

Abstract
Structure-specific DNA endonucleases have critical roles during DNA replication, repair and recombination, yet they also harbor the potential for causing genome instability. Controlling these enzymes may be essential to ensure efficient processing of ad hoc substrates and to prevent random, unscheduled processing of other DNA structures, but it is unknown whether structure-specific endonucleases are regulated in response to DNA damage. Here, we uncover DNA damage-induced activation of Mus81-Eme1 Holliday junction resolvase in fission yeast. This novel regulation requires both Cdc2CDK1 and Rad3ATR-dependent phosphorylations of Eme1. Mus81-Eme1 activation prevents gross chromosomal rearrangements in cells lacking the BLM-related DNA helicase Rqh1. We propose that linking Mus81-Eme1 DNA damaged-induced activation to cell cycle progression ensures efficient resolution of Holliday junctions that escape dissolution by Rqh1-TopIII while preventing unnecessary DNA cleavages.
Homologous recombination repair of damaged replication forks and double-strand breaks can lead to the formation of Holliday junctions (HJs). These 4-way DNA structures must be resolved to allow chromosome segregation during anaphase. Whereas the canonical bacterial HJ resolvase RuvC strongly prefers to process HJs, all three known eukaryotic nuclear HJ resolvases, Mus81-Eme1Mms4, Gen1Yen1 and Slx1-Slx4, efficiently process in vitro a variety of DNA structures that contain junctions between single-strand and double-strand DNA1–7. Given the multiplicity of DNA repair and recombination mechanisms that can generate these substrates, nuclear HJ resolvases must be tightly regulated in vivo 8,9. HJ resolution activity is particularly important in absence of Bloom (BLM)-related helicases, which are proposed to dissolve double Holliday junctions (HJs) in a non-endonucleolytic process that prevents sister-chromatid exchange10–12. In absence of BLM-related helicases, HJ resolvases are essential for viability1,11,13–16, but this comes at the expense of an increased risk of genomic instability and loss of heterozygosity due to crossovers. Thus, while crossovers are required for proper chromosome segregation and promoting genetic variation during meiosis, HJ resolving enzymes in vegetative cells need to be kept under tight control so that HJs are preferentially processed by BLM-related helicases. It was recently shown that Saccharomyces cerevisiae Mus81-Mms4Eme1 is activated during G2/M through phosphorylation of Mms4Eme1 by Cdc5PLK1 kinase 17 18. This regulation may act as a failsafe mechanism to ensure resolution of HJs formed by the occasional spontaneous collapse of replication forks. However, it is unclear whether Mus81-Eme1 is independently activated in response to DNA lesions caused by external DNA damaging agents or by the accumulation of HJs in mitotic cells lacking functional BLM-related helicases. To gain insight into this fundamental question we undertook to investigate the importance of regulating Mus81-Eme1 in absence of BLM-related helicases in the fission yeast Schizosaccharomyces pombe, where Mus81-Eme1 is the primary HJ resolvase and Rqh1 the major BLM-related helicase.
RESULTS
Stimulation of Mus81-Eme1 in response to DNA damage
To determine whether Mus81-Eme1 is regulated in response to DNA damage, we first assessed whether Eme1 undergoes post-translational modifications in cells lacking Rqh1 or in response to treatment with camptothecin (CPT), which is a prototypical DNA topoisomerase I inhibitor with antitumor activity. Mus81-Eme1 is essential for viability in both situations 13,19. A strong phosphatase-sensitive mobility-shift of Eme1 was observed by SDS-polyacrylamide gel electrophoresis (PAGE) following CPT treatment of both WT and rqh1Δ cells (Fig. 1a, b). This phosphorylation of Eme1 was notable because CPT treatment triggers a checkpoint arrest in G2 phase prior to activation of Plo1PLK1 kinase, and thus the CPT-induced phosphorylation of Eme1 likely involves other protein kinases. A pool of Eme1 was also hyper-phosphorylated in non-treated rqh1Δ cells, probably due to the high rate of spontaneous DNA damage that results from loss of Rqh1. This DNA damage-induced hyper-phosphorylation of Eme1 significantly increased the in vitro HJ resolvase activity of the partially affinity-purified Mus81-Eme1 complex, as well as its ability to process other secondary DNA structures (Figure 1c, d and Supplementary Fig. 1a, b). Use of a nuclease-dead mus81DD strain confirmed that all cuts were Mus81-Eme1 dependent (Supplementary Fig. 1c). These data suggest that DNA damage activates Mus81-Eme1 by a mechanism that might involve Eme1 hyper-phosphorylation.
Figure 1. Phosphorylation of Eme1 and hyper-activation of Mus81-Eme1 in response to DNA damage.

(a) Western blot assay on total lysates of wild-type (WT) or rqh1Δ cells producing TAP-tagged Eme1 (See Supplementary Figure 1 for structure of the TAP-tag). Cells were either non-treated or exposed to 40 μM camptothecin (CPT). Wild-type (WT) (PH423), rqh1Δ (PH454). (b) Western blot assay on TAP-Eme1 affinity purified from untreated or 40μM CPT-treated WT cells preincubated with active λ phosphatase (PPase) or heat inactivated λ phosphatase (PPase 95°C). (c) A 32P-labelled (red dot) Holliday Junction (HJ) was incubated with identical amounts of the Mus81-Eme1 samples purified from untreated or 40 μM CPT-treated cells shown in b. The reaction products were separated by neutral PAGE. (d) Average (+/−SEM) stimulation fold of HJ resolution by Mus81-Eme1 following CPT treatment calculated from n=8 independent experiments. The histogram shows the ratio of the HJ resolvase activity of the Mus81-Eme1 affinity-purified from CPT-treated cells over that of Mus81-Eme1 affinity purified from untreated cells.
CPT causes replication fork regression20 and collapse21, leading to activation of the checkpoint kinase Chk1 by the ATM-related (ATR) checkpoint kinase ortholog Rad322. While hyper-phosphorylation of Eme1 was observed in absence of the replication checkpoint kinase Cds1, deletion of the Chk1 DNA damage checkpoint kinase strongly diminished CPT-induced hyper-phosphorylation of Eme1 (Fig. 2a). Elimination of Chk1 also reduced the constitutive hyper-phosphorylation of Eme1 observed in non-treated rqh1Δ cells, confirming that accumulation of DNA damage in absence of Rqh1 leads to Chk1-dependent phosphorylation of Eme1 (Fig. 2b). DNA damage induced hyper-phosphorylation of Eme1 was also abolished when the Chk1 activation factors Rad3ATR and Crb2 were deleted (Fig. 2a). The phosphorylation pattern of Eme1 was analyzed by tandem mass-spectrometry to identify phosphorylated sites. Six serine residues were identified within an N-terminal domain of Eme1 (Supplementary Fig. 1d), which is essential for its in vivo functions but dispensable for the basal catalytic activity of the complex in vitro (unpublished, PMD, SS, PHLG). Mutating all six residues to alanine abolished CPT-induced phosphorylation of the resulting Eme16SA mutant protein and prevented stimulation of HJ resolution by Mus81-Eme1 (Fig. 2c and Supplementary Fig. 1e). Stimulation of Mus81-Eme1 following CPT treatment was also abolished in a rad3Δ mutant (Supplementary Fig. 1e), confirming that Mus81-Eme1 is stimulated in response to DNA damage through phosphorylation of Eme1. Five of the six serines were in CDK consensus sites (Supplementary Fig. 1c), suggesting that Eme1 phosphorylation might also be cell-cycle regulated in a CDK-dependent manner. DNA damage-independent phosphorylation of Eme1 was further suggested by the increased mobility of Eme1 from untreated cells following incubation with λ phosphatase (Fig. 1b).
Figure 2. Eme1 is phosphorylated by the DNA damage checkpoint.

(a) Western blot assay on total lysates derived from the indicated untreated or 40 μM CPT-treated strains producing TAP-tagged Eme1; WT: strain PH466, cds1Δ (PH113), chk1Δ (PH120), rad3Δ (PH132), crb2Δ (PH189). (b) Western blot assay on total lysates from untreated or 40 μM CPT-treated WT, rqh1Δ and chk1Δ rqh1Δ strains. (c), Upper panel: Western blot assay on affinity-purified Mus81-Eme1 from untreated or 40 μM CPT-treated WT (PH423) and eme16SA (PH481) cells, Lower panel: stimulation fold of HJ resolution. The histogram shows the ratio of the HJ resolvase activity of the Mus81-Eme1 affinity-purified from CPT-treated cells over that of Mus81-Eme1 affinity purified from untreated cells. The samples used in the in vitro nuclease assay are those shown on upper panel.
CDK-mediated phosphorylation of Eme1 in G2
Using “block and release” of a temperature-sensitive cdc25-22 strain to synchronize cells in late G2 and follow Eme1 phosphorylation throughout the cell cycle, we observed that fission yeast Eme1 undergoes cell-cycle dependent phosphorylation with hyper-phosphorylation in G2 and M, followed by a dip during S phase (Fig. 3a). Importantly, cell-cycle dependent phosphorylation of Eme1 was not abolished in a cdc25-22 rad3Δ mutant, indicating that Eme1 undergoes cell-cycle regulated phosphorylation independently of the DNA damage induced Rad3ATR-mediated phosphorylation described above (Fig. 3b). Using a chromatin fractionation assay23 we found that both Eme1 and Eme16SA were tightly associated with chromatin throughout the cell-cycle, indicating that cell-cycle controlled phosphorylation of Eme1 does not modulate its bulk chromatin association (Supplementary Fig. 2).
Figure 3. Eme1 is phosphorylated in a Cdc2CDK1-dependent manner.

(a) cdc25-22 cells producing TAP-tagged Eme1 (PH171) or Eme16SA (PH491) were blocked at 36°C for 4 hrs before release at 25°C. Top: Septation index measured for each strain. Middle: Western blot detection of TAP-tagged Eme1. Bottom: Western blot detection of TAP-tagged Eme16SA. (b) Western blot detection of TAP-Eme1 from block and released cdc25-22 rad3Δ (PH538) mutant cells. (c) cdc2as mutant cells (PH602) were incubated with 12 mM HU for 4 hrs to arrest cell cycle progression in early S-phase before release in fresh media with or without NmPP1. Top: FACS analysis of asynchronous (As), blocked and released cells. Bottom: Western blot detection of TAP-Eme1 from blocked and released (+ or − NmPP1) cells.
To confirm that Eme1 undergoes Cdc2CDK1-dependent phosphorylation, we compared the phosphorylation pattern of Eme1 in a cdc2as (Cdc2 analogue sensitive) mutant strain in presence or absence of the ATP analog NmPP124. When cells were synchronized in early S-phase with 12mM HU for 4hrs and released in fresh media in presence or absence of NmPP1, the increase of Eme1 phosphorylation observed upon entry into G2 was abolished in presence of NmPP1 (Fig. 3c), confirming that Eme1 undergoes Cdc2CDK1-dependent phosphorylation during G2 when Cdc2CDK1 has a moderate level of activity{Coudreuse:2010cr}. This cell-cycle regulated phosphorylation of Eme1 in G2 did not require the Cdc5PLK1 ortholog Plo1, in contrast to Mms4Eme1 phosphorylation in S. cerevisiae17,18 (Supplementary Fig. 3).
CDK primes Eme1 for phosphorylation after DNA damage
To discriminate whether DNA damage-induced phosphorylation of Eme1 mediated by Rad3ATR may operate in a Cdc2CDK1-dependent way as described for the checkpoint mediator Crb225,26, we first generated an eme14SA mutant strain where only CDK consensus target sites are mutated (Supplementary Fig. 1d). Eme14SA was no longer hyper-phosphorylated in response to CPT and the HJ resolvase activity of the resulting Mus81-Eme14SA complex was no longer stimulated (Fig. 4a). These data suggest that Cdc2CDK1-dependent phosphorylation of Eme1 is a prerequisite for the DNA damage-induced activation of Mus81-Eme1 HJ resolvase. The possibility remained that CPT-induced hyper-phosphorylation of Eme1 resulted from DNA damage checkpoint-dependent cell-cycle arrest in late G2, where Eme1 is hyper-phosphorylated. To rule this out, we tested whether activation of the DNA damage checkpoint in cells already arrested in G2 induced further phosphorylation of CDK-phosphorylated Eme1. Activation of the DNA damage checkpoint following CPT treatment of fission yeast requires ongoing DNA replication22. Therefore, to activate the DNA damage checkpoint of G2 arrested cells and assess whether Eme1 underwent additional DNA damage-induced phosphorylation, cdc25-22 cells synchronized in late G2 at 36°C were treated with the radiomimetic agent bleomycin. Importantly, bleomycin treatment induced a strong Chk1-dependent mobility shift of Eme1 that was abolished in eme16SA and eme14SA mutant cells, confirming that DNA damage induced phosphorylation of Eme1 is not limited to topoisomerase inhibition (Fig. 4b–d). Interestingly, bleomycin induced an additional mobility shift of Eme1 in cdc25-22 cells that had been first arrested in G2 at 36°C (Fig. 4d). Taken together our results show that Eme1 undergoes two phosphorylation events, with a first CDK-dependent phosphorylation that is needed for secondary DNA damage checkpoint-mediated phosphorylation following DNA damage.
Figure 4. Cdc2CDK1-dependent phosphorylation prime Eme1 for DNA damage dependent hyper-activation of Mus81-Eme1.
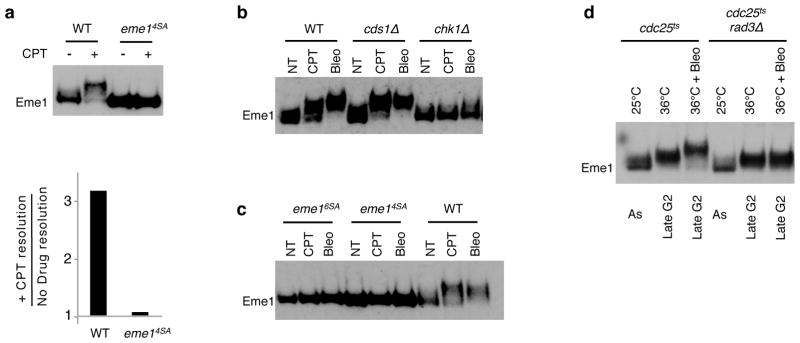
(a) Upper panel: Western blot detection of TAP-Eme1 from untreated or 40μM CPT-treated WT (PH466) and eme14SA (PH546) strains. Lower pannel: Hyper-activation of HJ resolvase activity of Mus81-Eme1 after 40 μM CPT-treatment of WT and eme14SA mutant cells. Mus81-Eme1 HJ resolvase activation was calculated by dividing the activity of Mus81-Eme1 affinity purified from CPT-treated cells by the activity of Mus81-Eme1 affinity purified from untreated cells. (b) Western blot detection of TAP-Eme1 in total lysates derived of the indicated untreated, 40 μM CPT or 5 mg/ml bleomycin-treated strains producing TAP-tagged Eme1; WT (PH466), cds1Δ (PH113), chk1Δ (PH120). (c) as in e, eme16SA (PH491), eme14SA (PH546), WT (PH466). (d) Western blot assay on the total lysates from the indicated strains grown asynchronously (As) at 25°C or synchronized and kept in late G2 at 36°C, without or with bleomycin. cdc25ts (PH171), cdc25ts rad3Δ (PH538).
Dual control of Mus81-Eme1 necessary in absence of Rqh1BLM
Considering that cells lacking Rqh1BLM rely on the Mus81-Eme1 HJ resolvase for survival 1, we anticipated that Cdc2CDK1- and Rad3ATR-dependent phosphorylation of Eme1 would be crucial in these cells. This prediction was confirmed by the synergistic growth defect caused by introducing the eme16SA and eme14SA mutations in the rqh1Δ background (Fig. 5a–c). This relationship contrasts with elimination of Cdc5PLK1 sites in S. cerevisiae Mms4, which in the absence of exogenous DNA damaging agents has no impact on the viability of cells lacking Sgs1Rqh1 DNA helicase or Yen1 HJ resolvase 18. Interestingly, the eme16SA and eme14SA mutants were insensitive to CPT and bleomycin, contrasting with the acute hypersensitivity of the eme1Δ mutant (Fig. 5c and Supplementary Fig. 4a). To our knowledge these are the first Mus81-Eme1 separation of function mutants that lack functions essential only in absence of Rqh1. The acute CPT and bleomycin sensitivity of the eme16SA rqh1Δ double mutant compared to an rqh1Δ mutant (Figure 5c and Supplementary Fig. 4a), suggests that stimulation of Mus81-Eme1 is also required to process HJs which accumulate in absence of Rqh1 following exogenously induced DNA damage. To test this assumption, we assessed whether the sensitivity to CPT of the eme16SA rqh1Δ double mutant could be rescued by over-producing the bacterial RusA resolvase. Wild type RusA, but not a catalytically inactive mutant that lacks HJ resolvase activity, substantially rescued the CPT sensitivity of an eme16SA rqh1Δ mutant (Fig. 5d). This genetic suppression contrasts with the inability of RusA to rescue the CPT-sensitivity of an rqh1Δ single mutant19. In fact, high expression of active RusA slows growth in an rqh1Δ mutant (Supplementary Figure 4b). This result supports the proposal that HJ resolvases must be properly regulated to ensure robust growth.
Figure 5. Hyper-activation of Mus81-Eme1 is critical in absence of Rqh1BLM.

(a) Western blot detection of TAP-Eme1 in cell lysates of the indicated untreated strains. (b) Left pannel: Tetrad analysis of a eme14SA x rqh1Δ mating germinated at 30°C. Right pannel: representative image of eme16SA rqh1Δ (PH462) colonies 16 hrs after plating on YES rich media. (c) 5-fold dilutions of cells with the indicated genotype were plated on medium supplemented or not with the indicated concentrations of CPT followed by incubation at 30°C. TAP-Eme1 (PH466); eme1Δ (PH419); rqh1Δ (SC3250); rqh1Δ TAP-Eme1 (PH454); rqh1Δ eme16SA (PH462). (c) 5-fold dilutions of eme1Δ (PH466) and eme16SA rqh1Δ (PH454) cells over-producing the bacterial wild-type RusA HJ resolvase or a nuclease dead RusAND version. Cells were plated on medium supplemented or not with the indicated concentrations of CPT followed by incubation at 30°C.
Gross chromosomal rearrangements in eme16SA rqh1Δ mutants
BLM-related helicases are required to maintain the integrity of specific genomic loci such as the ribosomal DNA (rDNA) tandem repeats 6,27. These rDNA repeats are located at both ends of chromosome III in fission yeast. To assess whether Cdc2CDK1- and Rad3ATR-mediated activation of Mus81-Eme1 is required for stability of the rDNA loci in absence of Rqh1, we used pulse-field gel electrophoresis (PFGE) to examine the chromosome structure in eme16SA rqh1Δ double-mutants (Fig. 6a). Whereas chromosomes I and II appeared normal, a striking plasticity of chromosome III (ChrIII) was observed in the eme16SA rqh1Δ clones we tested, with some clones containing two ChrIII of distinct sizes (Fig. 6b and Supplementary Fig. 5). An rDNA probe confirmed that the sizes of the rDNA loci were highly variable in eme16SA rqh1Δ cells (Supplementary Fig. 5). We also noticed that in strains containing two ChrIII, the smallest chromosome contained in some cases as much rDNA as the larger ChrIII, indicating that it was missing non-rDNA regions compared to the larger chromosome. To confirm this we used comparative genomic hybridization (CGH) to compare the genome of an eme16SA rqh1Δ clone with two ChrIII to that of a WT strain. This analysis established that the eme16SA rqh1Δ clone tested is disomic for ChrIII and showed that the smallest chromosome is lacking a large portion of its right arm (Fig. 6b). CGH analysis of other disomic eme16SA rqh1Δ clones indicated that the break-point was different from clone to clone. Taken together, our results confirm that Cdc2CDK1- and DNA damage checkpoint-dependent phosphorylation of Eme1 is essential for chromosome stability in absence of the BLM-related helicase Rqh1.
Figure 6. DNA damage induced phosphorylation of Eme1 prevents gross chromosomal rearrangements in absence of Rqh1BLM.

(a) Left: Schematic representation of S. pombe three chromosomes (Chr) and distribution of rDNA at both ends of ChrIII. Right: Genomic DNA samples of WT and the indicated mutant strains were subjected to pulse-field gel electrophoresis. The gel was stained with ethidium bromide. A dashed line indicates the position of WT ChrIII. WT (PH466); eme16SA (PH481); rqh1Δ (PH454); rqh1Δ eme16SA (PH462, PH463, PH464, PH465). (b) Comparative Genome Hybridization of the ChrIII disomic eme16SA rqh1Δ clone (PH463) indicated by “*” in panel a. (c) Model; see text for details.
DISCUSSION
Structure-specific DNA endonucleases are crucial for the repair of many types of DNA lesions, but it has remained unclear whether their activities are regulated in response to DNA damage. Here, we have found that Mus81-Eme1 activity significantly increases in cells treated with CPT, a drug that causes replication fork collapse. Unexpectedly, this regulation relies on both cell cycle driven phosphorylation of Eme1 by Cdc2CDK1 and Rad3ATR-dependent DNA damage-induced phosphorylation. Cdc2CDK1-dependent phosphorylation of Eme1 in G2 licenses Mus81-Eme1 for Rad3ATR-dependent catalytic stimulation in response to DNA damage. We propose that this mechanism ensures rapid activation of Mus81-Eme1 in response to DNA damage specifically in G2, thereby guaranteeing that Holliday junctions that have eluded processing by Rqh1BLM are resolved prior to the onset of mitosis. This DNA damaged-induced activation of Mus81-Eme1 differs from the Cdc5PLK1-mediated activation of Mus81-Mms4Eme1 that occurs during the merged G2/M phase in budding yeast17,18. Unlike budding yeast, fission yeast relies on the Cdc25-Wee1 network to regulate Cdc2CDK1 and thereby control transition from G2 to M phase. The moderate level of Cdc2CDK1 activity during G2 primes Mus81-Eme1 for activation in response to high levels of DNA damage (Fig. 6c). We further propose that this mechanism insures that Mus81-Eme1 activity is maintained at a low basal level except when its function is critical, thereby minimizing the possibility that it will create genome destabilizing lesions when its activity is not needed. The potential for genome destabilizing DNA cleavages by Mus81-Eme1 have been highlighted in several studies28–31.
Our phospho-mutant studies indicate that activation of Mus81-Eme1 is crucial in absence of the Rqh1BLM helicase. Phosphorylation of Eme1 in rqh1Δ cells is required to prevent gross chromosomal rearrangements even in absence of any exogenously induced DNA damage (Fig. 6). In contrast, loss of the Cdc5PLK1-mediated phosphorylation of Mms4 has no impact on the viability of sgs1Δ and sgs1Δ yen1Δ mutants unless they are also treated with DNA damaging agents 18. To our knowledge, this is the first report of a separation of function for Mus81-Eme1 where the essential functions that it fulfills in absence of Rqh1 have been dissociated from those that it fulfills in response to exogenous genotoxic stress in wild type cells.
The severe chromosome instability observed when Eme1 can no longer be phosphorylated in response to the endogenous DNA damage that accumulates in absence of Rqh1BLM underscores the importance of the adaptive control mechanism of Mus81-Eme1 unraveled in this study. Our PFGE and CGH analyses revealed chromosome rearrangements primarily on chromosome III with, in some cases, the appearance of disomic cells with two copies of ChrIII. In fission yeast, only ChrIII aneuploidy is viable. Therefore, it is likely that the severe growth defect phenotype of the eme16SA rqh1Δ and eme14SA rqh1Δ mutants results from unviable chromosomal rearrangements that also affect the other two chromosomes. We observed no phenotype associated with preventing DNA damaged induced phosphorylation of Eme1 in cells that have functional Rqh1. This contrasts with the growth defect of eme1Δ null mutants that have lost Mus81-Eme1 functions and suggests that the Cdc2CDK1 and Rad3ATR mediated control of Mus81-Eme1 in response to DNA damage acts as a fail-safe mechanism. Recombination structures in wild type cells are resolved by non-endonucleolytic activity of Rqh1BLM helicase (with its associated factors Top III and Rmi1) and the basal endonucleolytic processing activity of Mus81-Eme1. In absence of Rqh1BLM, the basal nuclease activity of Mus81-Eme1 is insufficient to cope with the extra burden of unprocessed recombination intermediates and DNA damage-induced hyper-activation of Mus81-Eme1 becomes necessary to limit chromosome instability. However, we found that over-expression of constitutively active bacterial RusA resolvase impaired the growth of rqh1Δ cells, indicating the importance of limiting resolvase activity even when it is essential for cell viability. The 2 to 3 fold activation of Mus81-Eme1 measured in our assays may be sufficient to ensure effective HJ resolution while still minimizing the risk of Mus81-Eme1 creating new DNA lesions by cleaving transient joint DNA molecules. Restricting Mus81-Eme1 activation to G2 phase may increase the probability that double HJs or regressed replication forks are dissolved or reversed by BLM-related helicases, respectively. There is a growing body of evidence that Mus81-Eme1 is subjected to a complex network of regulatory processes. Clearly, further investigations are needed to determine the precise underlying molecular mechanisms and how these processes are coordinated to maintain genome stability.
Online Methods
Yeast strains
Fission yeast strain genotypes are listed in Table S1. Standard fission yeast methods were used as described previously32,33.
The eme16SA and eme14SA phosphorylation mutants were generated as follows. The EME1 genomic locus from strain PH81 (h+, leu1-32 ura4-D18 TAP:Eme1 Mus81:13Myc-KanMX6) that produces N-terminally TAP-tagged Eme1, was sub-cloned into a TopoTA vector (Invitrogen). Point mutations were introduced on that TopoTA-EME1 vector using a Multiprime site directed mutagenesis kit (Stratagene). Mutations were confirmed by DNA sequencing. The mutated EME1 genomic locus from the TopoTA-EME1 vector was used to transform strain PH419, in which the entire EME1 gene is replaced by a URA4 cassette. 5′-FOA resistant clones were selected and confirmed as TAP:Eme16SA or TAP:Eme14SA producing strains by genomic DNA sequencing.
For synchronization of cells using cdc25-22 block and release, cells containing the temperature sensitive cdc25-22 allele were grown to exponential phase at permissive temperature (25°C), shifted at restrictive temperature (36°C) for 3.5 hours to arrest the cell cycle in G232. Upon release to permissive temperature (25°C), the cells synchronously enter the cell cycle. Cells were collected and processed every 20 minutes. Progression into S- phase was monitored microscopically by counting cells that contained septa, the appearance of which correlates with S-phase32.
For synchronization of cells in Figure 3c, cells containing the cdc2as allele were grown to exponential phase at 25°C before addition of 12mM of HU (Sigma-Aldrich) for 4 hrs to arrest the cells in early S-phase. Following extensive washes, cells were released in fresh medium at 25°C and collected every 20 min. FACS analysis confirmed HU-induced early S-phase arrest and allowed monitoring of cell cycle progression following release. Cdc2as was inhibited by addition of 1 μM NmPP1 (Carbosynth).
RusA complementation assays were performed by transforming eme1Δ and rqh1Δ eme16SA strains with pREP1-RusA or pREP1-RusAD70N plasmids for over-expression of RusA or the catalytically inactive RusAD70N mutant1.
Native and TCA protein extraction, immunoblotting
Cellular lysates were prepared from exponentially growing cell cultures treated or not with camptothecin (Sigma-Aldrich) or bleomycin (Calbiochem). Cells were lysed in lysis buffer1 using a Retsch mechanical grinder in presence of liquid nitrogen. The cellular lysate was clarified and stored at −80°C. Protein concentration of each sample was determined with a Nanodrop (Thermo-Scientific) and protein amounts were normalized between the different samples. Denatured cell lysates were prepared by TCA precipitation. Cells were resuspended in 20% TCA and vortexed. Following centrifugation, the TCA precipitate was resuspended in SDS-PAGE loading buffer (Invitrogen) containing Tris-Base.
Protein extracts were resolved on Tris-Acetate 3–8% polyacrylamide NUPAGE gels (Invitrogen). Proteins were transferred to a nitrocellulose Hybond-C membrane (Invitrogen). The membrane was blocked in PBS-T milk 5% and probed using anti-Flag (Sigma-Aldrich) antibody (1/5000 dilution).
Nuclease assays
TAP-Eme1 was affinity purified and used in nuclease assays on various DNA substrates as previously described for TAP:Mus811,2.32P-labelled DNA substrates were used in all nuclease assays except in Fig S1.c where fluorescent Cy5-labelled substrates were used. The HJ substrate used in this study was the X12 mobile HJ.
Pulse Field Gel Electrophoresis
PFGE of whole chromosomes was performed as previously described6. 5.108 cells were spheroplasted with 0.6 mg ml−1 Zymolase-100T (Seikagu) at 37 °C for 2h, resuspended in 1% low melting point agarose (SIGMA) at a concentration of 108 cells per plug. Agarose plugs were loaded onto 0.8% agarose gels. PFGE was performed on a BioRad CHEF DR-III system in 1× TAE (40 mM Tris-acetate buffer, 2 mM Na2EDTA pH 8.3) at 14 °C using the following program: 48 h at 2 V cm−1, 106° angle, 30 min switch time. After electrophoresis, DNA was visualized by ethidium bromide staining, and gels were processed for Southern blotting, which was performed using an rDNA probe and a Wee1 probe (used as a probe to visualize Chr III).
Comparative Genomic Hybridization
Agilent S. pombe ChIP on CHIP 4x44K G8410A Genome Microarrays were used to perform comparative genomic hybridizations. Genomic DNA from strains PH463 and PH466 was prepared using a Qiagen genomic kit and DNA was directly labeled using the Agilent Genomic DNA Enzymatic labeling Kit (5190-0453). Hybridizations were performed according to Agilent recommendations.
Proteomic identification of Eme1 phosphorylation sites
TAP-Eme1 was affinity purified from WT and cds1Δ + HU 10mM cells. Western blot analysis showed that Eme1 is hyper-phosphorylated to the same extent in CPT-treated and HU-treated cds1Δ cells (data not shown).
TCA precipitates were proteolytically digested by Lys-C and trypsin analyzed by MudPIT on a LTQ mass spectrometer (Thermofisher) as previously described 34. Data was analyzed with the SEQUEST and DTASelect algorithms using a differential modification search that considered +80 on serine, threonine, and tyrosine residues 35. Sites found with high confidence as constitutively phosphorylated or phosphorylated in response to DNA damage were further validated by manual inspection.
Supplementary Material
Acknowledgments
We thank M. Grenon, N. Rhind, C. Chahwan, N. Boddy, J.H. Guervilly and G. Almouzni for critical reading of the manuscript and stimulating discussions throughout the course of this study and L. Brossy for technical help. Many thanks to V. Simanis (ISREC, Lausanne, Switzerland) for kindly providing the cdc2as strain and to Y. Denis for technical help with the CGH analysis. PHLG acknowledges financial support from Fondation Arc pour la Recherche sur le Cancer (SF1052), Centre National de la Recherche Scientifique (ATIP program) and Agence Nationale de la Recherche (ANR-10-BLAN-1512-01). BL was supported by a grant from Agence Nationale de la Recherche (ANR-10-BLAN-1606-03). Work in the laboratory of PR was supported by the US National Institutes of Health (RO1 GM59447).
Footnotes
Author Contributions. PMD, SC, SS, AT, PS, JW, PHLG designed and performed experiments. PHLG, PR, BL, JRY designed experiments and supervised the work and PHLG wrote the paper. Experiments in Figure 1, 2 and 4 were performed by PMD and SS with initial input from PHLG, PS and SC in strain engineering. Experiments in Figure 3 were all performed by PMD with initial input from PHLG and PS in strain engineering 3a,b. In Figures 5a,b experiments were performed by PMD. PHLG performed experiments in 5c,d. Experiments in Figures 6a,b were carried out by SC.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Boddy MN, et al. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107:537–548. doi: 10.1016/s0092-8674(01)00536-0. [DOI] [PubMed] [Google Scholar]
- 2.Gaillard PHL, Noguchi E, Shanahan P, Russell P. The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell. 2003;12:747–759. doi: 10.1016/s1097-2765(03)00342-3. [DOI] [PubMed] [Google Scholar]
- 3.Whitby MC, Osman F, Dixon J. Cleavage of model replication forks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J Biol Chem. 2003;278:6928–6935. doi: 10.1074/jbc.M210006200. [DOI] [PubMed] [Google Scholar]
- 4.Rass U, et al. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010;24:1559–1569. doi: 10.1101/gad.585310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulon S, et al. Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol Biol Cell. 2004;15:71–80. doi: 10.1091/mbc.E03-08-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehmsen KT, Heyer WD. A junction branch point adjacent to a DNA backbone nick directs substrate cleavage by Saccharomyces cerevisiae Mus81-Mms4. Nucleic Acids Res. 2009;37:2026–2036. doi: 10.1093/nar/gkp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz-Galván S, et al. Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol Cell Biol. 2012;32:1592–1603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho CK, Mazón G, Lam AF, Symington LS. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashton TM, Mankouri HW, Heidenblut A, McHugh PJ, Hickson ID. Pathways for Holliday junction processing during homologous recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2011;31:1921–1933. doi: 10.1128/MCB.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wechsler T, Newman S, West SC. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature. 2011;471:642–646. doi: 10.1038/nature09790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 13.Boddy MN, et al. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20:8758–8766. doi: 10.1128/mcb.20.23.8758-8766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001;15:2730–2740. doi: 10.1101/gad.932201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen SL, Kuo HK, Savukoski D, Brodsky MH, Sekelsky J. Three structure-selective endonucleases are essential in the absence of BLM helicase in Drosophila. PLoS Genet. 2011;7:e1002315. doi: 10.1371/journal.pgen.1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2001;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matos J, Blanco MG, Maslen S, Skehel JM, West SC. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–172. doi: 10.1016/j.cell.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo-Fernandez M, Saugar I, Ortiz-Bazan MA, Vazquez MV, Tercero JA. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doe CL, Ahn JS, Dixon J, Whitby MC. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem. 2002;277:32753–32759. doi: 10.1074/jbc.M202120200. [DOI] [PubMed] [Google Scholar]
- 20.Ray Chaudhuri A, et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 21.Regairaz M, et al. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J Cell Biol. 2011;195:739–749. doi: 10.1083/jcb.201104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan S, Capasso H, Walworth NC. The topoisomerase I poison camptothecin generates a Chk1-dependent DNA damage checkpoint signal in fission yeast. Yeast. 1999;15:821–828. doi: 10.1002/(SICI)1097-0061(199907)15:10A<821::AID-YEA422>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Goshima G, Iwasaki O, Obuse C, Yanagida M. The role of Ppe1/PP6 phosphatase for equal chromosome segregation in fission yeast kinetochore. EMBO J. 2003;22:2752–2763. doi: 10.1093/emboj/cdg266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dischinger S, Krapp A, Xie L, Paulson JR, Simanis V. Chemical genetic analysis of the regulatory role of Cdc2p in the S. pombe septation initiation network. J Cell Sci. 2008;121:843–853. doi: 10.1242/jcs.021584. [DOI] [PubMed] [Google Scholar]
- 25.Esashi F, Yanagida M. Cdc2 phosphorylation of Crb2 is required for reestablishing cell cycle progression after the damage checkpoint. Mol Cell. 1999;4:167–174. doi: 10.1016/s1097-2765(00)80364-0. [DOI] [PubMed] [Google Scholar]
- 26.Caspari T, Murray JM, Carr AM. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002;16:1195–1208. doi: 10.1101/gad.221402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaliraman V, Brill SJ. Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr Genet. 2002;41:389–400. doi: 10.1007/s00294-002-0319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forment JV, Blasius M, Guerini I, Jackson SP. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS ONE. 2011;6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Froget B, Blaisonneau J, Lambert S, Baldacci G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol Biol Cell. 2008;19:445–456. doi: 10.1091/mbc.E07-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beck H, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domínguez-Kelly R, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forsburg SL. Growth and manipulation of S. pombe. Curr Protoc Mol Biol. 2003;Chapter 13(Unit 13.16) doi: 10.1002/0471142727.mb1316s64. [DOI] [PubMed] [Google Scholar]
- 33.Bähler J, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 34.Maccoss MJ, et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci USA. 2002;99:7900–7905. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabb DL, McDonald WH, Yates JR. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.