | MMP2 72 kDa type IV collagenase | 0.3991 | −0.03 | 1.0 |
| IPI00297284 | IGFBP2 insulin-like growth factor-binding protein 2 precursor |
0.2069 | −0.04 | 1.0 |
| IPI00016915 | IGFBP7 Insulin-like growth factor-binding protein 7 | 0.4361 | −0.06 | 1.0 |
| IPI00032293 | CST3 Cystatin-C | 0.4281 | −0.07 | 1.0 |
| IPI00011865 | PDGFD Isoform 2 of Platelet-derived growth factor D |
0.4448 | −0.08 | 0.9 |
| IPI00014572 | SPARC Secreted protein, acidic, cysteine-rich (Osteonectin) |
0.3801 | −0.1 | 0.9 |
| IPI00008780 | STC2 Stanniocalcin-2 | 0.3497 | −0.1 | 0.9 |
| IPI00844090 | COL5A1 Collagen alpha-1(V) chain | 0.4186 | −0.11 | 0.9 |
| IPI00029236 | IGFBP5 Insulin-like growth factor-binding protein 5 | 0.2876 | −0.12 | 0.9 |
| IPI00026944 | NID1 Isoform 1 of Nidogen-1 | 0.4153 | −0.13 | 0.9 |
| IPI00218539 | COL11A1 Isoform B of Collagen alpha-1(XI) chain I | 0.4589 | −0.14 | 0.9 |
| IPI00296534 | FBLN1 Isoform D of Fibulin-1 | 0.3628 | −0.15 | 0.9 |
| IPI00783665 | LAMA5 Laminin subunit alpha-5 | 0.5384 | −0.21 | 0.9 |
| IPI00009294 | CRIM1 Cysteine-rich motor neuron 1 protein | 0.2685 | −0.23 | 0.9 |
| IPI00005564 | STC1 Stanniocalcin-1 | 0.0249 | −0.23 | 0.9 |
| IPI00410487 | TWSG1 Isoform 1 of Twisted gastrulation protein homolog 1 |
0.3891 | −0.23 | 0.9 |
| IPI00009802 | VCAN Isoform V0 of Versican core protein | 0.152 | −0.23 | 0.9 |
| IPI00032292 | TIMP1 Metalloproteinase inhibitor 1 | 0.1998 | −0.25 | 0.8 |
| IPI00029723 | FSTL1;MIR198 Follistatin-related protein 1 | 0.1766 | −0.27 | 0.8 |
| IPI00018219 | TGFBI Transforming growth factor-beta-induced protein ig-h3 |
0.004 | −0.27 | 0.8 |
| IPI00029658 | EFEMP1 Isoform 1 of EGF-containing fibulin-like extracellular matrix protein 1 |
0.0004 | −0.29 | 0.8 |
| IPI00296537 | FBLN1 Isoform C of Fibulin-1 | 0.0291 | −0.3 | 0.8 |
| IPI00005292 | SPOCK1 Testican-1 | 0.205 | −0.32 | 0.8 |
| IPI00306046 | EDIL3 Isoform 1 of EGF-like repeat and discoidin I- like domain-containing protein 3 |
0.2225 | −0.33 | 0.8 |
| IPI00031510 | SEMA3A Semaphorin-3A | 0.2212 | −0.33 | 0.8 |
| IPI00019590 | PLAT Isoform 1 of Tissue-type plasminogen activator |
0.233 | −0.45 | 0.7 |
| IPI00296099 | THBS1 Thrombospondin-1 | 0.3548 | −0.45 | 0.7 |
| IPI00304962 | COL1A2 Collagen alpha-2(I) chain | 0.0169 | −0.48 | 0.7 |
| IPI00012503 | PSAP Isoform Sap-mu-0 of Proactivator polypeptide | 0.0295 | −0.51 | 0.7 |
| IPI00294839 | LOXL2 ;ENTPD4 Lysyl oxidase homolog 2 | 0.0012 | −0.53 | 0.7 |
| IPI00982395 | VEGFA 13 kDa protein | 0.2351 | −0.59 | 0.7 |
| IPI00306322 | COL4A2 Collagen alpha-2(IV) chain | 0.0854 | −0.62 | 0.7 |
| IPI00007118 | SERPINE1 Plasminogen activator inhibitor 1 | 0.0006 | −0.65 | 0.6 |
| IPI00020977 | CTGF Isoform 1 of Connective tissue growth factor | 0.0259 | −0.68 | 0.6 |
| IPI00032179 | SERPINC1 Antithrombin-III | 0.0766 | −0.91 | 0.5 |
| IPI00022431 | AHSG cDNA FLJ55606, highly similar to Alpha-2- HS-glycoprotein |
0.0963 | −0.94 | 0.5 |
| IPI00329482 | LAMA4 Isoform 1 of Laminin subunit alpha-4 | 0.5365 | −1.11 | 0.5 |
Mann-Whitney U-test,
Possible on-off differences in each group, TGFβ has been identified as a top upstream regulator with p-value of overlap 6.19×10−27 linking 21 proteins gray highlighted in the table1using Ingenuity Pathway upstream analysis, averages of relative protein quantity were listed in table1.
3. Results
3.1 Identification of the glioblastoma secretome
U251 cells were grown to ~90% confluence and incubated in media without serum overnight to free the overwhelming contamination from serum. Three each of control and IL-1-stimulated cultures were analyzed to ensure reproducibility. Equal amounts of proteins were used for the analyses (Supplementary Figure 1S). To ensure analytical reproducibility of the method, protein expressions in three biological replicates were compared using average spectral counts and number of peptides. Pearson correlation coefficient between average and replicate values from three measurements were all above 0.95 in each group. Sum of 190 proteins were identified in all three experiments from conditioned media from control and IL-1-stimulated U251 by LC-MS/MS analysis (Supplementary Table 1 and Figure 1). In addition to the correlation analysis, protein numbers from each group and replicates were illustrated to assess the reproducibility of the analysis. One hundred seventy two proteins were common between control and IL-1stimulated U251 secretome. Three replicates in control and IL-1 stimulated group also share 109 out of 184 and 125 out of 181 proteins, respectively, indicating a good reproducibility. Differential expressions were measured among common sets of proteins using deisotoped accurate mass retention time pairs. There are several proteins unique to each group which may work as “on-off” differences and they are noted in Table 1.
Figure 1. Venn diagrams of number of proteins detected in control vs. IL-1-treated U271 secretomes.

Protein numbers from each group and replicates are illustrated to assess the reproducibility of the analysis. (A) 172 proteins were common between control and IL-1-stimulated U251 secretomes. (B and C) Three replicates in control and IL-1 stimulated group also share 109 out of 184 and 125 out of 181 proteins, respectively.
Identified proteins were annotated with Gene Ontology (GO) functions using the Panther Classification system [25] and with Ingenuity Pathway® to identify subcellular location. The distribution of proteins among GO functions or predicted cellular locations did not differ between the two experimental conditions, i.e., control and IL-1 stimulation. Among these, 71 proteins have been previously annotated as extracellular proteins, supporting our findings of them as secreted proteins (highlighted gray in Supplementary Table 1 and Figure 2A). This does not preclude that the other proteins identified are secreted, only that they have not previously been identified as such.
Figure 2. (A) Subcellular localization of glioma secreted proteins. (B) Functional groups of secretome of IL-1-stimulated U251 cells.

Protein identification (n = 190) was performed with Genome Ontology classification tool, and represent as percent for each functional group.
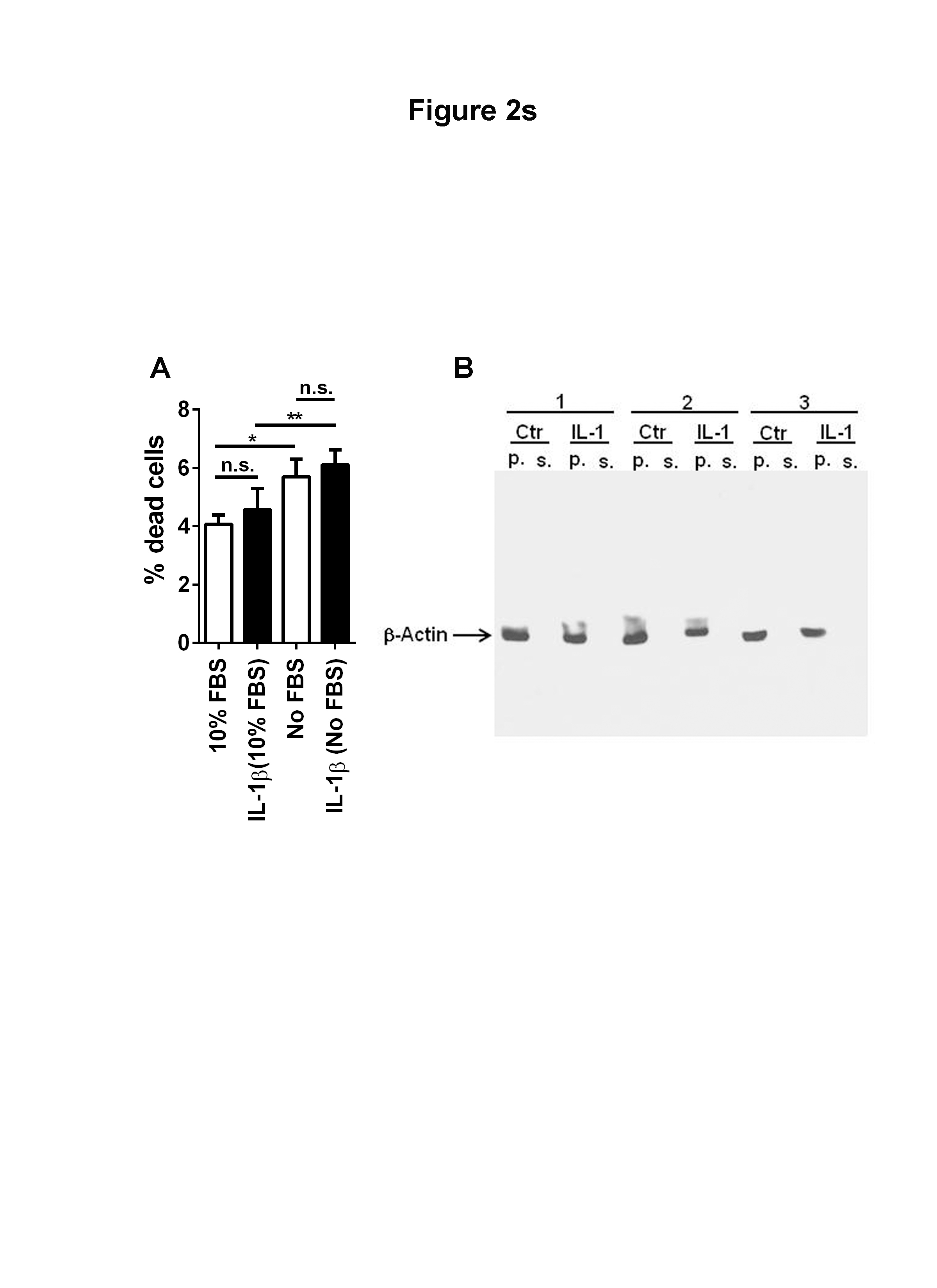
One of the challenges of isolating secreted proteins is the contamination of media from the proteins originated from dead cells or secondary to protein turnover. Proteins that had been pinocytosed or adsorbed from prior incubation with complete media can also contaminate the secretome. In spite of careful cell handling and sample preparation, several proteins annotated with intracellular as well as plasma membrane locations were detected using the current approach. These may not be solely originated from the leakage from dead or damaged cells, as some proteins are secreted through non-classical pathways [26] including the exosome (extracellular vesicle) mechanisms [27]. In our secretome study, IL-1 did not cause cell death (Supplementary Figure 2SA). In addition, western blot analysis for intracellular proteins such as β-actin failed to show its presence in the U251 secretome (Figure 2SB).
Before dismissing the remaining 119 proteins as contamination, analysis using SecretomeP software [24] was carried out. Most of the proteins identified as extracellular proteins (Supplementary Table 1) were predicted to proceed through the classical pathway, i.e., signal peptide triggered protein secretion. However, 28 of the proteins (highlighted light blue in Supplementary Table 1) identified in other cellular locations were found to have scores above the threshold (<0.5) suggesting their non-classical secretion, even without a predicted signal peptide. This analysis indicates that those proteins may have been secreted through non-classical pathways. For example, B4GALT1 is known to be located in the cytoplasm as is shown in the Supplementary Table 1. However, there exist long and short isoforms of B4GALT1 and the latter is known to be secreted [28]. This implies that one of the isoforms may have been secreted in a non-classical manner which confirms the prediction by Secretome P. Evidence for secretion of the other proteins by non-canonical pathways may simply not be available.
3.2 Functional classification of glioblastoma secreted proteins
Figure 2B shows the functional categories of the 190 identified proteins according to the GO Biological Process algorithm. Because of the focus of our study, protein groups such as cell communication (11%), cell adhesion (7%), response to stimulus (7%), and immune system process (10%) were of significant interest. Proteins from the cell communication group are important in term of the interaction of glioma cells with the brain microenvironment as well as with other tumor cells. Proteins from the cell adhesion group could play a role in glioma invasion into surrounding tissues.
3.3 IL-1 induced changes in the glioblastoma secretome
There have been several studies that attempted to quantitate the glioblastoma secretome [29-31]. Most of them utilized either spectral counting or number of peptides as a measure of relative quantitation between two groups. Those parameters have been frequently used as an indirect measure of the relative abundance in previous studies. However, these methods naturally require multiple MS/MS of several peptides to measure relative abundance, thus limiting the quantitation of low abundance proteins. Hence, we have employed label-free quantitation utilizing the measurement of area under the curve (AUC).
To measure AUC of peaks, a list of identified peptides was exported as Mascot DAT file. The m/z, z, and retention time from LC-MS/MS were used as identifiers to extract AUC of a peak from the total ion chromatogram recorded in mzXML. ProteoIQ® was used to extract the information to link this to the filtered protein list. Measured intensities of peptides were compared between two groups to produce log2 (IL-1β/control) abundance ratios. Both Student’s t-test and Mann-Whitney U test were performed in order to calculate p-values to determine the statistical significance of quantitation of proteins between control and IL-1 stimulated groups. The t-test was performed as implemented in ProteoIQ. However, though visualization of normalized peptide quantitation suggested a normal distribution, the use of a t-test is not always applicable for biological samples due to violation of the parametric assumption. For this reason, a Mann-Whitney analysis was performed. One of the prerequisites of the Mann-Whitney U test is separate lists of independent, ordinal measurements for each sample. Because abundance measurements of different peptides are not comparable, relative abundances, or fold-changes, must be utilized. In order to obtain separate lists using relative abundances, we randomly chose a biological replicate for each reference group to obtain independent measurements, and the analysis was repeated 1000 times and averaged in order to obtain an unbiased result. Extracellular proteins that are detected in the secretome (n = 71) are listed in Table 1. Of these, proteins that are significantly (p <0.05) increased in the IL-1β-stimulated secretome included the chemokines IL-8 and CCL2, pentraxin-related protein PTX3, tenascin-C (TNC) and galectin-1 (LGALS1). Additionally, a number of proteins were found to be decreased in IL-1β stimulated glioma secretome and these included collagen alpha-2(IV) chain (COL4A2), fibrillin-1 (FBN1), stanniocalcin-1 (STC1), metalloproteinase inhibitor 1 (TIMP1), isoform 1 of EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1), isoform C of fibulin-1 (FBLN1),, Lysyl oxidase homolog 2 (LOXL2),, plasminogen activator inhibitor 1 (SERPINE1), and TGFβ-induced protein ig-h3 (TGFBI) (Table 1).
3.4.1 Orthogonal validation of some proteins whose secretion is stimulated by IL-1
We used ELISA and western blotting to provide orthogonal validation of some of the proteins with increased expression induced by IL-1β. IL-8 and CCL2 were measured in the glioma conditioned supernatants by ELISA (Figure 3A). They indicate that the glioma secretome contains significantly increased amounts of IL-8 (52-fold on average) and CCL2 (4.6-fold on average) after stimulation with IL-1β. Figure 3B and 3C show western blot analysis of control and IL-1-stimulated glioma conditioned medium and confirmed the increase of TNC (3.3-fold on average) and PTX3 (5.8-fold on average). Additional experiments demonstrated that other factors such as galectin-1 (Gal-1) which was only marginally increased (30%, p = 0.0049) by MS showed larger increase (~130% on average, p<0.05) by Western blot analysis. We also validated two downregulated proteins, stanniocalcin-2 (STC2) and plasminogen activator inhibitor 1 (SERPINE1) (Figure 3B and 3C). MMP2, although it was not regulated proteins according to our MS data, Western blot analysis showed a more complicated picture. Two forms of MMP2 (proMMP2 and active MMP2) existed that can be detected by western in the IL-1 secretome, with only pro-MMP2 predominant in control secretome (Figure 3B and 3C, also see below section 3.5 post-translational modification). Otherwise, the blots illustrate the matching trend with MS data.
Figure 3. Validation of MS data.

(A) IL-8 and CCL2 ELISA of U251 cell culture supernatants: U251 cells were stimulated with IL-1β (10 ng/ml) for 15 min then cells were extensively washed to remove input cytokine. The cultures were further incubated for 16 h in serum-free DMEM. The levels of IL-8 and CCL2 in the culture supernatants were determined by ELISA. Mean ± SD from triplicate wells. Results from three separate experiments (1, 2 and 3) are shown. ***p<0.001 vs. control unstimulated cultures (Ctr). (B) Western blot analysis of TNC, PTX3, Gal-1, STC2, SERPINE1, proMMP2 and active MMP2. Concentrated culture supernatants (secretome) were separated in a 4-20% gradient gel and transferred to PVDF. Immunoblotting performed as described in Materials and Methods. Results from three separate experiments (1, 2 and 3) are shown except for MMP2 (n = 2). (C) Densitometric analyses of Western blots (n = 3) demonstrating significant increases and decreases of the factors shown in B (Mean ± SD, * p<0.05, **p <0.01) RU: relative units.
3.4.2 Functional implications of the glioblastoma secretome
Below, we provide a short literature review of the factors present in the GBM secretome in Table 1. We cover the general biology as well as information relevant to glioma and speculate the functional implications of our findings with respect to the pathogenesis of glioblastoma. Our simplified model and hypotheses based on our data and the literature is depicted in Figure 4. The secreted factors are classified into a few functional groups for discussion as follows:
Figure 4. Hypothetical model of the role of glioma secretome in GBM progression based on our results and potential role of IL-1.

We have generated a few categories of biologically identified pathways that impact on GBM progression and host damage, and speculate how the secreted factors identified in our secretome might contribute to these changes. See text for detail.
(A) Cytokines, chemokines and other inflammatory mediators
Among the most abundant secreted proteins upregulated by IL-1 are the chemokines IL-8 and CCL2. IL-8 (CXCL8) is a chemokine belonging to the alpha (α: CXC) subgroup, involved in neutrophil chemotaxis. IL-8 is also shown to be a pro-angiogenic factor. A recent study found that GBM-secreted IL-8 promotes angiogenesis and microvascular endothelial permeability [32]. The robust increase of secreted IL-8 following IL-1 stimulation of GBM cells shown here suggests that inflammation-induced IL-8 in tumor cells can have a major impact on GBM progression. CCL2 (aka MCP-1) is a chemokine belonging to the beta (β: CC) subgroup and is the principal chemoattractant involved in monocyte recruitment to inflamed tissues including the brain. A recent study found CCL2 expression by the majority of 16 human glioma lines and that over-expression of CCL2 in U87 cells increases glioma invasiveness by interacting with CCR2-bearing microglia [33]. Antibody-mediated blockade of CCL2 has been shown to prolong the survival of mice bearing murine or human glioma cells [34], suggesting that CCL2 might be a useful therapy target. Pentraxins (PTX) are a group of immune molecules that include C-reactive protein and serum amyloid P-component (short pentraxins) and pentraxin 3 (PTX3, long pentraxin). PTX3 is expressed by cells of the innate immune system and is involved in inflammation, angiogenesis, and ECM formation [35;36]. CNS glial cells and leukocytes express PTX3 following LPS injection [37;38]. Little is known about PTX3 in gliomas. Very recently, it has been shown that glioma exosomes derived under hypoxic conditions and GBM patient plasma-derived exosomes are enriched in hypoxia-regulated proteins including PTX3, IL-8 and lysyl oxidase (LOX) [27], all of which are present in our IL-1 induced glioma secretome. The identification of PTX3 as one of the highly increased secreted proteins by IL-1 in the GBM secretome therefore opens up a new research avenue.
(B) Growth factors and related proteins
There are several growth factors and related proteins detected in the GBM secretome in our study. Among them are vasculoendothelial cell growth factor (VEGFA), platelet-derived growth factor (PDGF-D), and leukemia inhibitory factor (LIF) all of which are implicated in glioma pathogenesis [39-41]. LIF belongs to the gp130 cytokine/growth factor family and activates Stat3, a transcription factor involved in multiple aspects of glioma progression [42;43]. LIF has also been shown to mediate TGFβ-induced glioma-initiating cell self-renewal [40]. Of interest is the presence of several insulin-like growth factor-binding proteins (IGFBPs 2, 5 and 7) in our secretome. IGFBP7 was found to be a highly selective biomarker of tumor vessels, as GBM-secreted factors have been shown to induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGFβ signaling [44]. Our secretome data suggest that IL-1 might reduce IGFBPs, but these require further validation studies.
(C) Extracellular matrix proteins (ECMs) and related proteins
A number of ECM proteins such as laminin (subunits α4, α5 and γ1), fibronectin, collagen alpha chain proteins (COL18A1, COL11A1, COL4A2, and CLO6A1), nidogen-1 (NID1), and extracellular matrix protein 1 (ECM1) are present in our secretome. Most of these proteins are part of the ECM proteins that make up the epithelial basement membrane and may be important in forming the barrier for tumor invasion. The effect of IL-1 is both stimulatory and downregulatory (COL4A2, for example) with many more being downregulated than upregulated. This is consistent with the well-known, generally stimulatory effects of TGFβ on ECM and integrin expression, and the proposed TGFβ-downregulatory effects of IL-1 (see below). Our data therefore suggest that IL-1 can induce GBM invasiveness/proliferation by modulating ECM proteins such as COL4A2. Furthermore, Ingenuity Pathway Upstream Regulator analysis identified 21 proteins in our secretome as TGFβ-downstream proteins (gray highlighted in Table 1). TGFβ is an important growth factor implicated in glioma biology. Several pieces of evidence also indicate that TGFβ and related proteins are major targets of IL-1 action in glioma secretome (see below).
TGFβ is secreted in a latent complex consisting of three proteins: TGFβ (for which there are three isoforms, 1-3), an inhibitor called latency-associated protein (LAP) which is derived from the TGFβ propeptide, and an ECM-binding protein called latent TGFβ binding protein (LTBP,1-4). LTBP interacts with fibrillins and other ECM components including fibulins to localize latent TGFβ in the ECM [45]. Humans and mice with fibrillin 1 (FBN1) gene mutations show the critical role of ECM and integrins in regulating TGFβ signaling. Mutation of human FBN1 gene causes Marfan’s syndrome as a result of overactive TGFβ [46;47]. Our data show that IL-1 suppresses fibrillin 1, indicating that IL-1 targets multiple groups of proteins involved in TGFβ-ECM interactions, further strengthening the idea that the ECM-TGFβ axis may be one of the major targets of IL-1 in glioma in vivo (Figure 4).
EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1 aka fibulin 3) was reported as a suppressor of malignant glioma growth by inhibiting angiogenesis and modulation of ECM. Its expression in GBM is associated with favorable prognosis [48]. IL-1-mediated suppression of EFEMP1 may thus contribute to glioma progression. TGFβ-induced protein (TGFBI, aka βig-h3) is a 68 kDa RGD motif-containing protein that binds to type I, II and IV collagens. This protein is induced by TGFβ and acts to inhibit cell adhesion, cell-ECM interaction and cell migration [49;50]. Various mutations are associated with corneal dystrophy. Experimental evidence indicates that MMP-9 mediated tumor cell and macrophage migration is mediated by its action on this migratory inhibitor protein [49]. TGFBI is also an angiogenesis inhibitor that inhibits endothelial cell migration and VEGFR-2 signaling [50]. Our data suggest that IL-1 can promote glioma progression by suppressing TGFBI.
Matricellular proteins including tenascin C, connective tissue growth factor, thrombospondin-1 and SPARC (aka osteonectin) are present in our secretome. Matricellular proteins are de novo deposited in the forefront of invading tumor cells and modulate structural, growth factor-binding and signaling functions of tumor and stromal cells [51]. Tenascin-C (TNC) is an ECM glycoprotein transiently induced upon tissue injury by the act of growth factors and cytokines [52;53]. Numerous studies show a key role that TNC plays in glioma progression including tumor cell proliferation, invasion, angiogenesis, mesenchymal differentiation and T cell suppression [42;54-56]. TNC expression is associated with overall worse prognosis and is among the nine genes that have predictive value in GBM malignancy [42]. We find that TNC is constitutively secreted by U251 cells similar to another study [30] and that it is highly upregulated by IL-1 (Table 1 and Figure 2). These results reinforce the idea that multiple secreted proteins including TNC are aberrantly expressed by GBM cells, and that IL-1 upregulates these factors further contributing to tumor progression.
Connective tissue growth factor (CTGF aka CCN2) is a matricellular protein identified as a secreted factor that stimulates the migration and invasion of GBM cells [57]. CTGF is over-expressed in many cancers, and its expression in glioma is reportedly to correlate with tumor grade and patient survival. CTGF is induced by growth factors, typically by TGFβ. IL-1-mediated suppression of CTGF may thus indicate that IL-1 can suppress TGFβ signaling in our secretome. In support of this, opposing effects of IL-1 on tenascin-C and CTGF (similar to ours) have been reported [58]. Thrombospondin 1 (THBS-1) is an adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions. This protein can bind to fibrinogen, fibronectin, laminin, type V collagen and integrins αV and β1. THBS-1 has been shown to play roles in platelet aggregation, angiogenesis, and tumorigenesis [59;60]. THBS has also been shown to activate TGFβ.
(D) Extracellular heparan/chondroitin sulfate proteoglycans (H/CSPG)
Several extracellular matrix proteoglycans are present in our glioma secretome including testican-1 (SPOCK1), an inhibitor of membrane type (MT)-MMPs [61], secretogranin-2 (SCG2, aka chromogranin-C) a neurosecretory molecule, and versican (VCAN, isoform V0). Versicans are large CSPG found in glioma and tumor vessels [62]. The isoforms V0/V1 have been shown to be induced by TGFβ2 and involved in glioma invasion [63].
(E) ECM- and TGFβ-modulating enzymes
A number of proteins involved in blood coagulation that also play important roles in glioma invasiveness are present in our GBM secretome. Among these are plasminogen activator inhibitor 1 (PAI-1 aka SERPINE1), isoform 2 of glia-derived nexin (SERPINE2), urokinase-type plasminogen activator (PLAU), plasma serine protease inhibitor (SERPINA5), tissue-type plasminogen activator (PLAT), and antithrombin-III (SERPINC1). Plasmin and MMPs play a key role in promoting tumor invasion and tissue remodeling by inducing proteolysis of several ECM components [64-66]. Serum proteases such as plasmin catalyze the release of active TGFβ from the matrix. MMP2 and MMP9 are also known to cleave latent TGFβ. Therefore, the actions of these enzymes are intricately linked to their activity on TGFβ.
Plasminogen activator inhibitor 1 (PAI-1 aka serine protease inhibitor SERPINE1, aka protease nexin-1): SERPINE1 is the principal inhibitor of tissue plasminogen activator (tPA) and urokinase (uPA) hence is an inhibitor of fibrinolysis. Previous studies have shown that IL-1 induces SERPINE1 in GBM cells and increases their invasiveness [67]. SERPINE1 is a well-known TGFβ-induced gene, and its expression in glioma is a predictor of poor prognosis [68]. We find that SERPINE1 is suppressed by IL-1 in our glioma secretome (Table 1 and Figure 3), perhaps reflective of its action on TGFβ. Matrix metalloproteinase 2 (MMP2): In spite of the mass spectrometry data showing MMP2 unchanged by IL-1, our Western blot data show that U251 cells secrete large amounts of proMMP2 and that IL-1 might slightly increase proMMP2 secretion. Furthermore, active MMP2 is produced almost exclusively in the presence of IL-1 (Figure 3B, C). MMP2 has been shown to be an important enzyme involved in glioma invasiveness. Interestingly, the MMP inhibitors TIMP1 and TIMP2 are both present in our secretome and the TIMP1is suppressed by IL-1. These results together suggest that IL-1 upregulates the expression and activity of crucial enzymes involved in ECM degradation and glioma invasion. Antithrombin-III (SERPINC1): The expression of antithrombic molecules have been found to be altered in glioma vessels. Specifically, reduction of antithrombin-III immunoreactivity was reported in GBM vessels. Furthermore, thrombin has been suggested to promote tumor growth and angiogenesis, as well as vascular complications [69-71]. Thus, IL-1-mediated suppression in our secretome suggests that the thrombin system may be yet another novel tumor target.
(F) Miscellaneous
Galectin-1 (Gal-1) is a member of galectins, a family of β-galactoside-binding proteins which participate in cell growth and adhesion. The involvement of galectin-1 and 3 in different steps of glioma malignant progression including cell migration, angiogenesis or chemoresistance have been demonstrated [72], making them potentially good drug targets. Importantly, Gal-1 induces alternative activation of macrophages and microglia via interacting with the surface CD45 molecule [73]. We find that IL-1 upregulates glioma Gal-1 secretion (Figure 3B, C). These data suggest that IL-1 can suppress tumor-specific immunity by inducing Gal-1. Stanniocalcin 1 and 2 (STC1 and 2): STCs are originally described in fish as hormones that regulate calcium and phosphate homeostasis. Human STC2 is recently found to be a heat shock- and HIF-1α-induced protein that interacts with heme oxygenase-1 (HO-1) in the degradation of heme [74]. STC1 is neuroprotective and is induced by hypoxic preconditioning through IL-6 signaling [75]. Inflammation-mediated suppressions of STCs may hamper the ability of brain to cope with oxidative stress. Virtually nothing is known about STC in gliomas. Importantly, a glioma secretome study identified STC1 as a significantly upregulated protein following cell treatment with cAMP [76], which together with our data showing downregulation by IL-1 (Figure 3B, C) suggest a potentially important role that STC1 plays in inflammation-mediated modulation of glioma pathogenesis. Follistatin-like 1 (FSTL1) was identified as a TGFβ-inducible gene as well as a suppressor of TGFβ activity [77]. In epithelial cancer, FSTL1 has been suggested to act as a potential tumor suppressor. FSTL1 was shown to be upregulated in GBM [78]. Intriguingly, a prior study of cAMP-modulated glioma secretome identified FSTL1 among the handful of proteins upregulated by cAMP [76], in contrast to the downregulatory effect of IL-1 that we find in our secretome. Semaphorin 3A (SEMA3A) is a secreted chemo-repellent that facilitates axon guidance during neural development through interaction with its receptor neuropilin-1 [79;80]. Recent evidence indicates that semaphorins modulate the adhesive and migratory properties of malignant tumor cells. One study reports that GBM cells endogenously secrete SEMA3A and affects cell migration, proliferation, and tumor development [81]. These studies contrast with those that report semaphorins as suppressors of tumor progression [80]. Broadly, SEMA3A is featured as an inhibitor of angiogenesis and T cell proliferation. Carboxypeptidase A4 (CPA4) is a rarely studied enzyme with known functions in neuropeptide processing and regulation in the extracellular environment [82]. CPA4 substrates also have known functions in cell proliferation and differentiation [83]. Interestingly, CPA4 was one of the few significantly downregulated proteins by cAMP in the previous glioma secretome study [76], suggesting as yet unknown but possibly significant role that CPA4 plays in glioma pathogenesis. Clusterin (CLU) also known as apolipoprotein J is a versatile protein expressed by reactive astrocytes in vivo and has been implicated in the pathogenesis of Alzheimer’s disease. Clusterin can also bind Smad2/3 proteins and potentiates TGFβ signaling. Its role in cancer is unclear with both oncogenic as well as tumor suppressor activities assigned [84].
3.4.3 TGFβ induces TGFβ in human GBM cells: effect of IL-1
Our data strongly suggest interactions between the TGFβ and the IL-1 systems in glioma cells, yet very little information is available on the details of this interaction. We thus performed initial analyses of this relationship by posing questions such as (1) how is TGFβ production regulated in glioma cells; (2) what are the roles of IL-1 and TGFβ on the expression of ECM and related proteins? Our ELISA results shown in Figure 5 demonstrate that TGFβ is a strong inducer of TGFβ itself in glioma cells (though this required high concentrations of recombinant TGFβ) and that IL-1 suppresses TGFβ-induction of TGFβ. Interestingly, while IL-1 is also a strong inducer of IL-1 itself in glioma cells, TGFβ failed to show any effect on this. The results were virtually identical in both U251 and U87 cells. Furthermore, the expression of COL4A2 and CTGF in the glioma secretome was enhanced by TGFβ and this was also reduced by IL-1 as determined by western blot analyses of the concentrated U251 and U87 secretomes. These results directly demonstrate some of the antagonistic interactions between IL-1 and TGFβ using high concentrations of recombinant cytokines.
Fig. 5. Interactions between TGFβ1 and IL-1 in glioma cells.

(A) TGFβ induces TGFβ in U251 and U87 glioma cells: suppression by IL-1. Cultures were treated with two different concentrations of TGFβ1 (100 ng/ml and 10 ng/ml), IL-1α (10 ng/ml) or both for 6 h in DMEM without serum then extensively washed to remove input cytokines. Cultures were refed with DMEM and incubated for another 18 h. Culture supernatants (sup.) and cell lysates (i.c.) were subjected to ELISA for TGFβ1 using the DuoSet ELISA from R&D Systems with the sensitivity range of 31.2 - 2,000 pg/ml. Ctr = medium alone. Results are mean ± SD from triplicate cultures (* p <0.05, ** p <0.01, *** p <0.001, n.s. not significant). TGFβ was induced in glioma cells by TGFβ but not by IL-1. IL-1 suppressed TGFβ induction by TGFβ. The findings were similar in both U251 and U87 cells. (B) Time-dependent production of TGFβ. Experiments were performed exactly as in (A) above using 100 ng/ml of recombinant TGFβ1, except cytokine exposure was limited to first 30 min and culture supernatants were harvested at indicated time points (0.5 h - 22 h). (C) IL-1 induces IL-1 in glioma cells: TGFβ has no effect on IL-1 production. IL-1β levels were determined by ELISA in the same cell lysates (i.c.) and culture supernatants (s.) as in (A). In glioma cells, IL-1(α) induced robust amounts of intracellular IL-1(β) with small fractions being secreted (s.). TGFβ did not show significant effects on IL-1 production. (D) IL-1 suppresses TGFβ-induced ECM and matricellular proteins: Cultures were treated as above for 16 h, then culture supernatants were concentrated (~x25) and were separated by SDS-PAGE using a 4-20% gradient gel. Immunoblotting was performed using anti-COL4A2 and anti-CTGF antibodies.
3.5 Post translational modifications of secreted proteins
We have identified and quantified secreted proteins based on the detection of tryptic peptides. Although this allowed detection of many secreted proteins and facilitates accurate quantitation, there are extra layers of regulation (unaccounted for the study) that control secretion of proteins to the extracellular space. One of the most important controlling mechanisms is perhaps the post-translational modification (PTM) including proteolytic cleavage, phosphorylation and glycosylation. One of the advantages of using MS based proteomic strategy is its capability in further characterization of PTMs during the secretion as described above. To take advantage of this capability, additional Mascot search was performed to test the feasibility of using this method in detecting PTMs uniquely involved in the secretion of proteins. The same set of peak lists generated was subjected to the search against IPI-Human database with the semitryptic restriction and variable phosphorylation (STY). Several candidates were matched. Among those, two cases were manually curated and reported below.
3.5.1 Activation of plasminogen activator inhibitor 1 (SERPINE1: serpin peptidase inhibitor, clade E, aka nexin)
SERPINE1 is a serine protease inhibitor that consists of 402 Amino acids (AA) and works as a principal inhibitor of tissue plasminogen activator and urokinase. It is synthesized as a larger protein which is activated by cleavage of the N-terminal portion, known to be 1-23AA, by a second peptidase or by auto-cleavage. Interestingly, we detected two different N-terminal tryptic peptides of SERPINE1 as shown in Figure 6A. Both are slightly decreased upon IL-1β treatment.
Figure 6. Post translational modification of secreted proteins.

(A) The known active form of SERPINE1 missing signal peptides 1-23 confirmed with MS/MS. (B) APSPIIKFPGDVAPK from MMP2 which demonstrates signal peptide cleavage at the N-terminus. (C) Phosphorylated AHHGEAGHHLPEPSSRETGR from STC2. Either S250 or S251 is phosphorylated.
3.5.2 Activation of MMP2
MMP2 is produced as pro-forms with 660 amino acids and its molecular weight is 73.9 kDa. Activation of MMP-2 requires proteolytic processing. First, signal peptide from N-terminus and consequently propeptides (30A-109N) can also be cleaved off prior activation. The latter process is clearly shown in Figure 2B as pro-MMP2 and MMP-2. However, removal of signal peptides cannot be detected using Western blot alone. In addition, the C-terminal non-catalytic fragment of MMP2 (which has a reported suppressive role in glioma progression) can be generated by cleavage between 444L and 445Y [85]. Hence, four types of MMP2 fragments can be mixed in both conditions in perhaps different abundance upon IL-1 stimulation. Once analyzed, detected tryptic peptides from all four forms are grouped into the pro-MMP2 and quantitated to produce one average ratio. Therefore, discordant results can be generated between Western and MS data and the averaged ratio of pro-MMP2 cannot represent the accurate values in this case.
N-terminal tryptic peptides (29APSPIIKFPGDVAP44K) without the signal peptides are indeed detected as shown in Figure 6B. This has been enabled by re-searching of the data with semitryptic specificity as described above. This N-terminal peptide seems to increase upon IL-1β stimulation in some cases, although further studies are required to confirm this finding, since quantitation using single peptide is less reliable.
3.5.3 Phosphorylation of stanniocalcin-2 (STC2)
STC2 is a ubiquitously expressed enzyme regulating calcium and phosphate homeostasis. As is shown in Figure 6C, a tryptic peptide, 237AHHGEAGHHLPEPSSRETG256R, was detected with the possible phosphorylation at either S250 or S251. Phosphorylation of STC2 has not been reported in PHOSIDA [86] probably because it is an extensive database reporting PTM of “intracellular” proteins. It has previously been shown to exist only in the secreted STC1 and STC2 of human fibrosarcoma cells [87]. This demonstrates the necessity of focusing on extracellular proteins to understand the cell-cell interaction. Although its function has yet to be determined, phosphorylation may regulate its activity.
4. Discussion
This is the first comprehensive unbiased analysis of tumor cell secretome that identifies secreted targets of IL-1 to our knowledge. Our quantitative MS analysis and subsequent validation of a few selected factors suggest that AUC measurements may underestimate the fold-induction and reduction as determined by ELISA or immunoblotting, also diminishing the statistical power. This can be explained based on the fact that while the measurement of AUC is linearly proportional to the concentration of peptides in the range of 10 fmol to 100 pmol [88], when peaks in low fmol concentration are absent from one of the experimental conditions, fold changes are measured from the values acquired from the baseline resulting in underestimation. Furthermore, we note that certain secreted proteins known to be present in the IL-1 conditioned media are conspicuously absent in our MS data, probably due to the limitation in dynamic range of detection. For example, human glioblastoma cells secrete IL-1α and IL-1β in response to IL-1 stimulation, which is detectable by ELISA (Tarassishin and Lee, manuscript in preparation, also see Figure 5). Proteomics analysis can miss certain critical components of the secretome due to their low abundance and this can be improved by further fractionation. Nevertheless, this technique provides an unbiased and non-targeted approach that can enable discovery of secreted proteins engaged in important cell-cell communications.
We find a significant overlap in the types of proteins detected in our secretome with those in published GBM secretome studies [29-31;89], confirming the validity of our system and approach. As inflammation has been found to be important in tumorigenesis and tumor progression, our IL-1 activated secretome study has a number of implications for understanding and treatment of malignant gliomas. Specifically, our data indicates that IL-1-induced tumor secretome can modulate glioma and the tumor microenvironment including tumor cell survival, invasion, tumor angiogenesis, and anti-tumor immunity (Figure 4). Limited amount of information is available on GBM secretome [29-31;89] and our results both resemble and differ from the published studies by several accounts. First, the percentage of proteins that are extracellular is much higher in our study than published, which may reflect the purity of our cultures and samples. We used U251 cells where others most often used U87 cells which grow in a non-cohesive manner and are shown to be highly invasive, corresponding to more a “mesenchymal” character of GBM cells [42;90;91]. For example, the mesenchymal subtype marker chitinase-3-like protein 1 was absent from our secretome, whereas it was often detected in the U87 secretome. Otherwise, we see high overlaps in the types of proteins detected in our vs. published GBM secretome. Ours is the first quantitative glioblastoma secretome study using a cytokine as a stimulus. In our analysis, treatment with IL-1 led to changes in the levels of proteins, several of them with statistical significance (Table 1). We surmise that transcriptional activation as well as transcriptional repression is probably the major underlying mechanisms, but post-transcriptional mechanisms (such as translational and post-translational modifications) as well as proteolytic cleavage and modulation of protein secretory pathways also likely account for those changes.
Few secretome studies address the quantitative changes. In one study, the effect of WT-p53 expression was determined in a glioma (LN-Z308) cells [31]. This study found 18 enhanced proteins among which were Gal-1 and CTGF and 16 reduced proteins which included VEGF, TGFβ, progranulin (PGRN), IL-8, and IGFBP6. Among 34 unchanged proteins were fibulin 1A, follistatin-like 1, cathepsin B, filamin, STC2 and IGFBP5. As p53 mutation is found in a major glioma subtype, the list of modulated proteins provides insight into the mechanism/role of p53 mutation in glioma pathogenesis.
Another example of quantitative secretome study is Hill et al., which examined the effect of cAMP [76]. The list of increased (n = 14) and decreased (n = 21) N-linked glyco-proteins mirrors the effect of IL-1 in our secretome. For example, cAMP increased STC1 and FSTL1 but reduced TNC. These results indicate the contrasting mechanisms used by cAMP and IL-1 in modulating the expression/secretion of these proteins. As cAMP reduces the angiogenic, invasive and proliferative potential of GBM cells, the results also strongly support that IL-1 promotes glioma progression. cAMP is stimulated by endogenous brain peptides and provokes anti-inflammatory signaling through protein kinase A activation [92], providing a larger picture that anti-inflammatory signaling might benefit GBM patients [93].
A number of proteins in our secretome that are modulated by IL-1 belong to the ECM proteins (procollagen, laminin, fibronectin, etc) and ECM modulating proteins. The ability to produce ECM proteins and form basal lamina is a unique property of astrocytes facing the blood vessels and the meninges (“glia limitans”). The spaces created (perivascular and subpial spaces) are also the major routes for GBM spread. The ECM-related proteins were mostly downregulated by IL-1 in our study, with some exceptions. As TGFβ is a major component as well as the regulator of ECM-related proteins and integrins (with the generally positive effects on them), our study reveals the TGFβ system as a probable target of IL-1 in the glioma secretome. IL-1 also appears to be a major regulator of TGFβ activity, since fibrillin 1 (crucial proteins known to suppress TGFβ activation in ECM) are reduced in IL-1 secretome. Together, these results strongly suggest that in in vivo glioma, the ECM-TGFβ axis may be the main target of IL-1 action. We demonstrate this possibility in principle by use of high concentrations of exogenous recombinant cytokines, although we see little or no TGFβ (limited to TGFβ1 investigation) in the glioma secretome in either the control or IL-1 treated conditions (without the addition of recombinant TGFβ) (see Figure 5).
Plasmin and MMPs play a key role in promoting tumor invasion and tissue remodeling by inducing proteolysis of several ECM components [64-66]. Serum proteases such as plasmin catalyze the release of active TGFβ from the matrix. MMP2 and MMP9 are also known to cleave latent TGFβ. Therefore, the actions of these enzymes are intricately linked to their activity on TGFβ. Not surprisingly, we find over-representation of these enzymes in the GBM secretome and their modulation by IL-1 (activation of MMP2, suppression of TIMPs), once again reinforcing the idea that modulation of the ECM-TGFβ axis may be the main downstream effect of IL-1 in the glioma microenvironment.
Based on the available evidence, we speculate that IL-1 is a critical positive regulator of glioma progression. IL-1 produced by the tumor cells as well as by microglia and inflammatory cells can function as the central molecule that connects and impacts on several important facets of glioma progression, including glioma invasiveness, angiogenesis and tumor immune surveillance. We further speculate that anti-IL-1 therapy can reverse many of these processes through modulation of cellular transcriptional reprograms, suppression of toxic inflammation, and inhibition of immune modulators such as miR-155 [94-99]. We propose that anti-IL-1 therapy directly targeting the IL-1 system can benefit a subset of GBM patients. Currently there are three FDA-approved anti-IL-1 agents: IL-1ra (anakinra), a neutralizing mAb to IL-1β (canakinumab), and a soluble IL-1R (rilonacept). The availability of approved reagents with few side effects and the newly emerging evidence that links IL-1 and human cancer [18;93;100] provide further arguments for the development of IL-1 blockade therapies for gliomas. Identification of the innate immune pathway as potential therapy is also novel, as most current immune therapies are aimed at specific anti-tumor immunity.
Conclusions
In present study label free quantitation using AUC has been employed to reliably measure IL-1 regulated glioblastoma secretome. This simple method which does not require isotope labeling can easily be applied to study other secretomes. Its utility in quantitating secreted proteins upon IL-1 stimulation has been verified with ELISA and western blot analysis.
Analyses of the MS data suggest that IL-1 may contribute to glioma progression in part by modulating TGFβ and extracellular matrix protein interactions.
Supplementary Material
Figure 1S: Protein loading in each sample as shown in Coomassie blue-stained gel: Culture supernatants from untreated (Ctr) and IL-1-treated U251 cells in triplicates were separated by SDS-PAGE (4-15% gradient gel from Bio-Rad) and the gel was stained with Bio-Safe Coomassie G-250 (Bio-Rad). Molecular weight markers are shown on the left.
{kind=link}
Figure 2S (A) IL-1 does not increase U251 cell death: U251 cells were grown in DMEM with or without 10% FBS and untreated or treated with IL-1β (10 ng/ml) for 22 h. Cells were then lifted by trypsinization followed by wash in DMEM with 10% FBS twice. An aliquot (50 μl) of cell suspension was subjected to trypan blue exclusion test in a hemocytometer and the number of total and dead (TB) cells per ml were calculated. The numbers indicate the percentages of dead cells in each condition. Data are mean ± SD from triplicate cultures. The results show that while significance increase in cell death occurred following serum withdrawal, treatment with IL-1β did not affect cell death. n.s. = not significant, * p <0.05, ** p<0.01
Figure 2S (B): Lack of detectable β-actin in concentrated U251 secretome: The pellets (p.) and the concentrated supernatants (s., the same samples subjected to secretome analysis) of untreated and IL-1-treated U251 cultures (n = 3) were analyzed for β-actin expression by immunoblotting following 10% SDS-PAGE. Thirty μg of total protein was loaded in each lane. Beta-actin (intracellular protein) was not detectable in this highly concentrated tumor secretome (see Materials and Methods), suggesting the lack of significant cell death and/or contamination of the secretome by dead cell debris/intracellular protein.
{kind=link}
Significance.
Present study is on an unbiased screening of the glioblastoma secretome stimulated by IL-1 which triggers neuroinflammatory cascades in the central nervous system. Network of secreted proteins were shown to be regulated revealing their possible contribution to glioma progression. Label free quantitative proteomics has provided unique novel targets for potential glioblastoma intervention.
Acknowledgement
This study was supported by NIH RO1 MH55477, Einstein CFAR, NCRR 1S10RR021056, and NCRR 1S10RR15859. The authors are grateful to Dr. Yungtai Lo for many helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Sul J, Fine HA. Malignant gliomas: new translational therapies Mt Sinai. J Med. 2010;77:655–666. doi: 10.1002/msj.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henriksson R, Asklund T, Poulsen HS. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: a review. J Neurooncol. 2011;104:639–646. doi: 10.1007/s11060-011-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 4.Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211–221. doi: 10.1002/glia.440150303. [DOI] [PubMed] [Google Scholar]
- 5.Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011;13:3–13. doi: 10.1093/neuonc/noq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu A, Krady JK, Levison SW. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res. 2004;78:151–156. doi: 10.1002/jnr.20266. [DOI] [PubMed] [Google Scholar]
- 7.Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22:903–908. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 8.Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans. 2007;35:1122–1126. doi: 10.1042/BST0351122. [DOI] [PubMed] [Google Scholar]
- 9.John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 2005;49:161–176. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- 10.Tarassishin L, Lee SC. Interferon regulatory factor 3 alters glioma inflammatory and invasive properties. J Neurooncol. 2013;113:185–194. doi: 10.1007/s11060-013-1109-3. [DOI] [PubMed] [Google Scholar]
- 11.Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104:4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki A, Tamura M, Hasegawa M, Ishiuchi S, Hirato J, Nakazato Y. Expression of interleukin-1beta mRNA and protein in human gliomas assessed by RT-PCR and immunohistochemistry. J Neuropathol Exp Neurol. 1998;57:653–663. doi: 10.1097/00005072-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- 14.John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist. 2003;9:10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- 15.Basu A, Krady JK, O’Malley M, Styren SD, DeKosky ST, Levison SW. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–6082. doi: 10.1523/JNEUROSCI.22-14-06071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Downen M, Amaral TD, Hua LL, Zhao ML, Lee SC. Neuronal death in cytokine-activated primary human brain cell culture: role of tumor necrosis factor-alpha. Glia. 1999;28:114–127. [PubMed] [Google Scholar]
- 17.Rothwell N. Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun. 2003;17:152–157. doi: 10.1016/s0889-1591(02)00098-3. [DOI] [PubMed] [Google Scholar]
- 18.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–351. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 19.Sen E. Targeting inflammation-induced transcription factor activation: an open frontier for glioma therapy. Drug Discov Today. 2011;16:1044–1051. doi: 10.1016/j.drudis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Morgan AC, Chang HY, Liu JS, Hua LL, Lee SC. High extracellular potassium modulates nitric oxide synthase expression in human astrocytes. J Neurochem. 2000;74:1903–1912. doi: 10.1046/j.1471-4159.2000.0741903.x. [DOI] [PubMed] [Google Scholar]
- 21.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 22.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, Eng JK, Martin DB, Nesvizhskii AI, Aebersold R. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38:D204–D210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bendtsen JD, Jensen LJ, Blom N, von HG, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 25.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic. 2005;6:607–614. doi: 10.1111/j.1600-0854.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 27.Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, Belting M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A. 2013;110:7312–7317. doi: 10.1073/pnas.1220998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez LC, Youakim A, Evans SC, Shur BD. Evidence for a molecular distinction between Golgi and cell surface forms of beta 1,4-galactosyltransferase. J Biol Chem. 1991;266:15984–15991. [PubMed] [Google Scholar]
- 29.Formolo CA, Williams R, Gordish-Dressman H, MacDonald TJ, Lee NH, Hathout Y. Secretome signature of invasive glioblastoma multiforme. J Proteome Res. 2011;10:3149–3159. doi: 10.1021/pr200210w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polisetty RV, Gupta MK, Nair SC, Ramamoorthy K, Tiwary S, Shiras A, Chandak GR, Sirdeshmukh R. Glioblastoma cell secretome: analysis of three glioblastoma cell lines reveal 148 non-redundant proteins. J Proteomics. 2011;74:1918–1925. doi: 10.1016/j.jprot.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Khwaja FW, Svoboda P, Reed M, Pohl J, Pyrzynska B, Van Meir EG. Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene. 2006;25:7650–7661. doi: 10.1038/sj.onc.1209969. [DOI] [PubMed] [Google Scholar]
- 32.Dwyer J, Hebda JK, Le GA, Galan-Moya EM, Smith SS, Azzi S, Bidere N, Gavard J. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS ONE. 2012;7:e45562. doi: 10.1371/journal.pone.0045562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33:312–319. doi: 10.1093/carcin/bgr289. [DOI] [PubMed] [Google Scholar]
- 34.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cieslik P, Hrycek A. Long pentraxin 3 (PTX3) in the light of its structure, mechanism of action and clinical implications. Autoimmunity. 2012;45:119–128. doi: 10.3109/08916934.2011.611549. [DOI] [PubMed] [Google Scholar]
- 36.Deban L, Jaillon S, Garlanda C, Bottazzi B, Mantovani A. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res. 2011;343:237–249. doi: 10.1007/s00441-010-1018-0. [DOI] [PubMed] [Google Scholar]
- 37.Ravizza T, Moneta D, Bottazzi B, Peri G, Garlanda C, Hirsch E, Richards GJ, Mantovani A, Vezzani A. Dynamic induction of the long pentraxin PTX3 in the CNS after limbic seizures: evidence for a protective role in seizure-induced neurodegeneration 1. Neuroscience. 2001;105:43–53. doi: 10.1016/s0306-4522(01)00177-4. [DOI] [PubMed] [Google Scholar]
- 38.Polentarutti N, Bottazzi B, Di SE, Blasi E, Agnello D, Ghezzi P, Introna M, Bartfai T, Richards G, Mantovani A. Inducible expression of the long pentraxin PTX3 in the central nervous system 3. J Neuroimmunol. 2000;106:87–94. doi: 10.1016/s0165-5728(00)00214-9. [DOI] [PubMed] [Google Scholar]
- 39.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, Garcia-Dorado D, Poca MA, Sahuquillo J, Baselga J, Seoane J. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–327. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 41.Dieterich LC, Mellberg S, Langenkamp E, Zhang L, Zieba A, Salomaki H, Teichert M, Huang H, Edqvist PH, Kraus T, Augustin HG, Olofsson T, Larsson E, Soderberg O, Molema G, Ponten F, Georgii-Hemming P, Alafuzoff I, Dimberg A. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J Pathol. 2012;228:378–390. doi: 10.1002/path.4072. [DOI] [PubMed] [Google Scholar]
- 42.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res. 2008;6:675–684. doi: 10.1158/1541-7786.MCR-07-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pen A, Moreno MJ, Durocher Y, Deb-Rinker P, Stanimirovic DB. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-beta signaling. Oncogene. 2008;27:6834–6844. doi: 10.1038/onc.2008.287. [DOI] [PubMed] [Google Scholar]
- 45.Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3:a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 47.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012;586:2003–2015. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Y, Pioli PD, Siegel E, Zhang Q, Nelson J, Chaturbedi A, Mathews MS, Ro DI, Alkafeef S, Hsu N, Hamamura M, Yu L, Hess KR, Tromberg BJ, Linskey ME, Zhou YH. EFEMP1 suppresses malignant glioma growth and exerts its action within the tumor extracellular compartment. Mol Cancer. 2011;10:123. doi: 10.1186/1476-4598-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YH, Kwon HJ, Kim DS. Matrix metalloproteinase 9 (MMP-9)-dependent processing of betaig-h3 protein regulates cell migration, invasion, and adhesion. J Biol Chem. 2012;287:38957–38969. doi: 10.1074/jbc.M112.357863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nam JO, Son HN, Jun E, Cha K, Lee BH, Park RW, Kim IS. FAS1 domain protein inhibits VEGF165-induced angiogenesis by targeting the interaction between VEGFR-2 and alphavbeta3 integrin. Mol Cancer Res. 2012;10:1010–1020. doi: 10.1158/1541-7786.MCR-11-0600. [DOI] [PubMed] [Google Scholar]
- 51.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 52.Rettig WJ, Erickson HP, Albino AP, Garin-Chesa P. Induction of human tenascin (neuronectin) by growth factors and cytokines: cell type-specific signals and signalling pathways. J Cell Sci. 1994;107(Pt 2):487–497. [PubMed] [Google Scholar]
- 53.Midwood KS, Hussenet T, Langlois B, Orend G. Advances in tenascin-C biology. Cell Mol Life Sci. 2011;68:3175–3199. doi: 10.1007/s00018-011-0783-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12 15. Cancer Res. 2006;66:11771–11780. doi: 10.1158/0008-5472.CAN-05-0470. [DOI] [PubMed] [Google Scholar]
- 55.Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH. Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer. 2002;98:362–369. doi: 10.1002/ijc.10233. [DOI] [PubMed] [Google Scholar]
- 56.Huang JY, Cheng YJ, Lin YP, Lin HC, Su CC, Juliano R, Yang BC. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J Immunol. 2010;185:1450–1459. doi: 10.4049/jimmunol.0901352. [DOI] [PubMed] [Google Scholar]
- 57.Wuchty S, Arjona D, Li A, Kotliarov Y, Walling J, Ahn S, Zhang A, Maric D, Anolik R, Zenklusen JC, Fine HA. Prediction of Associations between microRNAs and Gene Expression in Glioma Biology. PLoS ONE. 2011;6:e14681. doi: 10.1371/journal.pone.0014681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nowinski D, Koskela A, Kiwanuka E, Bostrom M, Gerdin B, Ivarsson M. Inhibition of connective tissue growth factor/CCN2 expression in human dermal fibroblasts by interleukin-1alpha and beta. J Cell Biochem. 2010;110:1226–1233. doi: 10.1002/jcb.22637. [DOI] [PubMed] [Google Scholar]
- 59.Chong HC, Tan CK, Huang RL, Tan NS. Matricellular proteins: a sticky affair with cancers. J Oncol. 2012;2012:351089. doi: 10.1155/2012/351089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: multiple paths to inflammation Mediators. Inflamm. 2011;2011:296069. doi: 10.1155/2011/296069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakada M, Miyamori H, Yamashita J, Sato H. Testican 2 abrogates inhibition of membrane-type matrix metalloproteinases by other testican family proteins. Cancer Res. 2003;63:3364–3369. [PubMed] [Google Scholar]
- 62.Paulus W, Baur I, Dours-Zimmermann MT, Zimmermann DR. Differential expression of versican isoforms in brain tumors. J Neuropathol Exp Neurol. 1996;55:528–533. doi: 10.1097/00005072-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 63.Arslan F, Bosserhoff AK, Nickl-Jockschat T, Doerfelt A, Bogdahn U, Hau P. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-beta2. Br J Cancer. 2007;96:1560–1568. doi: 10.1038/sj.bjc.6603766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaminska B, Kocyk M, Kijewska M. TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol. 2013;986:171–187. doi: 10.1007/978-94-007-4719-7_9. [DOI] [PubMed] [Google Scholar]
- 65.Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 67.Bryan L, Paugh BS, Kapitonov D, Wilczynska KM, Alvarez SM, Singh SK, Milstien S, Spiegel S, Kordula T. Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: implications for invasiveness. Mol Cancer Res. 2008;6:1469–1477. doi: 10.1158/1541-7786.MCR-08-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colin C, Voutsinos-Porche B, Nanni I, Fina F, Metellus P, Intagliata D, Baeza N, Bouvier C, Delfino C, Loundou A, Chinot O, Lah T, Kos J, Martin PM, Ouafik L, Figarella-Branger D. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol. 2009;118:745–754. doi: 10.1007/s00401-009-0592-2. [DOI] [PubMed] [Google Scholar]
- 69.Yamaguchi Y, Shao Z, Sharif S, Du XY, Myles T, Merchant M, Harsh G, Glantz M, Recht L, Morser J, Leung LL. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J Biol Chem. 2013;288:3097–3111. doi: 10.1074/jbc.M112.362954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamahata H, Takeshima H, Kuratsu J, Sarker KP, Tanioka K, Wakimaru N, Nakata M, Kitajima I, Maruyama I. The role of thrombin in the neo-vascularization of malignant gliomas: an intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int J Oncol. 2002;20:921–928. [PubMed] [Google Scholar]
- 71.Isaka T, Yoshimine T, Maruno M, Kuroda R, Ishii H, Hayakawa T. Altered expression of antithrombotic molecules in human glioma vessels. Acta Neuropathol. 1994;87:81–85. doi: 10.1007/BF00386257. [DOI] [PubMed] [Google Scholar]
- 72.Le MM, Fortin S, Mathieu V, Kiss R, Lefranc F. Galectins and gliomas. Brain Pathol. 2010;20:17–27. doi: 10.1111/j.1750-3639.2009.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D, Wang Y, Elyaman W, Khoury SJ, Rabinovich GA. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37:249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang J, Westberg JA, Andersson LC. Stanniocalcin 2, forms a complex with heme oxygenase 1, binds hemin and is a heat shock protein. Biochem Biophys Res Commun. 2012;421:274–279. doi: 10.1016/j.bbrc.2012.03.151. [DOI] [PubMed] [Google Scholar]
- 75.Westberg JA, Serlachius M, Lankila P, Penkowa M, Hidalgo J, Andersson LC. Hypoxic preconditioning induces neuroprotective stanniocalcin-1 in brain via IL-.6 signaling. Stroke. 2007;38:1025–1030. doi: 10.1161/01.STR.0000258113.67252.fa. [DOI] [PubMed] [Google Scholar]
- 76.Hill JJ, Moreno MJ, Lam JC, Haqqani AS, Kelly JF. Identification of secreted proteins regulated by cAMP in glioblastoma cells using glycopeptide capture and label-free quantification. Proteomics. 2009;9:535–549. doi: 10.1002/pmic.200800257. [DOI] [PubMed] [Google Scholar]
- 77.Sepporta MV, Tumminello FM, Flandina C, Crescimanno M, Giammanco M, La GM, di MD, Leto G. Follistatin as potential therapeutic target in prostate cancer. Target Oncol. 2013 doi: 10.1007/s11523-013-0268-7. [DOI] [PubMed] [Google Scholar]
- 78.Reddy SP, Britto R, Vinnakota K, Aparna H, Sreepathi HK, Thota B, Kumari A, Shilpa BM, Vrinda M, Umesh S, Samuel C, Shetty M, Tandon A, Pandey P, Hegde S, Hegde AS, Balasubramaniam A, Chandramouli BA, Santosh V, Kondaiah P, Somasundaram K, Rao MR. Novel glioblastoma markers with diagnostic and prognostic value identified through transcriptome analysis. Clin Cancer Res. 2008;14:2978–2987. doi: 10.1158/1078-0432.CCR-07-4821. [DOI] [PubMed] [Google Scholar]
- 79.Vadasz Z, Toubi E. Semaphorins: Their Dual Role in Regulating Immune-Mediated Diseases. Clin Rev Allergy Immunol. 2013 doi: 10.1007/s12016-013-8360-4. [DOI] [PubMed] [Google Scholar]
- 80.Tamagnone L. Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer. Cancer Cell. 2012;22:145–152. doi: 10.1016/j.ccr.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 81.Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene. 2009;28:3537–3550. doi: 10.1038/onc.2009.204. [DOI] [PubMed] [Google Scholar]
- 82.Tanco S, Zhang X, Morano C, Aviles FX, Lorenzo J, Fricker LD. Characterization of the substrate specificity of human carboxypeptidase A4 and implications for a role in extracellular peptide processing. J Biol Chem. 2010;285:18385–18396. doi: 10.1074/jbc.M109.060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ross PL, Cheng I, Liu X, Cicek MS, Carroll PR, Casey G, Witte JS. Carboxypeptidase 4 gene variants and early-onset intermediate-to-high risk prostate cancer. BMC Cancer. 2009;9:69. doi: 10.1186/1471-2407-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shannan B, Seifert M, Leskov K, Willis J, Boothman D, Tilgen W, Reichrath J. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006;13:12–19. doi: 10.1038/sj.cdd.4401779. [DOI] [PubMed] [Google Scholar]
- 85.Bello L, Lucini V, Carrabba G, Giussani C, Machluf M, Pluderi M, Nikas D, Zhang J, Tomei G, Villani RM, Carroll RS, Bikfalvi A, Black PM. Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res. 2001;61:8730–8736. [PubMed] [Google Scholar]
- 86.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jellinek DA, Chang AC, Larsen MR, Wang X, Robinson PJ, Reddel RR. Stanniocalcin 1 and 2 are secreted as phosphoproteins from human fibrosarcoma cells. Biochem J. 2000;350(Pt 2):453–461. [PMC free article] [PubMed] [Google Scholar]
- 88.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, Van Sluyter SC, Haynes PA. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics. 2011;11:535–553. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- 89.Venugopal C, Wang XS, Manoranjan B, McFarlane N, Nolte S, Li M, Murty N, Siu KW, Singh SK. GBM secretome induces transient transformation of human neural precursor cells. J Neurooncol. 2012;109:457–466. doi: 10.1007/s11060-012-0917-1. [DOI] [PubMed] [Google Scholar]
- 90.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 91.Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- 92.Feinstein DL, Heneka MT, Gavrilyuk V, Dello RC, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002;41:357–365. doi: 10.1016/s0197-0186(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 93.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–329. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 95.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 99.Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, Croce CM. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci U S A. 2011;108:4908–4913. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, Dinarello CA, Fujita M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem. 2010;285:6477–6488. doi: 10.1074/jbc.M109.064907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S: Protein loading in each sample as shown in Coomassie blue-stained gel: Culture supernatants from untreated (Ctr) and IL-1-treated U251 cells in triplicates were separated by SDS-PAGE (4-15% gradient gel from Bio-Rad) and the gel was stained with Bio-Safe Coomassie G-250 (Bio-Rad). Molecular weight markers are shown on the left.
Figure 2S (A) IL-1 does not increase U251 cell death: U251 cells were grown in DMEM with or without 10% FBS and untreated or treated with IL-1β (10 ng/ml) for 22 h. Cells were then lifted by trypsinization followed by wash in DMEM with 10% FBS twice. An aliquot (50 μl) of cell suspension was subjected to trypan blue exclusion test in a hemocytometer and the number of total and dead (TB) cells per ml were calculated. The numbers indicate the percentages of dead cells in each condition. Data are mean ± SD from triplicate cultures. The results show that while significance increase in cell death occurred following serum withdrawal, treatment with IL-1β did not affect cell death. n.s. = not significant, * p <0.05, ** p<0.01
Figure 2S (B): Lack of detectable β-actin in concentrated U251 secretome: The pellets (p.) and the concentrated supernatants (s., the same samples subjected to secretome analysis) of untreated and IL-1-treated U251 cultures (n = 3) were analyzed for β-actin expression by immunoblotting following 10% SDS-PAGE. Thirty μg of total protein was loaded in each lane. Beta-actin (intracellular protein) was not detectable in this highly concentrated tumor secretome (see Materials and Methods), suggesting the lack of significant cell death and/or contamination of the secretome by dead cell debris/intracellular protein.