

Abstract
Ribosomes, as the center of protein translation in the cell, require careful regulation via multiple pathways. While regulation of ribosomal synthesis and function has been widely studied on the transcriptional and translational “levels,” the biological roles of ribosomal post-translational modifications (PTMs) are largely not understood. Here, we explore this matter by using quantitative mass spectrometry to compare the prevalence of ribosomal methylation and acetylation for yeast in the log phase and the stationary phase of growth. We find that of the 27 modified peptides identified, two peptides experience statistically significant changes in abundance: a 1.9-fold decrease in methylation for k(Me)VSGFKDEVLETV of ribosomal protein S1B (RPS1B), and a 10-fold increase in dimethylation for r(DiMe)GGFGGR of ribosomal protein S2 (RPS2). While the biological role of RPS1B methylation has largely been unexplored, RPS2 methylation is a modification known to have a role in processing and export of ribosomal RNA. This suggests that yeast in the stationary phase increase methylation of RPS2 in order to regulate ribosomal synthesis. These results demonstrate the utility of mass spectrometry for quantifying dynamic changes in ribosomal PTMs.
Keywords: Ribosome, Post-translational Modifications, Mass Spectrometry, Proteomics, Stationary Phase, Growth Conditions
Introduction
Ribosomes are complex molecular machines consisting of about 79 proteins and 4 rRNA molecules (in Saccharomyces cerevisiae),1 and they serve as the center of protein translation in the cell. Protein translation and the synthesis of new ribosomes are energy-intensive processes; ribosome synthesis utilizes 90% of cell energy in yeast during exponential growth.2 Thus, it is important for the cell to carefully regulate ribosomal synthesis and function. Ribosomal regulation has been found to be influenced by a number of environmental conditions, including nutrient availability, environmental stressors, and growth phase. Here, we examine regulation of ribosomes by using mass spectrometry to quantify the PTMs of yeast ribosomal proteins under different growth phase conditions.
The yeast population growth curve is generally characterized by three phases: the lag phase, the log phase, and the stationary phase.3 As yeast transition from log phase into stationary phase, the starvation conditions prompt the cells to enter a quiescent state, part of which involves regulating ribosomal synthesis and function.4 This regulation is an immensely complex process involving changes on multiple “levels” including: transcription,5–8 translation,9 small molecule10 and protein binding partners for the ribosome,11 as well as post-translational modifications to ribosomal proteins.12
While the regulatory pathways at the transcriptional and translational levels have been widely explored, the role of PTMs of the ribosomal proteins has undergone substantially less investigation. Studies of the biological role of ribosomal protein PTMs generally involve creating binding assays or deletion mutants for enzymes known to modify other proteins in the cell, and then observing changes in ribosomal protein PTMs by gel electrophoresis and/or mass spectrometry.13–23 However, this approach requires prior knowledge of target enzymes and is limited to only identifying drastic PTM changes. One exception to this enzyme-focused approach is a study of phosphorylation in quiescent rat fibroblasts using a qualitative 2D-PAGE gel assay.12 To our knowledge, there has yet to be a quantitative analysis of how ribosomal protein PTMs change under different environmental conditions. Here, we employ mass spectrometry to quantify the dynamic effect of growth phase on ribosomal protein methylation and acetylation.
Mass spectrometry is one of the foremost tools of the field of proteomics, which endeavors to identify all of the protein variants, or proteoforms, found in an organism.24 Mass spectrometry is often coupled with on-line fractionation and pre-fractionation methods in order to lessen co-elution of analytes into the mass spectrometer, improving the number of peptides and proteins identified. Mass spectrometry is especially useful for elucidating the post-translational modifications of a protein, such as methylation, acetylation, and phosphorylation, which serve to alter or regulate the behavior of a protein. Mass spectrometry-based proteomics has been used previously to analyze ribosomal protein PTMs, although mostly by using the ribosome as a convenient model system for developing proteomics approaches that identify as many proteins and PTMs as possible.25–33 These proteomics papers have not focused on understanding the role of post-translational modifications on ribosomal proteins. Conversely, biological studies have delved into the role of specific enzymes or PTMs, but have not quantitatively monitored dynamic changes in protein modification. In this study, we combine these two approaches to quantify differences in PTM prevalence on yeast ribosomal proteins in the log phase of cell culture versus those in the stationary phase.
Materials and Methods
Yeast Growth
The YIT613 FLAG-tagged ribosome yeast strain used was a generous gift from Professor Toshifumi Inada at Nagoya University, Nagoya, Japan. The strain is engineered to express a FLAG tag (amino acid sequence DYKDDDDK) on ribosomal protein L25. YIT613 strain yeast was grown in YEPD media at 30°C, shaken at 200 RPM. Cultures (250 mL) were grown in the presence of 0.0045 g tetracycline and 0.0045 g chloramphenicol, and then transferred into 2 L cultures. Log phase yeast was harvested at OD600 = 0.8, while stationary phase yeast was harvested after incubating for 48 hours. Yeast pellets were washed with lysis buffer (20 mM Tris-HCl, 2 mM magnesium acetate, 100 mM potassium acetate, 100 μg/mL cycloheximide), flash frozen with liquid nitrogen, and stored at −80°C. For this experiment, 3 biological replicates were grown to log phase, and 3 biological replicates were grown to stationary phase.
Ribosome Preparation
Purification of ribosomes was adapted from published reports.34, 35 One gram of wet yeast cells were resuspended in lysis buffer containing 10 μL/mL HALT protease inhibitor (Thermo Scientific) and 425–600 μm acid-washed glass beads (Sigma) and lysed with a Biospec Products Mini-Beadbeater-1. Cell debris was pelleted at 13,000 g for 5 minutes and the supernatant lysate was loaded onto a 2.5 mL column of ANTI-FLAG M2 Affinity Gel (Sigma). Bound ribosomes were washed with running buffer (50 mM Tris-HCl, 12 mM magnesium acetate, 100 mM ammonium chloride, 0.02% sodium azide, pH 7.4) and eluted with 100 μg/mL FLAG peptide. Ribosomes were partially concentrated by vacuum evaporation and pelleted by ultracentrifugation using a Type 70 Ti rotor at 50,000 rpm (max 257,000 g) for 3 hours. Concentrated ribosome samples were stored at −80°C. This procedure was found to yield ~100 μg of ribosomal protein by BCA assay.
Peptide Preparation
Ribosomal proteins were isolated using the acetic acid method.36 Briefly, ribosomes were treated with 66% acetic acid and 0.1 M magnesium acetate for 2 hours and then centrifuged to pellet ribosomal RNA, after which ribosomal protein was recovered from the supernatant by precipitation with acetone. Figure 1 shows a representative SDS-PAGE analysis of the ribosomal protein preparation. Ribosomal proteins were dissolved in 8M urea, reduced with dithiothreitol, and alkylated with iodoacetamide, followed by a 7-fold dilution with 25 mM ammonium bicarbonate and digestion with trypsin overnight. Tryptic digests were desalted using Waters Sep-pak 1cc C18 cartridges, dried under vacuum, and dissolved in 0.1% formic acid.
Figure 1.
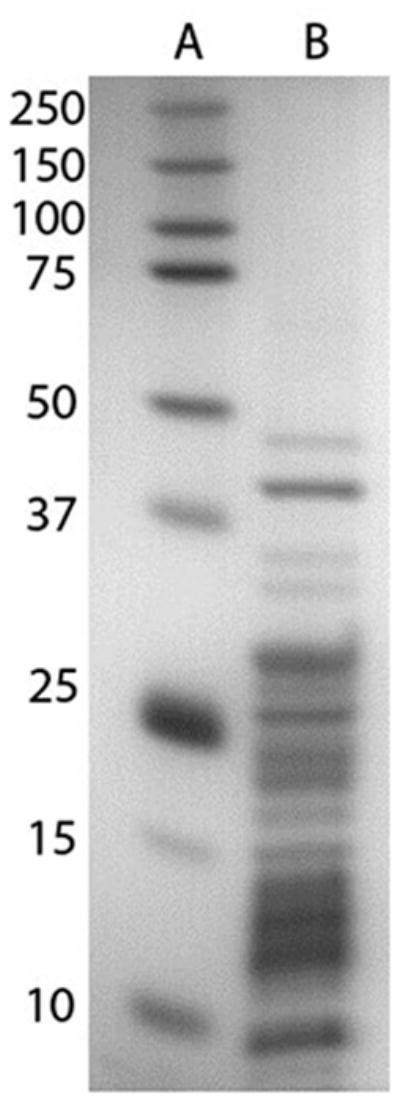
SDS-PAGE gel of Saccharomyces cerevisiae ribosomal proteins. Lane A contains molecular weight markers. Lane B contains ribosomal proteins after acetic acid wash, acetone precipitation, and resuspension in SDS-PAGE buffer. Multiple proteins are visible, ranging from 5 kDa to 45 kDa.
LC-MS
All LC-MS experiments employed a capillary HPLC-ESI-MS/MS system consisting of a Waters nanoAcquity HPLC (Milford, MA) connected to an electrospray ionization (ESI) ion-trap/orbitrap mass spectrometer (LTQ Orbitrap Velos, Thermo Scientific, San Jose, CA). Analytical columns (15 cm) were prepared by packing 3 μm-diameter 100Å-pore C18 beads (Michrom Bioresources Inc., Auburn, CA) in 100 μm ID × 365 μm OD fused silica capillaries, with the capillary tip pulled to ~1 μm with a P-2000 laser puller (Sutter Instruments, Novato, CA). For each sample, 0.15 μg of ribosomal peptides were loaded onto the analytical column at 0.5 μL/min for 30 min in an aqueous mobile phase containing 0.1% formic acid. Peptides were then eluted at 0.3 μL/min with a gradient of increasing acetonitrile (2%–30% in the first 120 min, increasing to 70% in the next 6 min). A full-mass scan (300–1500 m/z) was performed in the orbitrap at a resolution of 60,000. The ten most intense peaks with z > 1 from the full scan were selected for fragmentation by either higher-energy collisional dissociation (HCD) or electron transfer dissociation (ETD) fragmentation. HCD employed 35% collision energy, while ETD employed 3.5 × 105 fluoranthene reagent ions and 5 × 104 precursor ions with a reaction time of 50 ms. The isolation width for the precursor ions was 2.5 m/z, and the product ions from fragmentation were analyzed in the orbitrap detector at a resolution of 7500. Dynamic exclusion was enabled with a repeat count of three over 30 s and an exclusion window of 120 s. Each biological replicate was run in triplicate for each fragmentation type (HCD & ETD), producing a total of 18 log phase ribosome runs, and 18 stationary phase ribosome runs.
Data Analysis
MS runs were analyzed individually with Thermo Proteome Discoverer 1.3. Peptides were searched against a S. cerevisiae database from Uniprot (downloaded 5/5/2011) with the following modifications: variable methionine oxidation (+15.995 Da), static carbamidomethylation of cysteines (+57.021 Da), variable lysine, arginine, and n-terminal methylation (+14.016) and dimethylation (+28.031), and variable lysine and n-terminal acetylation (+42.011). Mono-isotopic masses were used for both precursor and fragment ions with tolerances of 15 ppm and 0.02 Da, respectively. The searches allowed for up to two missed trypsin cleavages. The results were filtered using a 1% peptide false discovery rate. Methylated and acetylated ribosomal peptides were quantified in Xcalibur Qualbrowser using peak areas of the unfragmented peptide from extracted ion chromatograms (XIC) (+/−0.02 Da of the theoretical mass).
Peak areas were normalized using the total peak area of the entire total ion chromatogram (TIC). Peak area data were grouped by modified peptide and tested for significance with the student’s t-test and the Benjamini-Hochberg correction.37
Results
The experimental workflow was designed to produce highly pure samples with minimal steps (Figure 2). Purity of ribosomal proteins prior to tryptic digestion was demonstrated by the SDS-PAGE gel shown in Figure 1, which matches similar separations of pure ribosomal protein samples as seen in the literature.36 This coincides with the proteomics results, which demonstrated sample purity on the peptide level. A representative LC-MS experiment on log phase ribosomes detected 96 ribosomal proteins, with 93% of the 7991 peptide spectral matches (PSMs) corresponding to yeast ribosomal peptides; for stationary phase ribosomes, we detected 98 ribosomal proteins with 90% of the 8615 PSMs corresponding to ribosomal peptides (Supplementary Material). PSMs refer to every MS/MS spectrum that can be matched to a peptide, making PSMs the best gauge of sample purity in a quantitative LC-MS study. This high quality for both the log phase and stationary phase ribosomes is invaluable for the present quantitative comparison study.
Figure 2.

Experiment workflow. Three replicates each of YIT613 strain yeast were grown to log phase and stationary phase. Yeast were lysed by bead beating and loaded onto an ANTI-FLAG column. FLAG-tagged ribosomes were retained on the column and eluted with FLAG peptide. Ribosomal RNA was precipitated in 66% acetic acid and 0.1M magnesium acetate and dissolved ribosomal proteins were collected by acetone precipitation. Ribosomal proteins were resuspended in 8M urea and digested with the proteolytic enzyme trypsin, and the resulting tryptic peptides were analyzed by LC-MS.
Sample purity was also important for optimizing the number of PTMs observed. On average, LC-MS analysis of the FLAG-tag affinity purified ribosomes detected 20 methylated or acetylated peptides per experiment, with 27 modified peptides detected across several replicate experiments (Table 1). While ribosomes are regularly seen in proteomic analyses of full yeast digests, a search for methylated and acetylated ribosomal peptides in a full yeast digest experiment yielded only 8 modified peptides on average (data not shown). Pre-fractionating yeast digest peptides into eight fractions using off-line high-pH reverse phase chromatography was found to improve the number of methylated or acetylated peptides identified to 24, at the cost of eight times the instrument analysis time. Thus, utilizing FLAG-tag affinity purification allowed us to improve the quality of our data while shortening required instrument time, allowing for analysis of multiple biological and technical replicates.
Table 1.
Methylated and acetylated ribosomal peptides detected by mass spectrometry. Peptides were fragmented with HCD and/or ETD fragmentation, and then searched against a Uniprot S. cerevisiae database. Twenty-seven modified peptides were identified and quantified XIC peak area across all biological and technical replicates. The average peak areas were used to find the stationary phase/log phase ratio, and tested for statistical significance with the student’s t-test. Resulting p-values were adjusted with the Benjamini-Hochberg correction, and two peptides, rGGFGGR and kVSGFKDEVLETV were found to exhibit statistically significant changes in modified peptide quantity.
| Protein | Peptide Sequence | Modifications | Fragment | Ratio | P Value | Adj. P Value |
|---|---|---|---|---|---|---|
| S2 | rGGFGGR | R1(Dimethyl) | HCD/ETD | 10.54621 | 0.0003 | 0.00954 |
| S1-B | kVSGFKDEVLETV | K1(Methyl) | HCD/ETD | 0.532633 | 0.0005 | 0.00726 |
| L7-A | TAEQVAAEr | R9(Methyl) | HCD | 0.752925 | 0.0069 | 0.06706 |
| S1-B | VSGFkDEVLETV | K5(Methyl) | HCD | 0.738775 | 0.0507 | 0.36743 |
| L8-A | KMGVPYAIVk | K10(Methyl) | HCD/ETD | 0.782643 | 0.0569 | 0.32985 |
| L12 | IQNrQAAASVVPSASSLVITALK | R4(Methyl) | HCD/ETD | 1.407789 | 0.0805 | 0.38913 |
| S21-A | AIPGEYVTYALSGYVr | R16(Methyl) | HCD/ETD | 0.71243 | 0.0982 | 0.40684 |
| L9-A | YIQTEQQIEVPEGVTVSIk | K19(Methyl) | HCD | 1.783777 | 0.1166 | 0.42264 |
| L33-A | IEGVATPQDAQFYLGk | K16(Methyl) | HCD/ETD | 0.825141 | 0.1212 | 0.39053 |
| S29-B | VCSSHTGlVrK | C2(Carbamidomethyl); R10(Methyl) | HCD | 0.537515 | 0.1282 | 0.37184 |
| S21-A+B | MENDkGQLVELYVPR | K5(Acetyl) | HCD/ETD | 0.785795 | 0.153 | 0.40337 |
| L42 | KQSGFGGQTkPVFHK | K10(Methyl) | HCD/ETD | 0.698218 | 0.1846 | 0.44621 |
| L33-A | IEGVATPQDAQFYLGkR | K16(Methyl) | HCD | 1.306633 | 0.2736 | 0.61041 |
| L42 | KQSGFGGQTkPVFHKK | K10(Methyl) | ETD | 0.645514 | 0.2845 | 0.54997 |
| L23 | DGVFLYFEDNAGVIANPkGEMkGSAITGPVGK | K18(Dimethyl); K22(Dimethyl) | HCD | 2.597351 | 0.3172 | 0.57495 |
| L42 | QSGFGGQTkPVFHKK | K9(Methyl) | HCD/ETD | 0.660266 | 0.3593 | 0.61289 |
| L31-A | LHMGTDDVr | R9(Methyl) | HCD | 0.901475 | 0.382 | 0.61539 |
| S8 | KNVkEEETVAK | K4(Acetyl) | ETD | 0.64605 | 0.3997 | 0.61007 |
| L42 | QSGFGGQTkPVFHK | K9(Methyl) | HCD/ETD | 0.696164 | 0.4622 | 0.67022 |
| L24-B | MkVEVDSFSGAK | K2(Methyl) | HCD/ETD | 0.890899 | 0.5313 | 0.73371 |
| L16-A | LkVFEGIPPPYDK | K2(Methyl) | ETD | 0.905814 | 0.5475 | 0.72173 |
| L16-A | LkVFEGIPPPYDKK | K2(Methyl) | HCD/ETD | 0.909256 | 0.6444 | 0.77869 |
| L42 | ASLFAQGkR | K8(Methyl) | HCD/ETD | 0.862425 | 0.802 | 0.93037 |
| S21-A+B | MENDkGQLVELYVPRK | M1(Oxidation); K5(Acetyl) | HCD | 0.904987 | 0.8059 | 0.89888 |
| S29-B | VCSSHTGlVr | C2(Carbamidomethyl); R10(Methyl) | HCD | 0.928612 | 0.8303 | 0.89179 |
| S28-A | MDNkTPVTLAK | K4(Acetyl) | HCD | 1.013954 | 0.9499 | 0.98383 |
| S21-A+B | MENDkGQLVELYVPRK | K5(Acetyl) | HCD | 0.997643 | 0.9786 | 0.97858 |
To further expand the range of peptides detected, we analyzed each sample using both higher-energy collisional dissociation (HCD) and electron transfer dissociation (ETD) fragmentation, which have been found to produce complementary data.38 As shown in Table 1, eleven modified peptides were only detected using HCD, while three peptides were detected only using ETD. Thirteen peptides were detected using both methods. Thus, while ETD fragmentation allowed us to detect a few additional modified peptides, it may not be worth the additional instrument time in future experiments.
Quantitative data showing the relative amounts of modified ribosomal peptides in log phase versus stationary phase yeast is also shown in Table 1. Modified peptides were quantified by constructing the XIC of the most intense isotopic peak in the full scan and then measuring the peak area (Figure 3). While spectral counting is commonly used for label-free quantification of proteins, our goal was to quantify PTM prevalence on the peptide level. Furthermore, label-free quantification by peak area has been shown to be a useful tool for analyzing complex samples.39 Peak areas were normalized using the TIC peak area to correct for differences in injected sample amount. When comparing the log phase to the stationary phase, some PTMs were seen to increase in abundance, while others decreased. Most changes were minor, ranging between a 1.5-fold decrease and a 1.5-fold increase, but occasionally larger changes were observed, including a 10-fold increase for the dimethylated rGGFGGR peptide in the stationary phase.
Figure 3.

A representative LC-MS experiment showing data for dimethylated peptide rGGFGGR. In Fig. 3A, the TIC represents all of the yeast ribosomal peptides detected by the mass spectrometer. Several full scan mass spectra are taken per second, each of which may contain peaks from multiple peptides. Fig. 3B shows the isotopic distribution of the +2 charge state for rGGFGGR as seen in a full scan mass spectrum. In Fig. 3C, the XIC corresponds to the signal intensity over time of the 367.71 m/z ion, the most intense isotopic peak for rGGFGGR. The peak area is integrated to yield a quantitative measure of the amount of peptide in the sample. Finally in Fig. 3D, the 367.71 m/z ion was isolated for HCD fragmentation and the MS/MS spectrum is shown. The peptide is identified by comparison to a Uniprot S. cerevisiae database, and characteristic fragmentation peaks are checked for mass shifts to identify PTMs. A mass shift of +42.01 indicates the presence of an acetyl group, while a +14.02 or +28.03 mass shift corresponds to a methyl or dimethyl group, respectively. Trimethylations (+42.04) were not detected.
Statistically significant changes in modified peptide quantity between log phase and stationary phase samples were found using the student’s t-test40 with the Benjamini-Hochberg correction.37 Two peptides met the p-value cutoff of 0.05: a 1.9-fold decrease in methylation for k(Me)VSGFKDEVLETV of RPS1B, and a 10-fold increase in dimethylation for r(DiMe)GGFGGR of RPS2. We will henceforth refer to these modifications as RPS1B K243 methylation and RPS2 R10 dimethylation, which indicate the protein and the amino acid position of the PTM. While we did not find published reports in the literature on the biological role of RPS1B K243 methylation, RPS2 R10 dimethylation is a known modification, and we discuss below its biological roles.
Discussion
It is interesting to find two cases where methylation depends on growth phase, as methylation has been a recent focus of studies of ribosome regulation.13, 15, 16, 18–23 RPS2 R10 dimethylation is of particular note, in that it is 10-fold more abundant in the stationary phase. Lipson et al. informally observed that RPS2 methylation varied depending on growth conditions, although they did not publish this data.22
Methyltransferase studies have identified Rmt3 in S. pombe, Rmt1 in S. cerevisiae, and Prmt3 in mouse as catalysts for RPS2 R10 methylation, but gene deletion studies of these three enzymes have found differing phenotypic results. S. pombe Rmt3 was found to exhibit a methyltransferase-independent function that plays a role in regulating the production of the 40S subunit.20, 41–43 In mammalian cells, mice lacking Prmt3 exhibit smaller embryos during gestation, but ribosome stability and function are not affected.21, 44 Lastly, deletion of Rmt1 in S. cerevisiae causes no phenotypic difference in ribosome stability.
However, Rmt1 is known to bind and/or methylate a number of proteins that process and export mRNA; methylation of Rmt1 target proteins Npl3, Hrp1, and Nab2 promotes their dissociation from nuclear factors and facilitates their export from the nucleus.46–48 This is consistent with RPS2’s known functions of processing and exporting 20S rRNA, a precursor for 40s ribosomal subunits.42, 45 Our observation that RPS2 R10 dimethylation increases tenfold in stationary phase suggests that the cell may be facilitating export of RPS2 from the nucleus. This would result in slowing processing and export of 20S rRNA, which in turn restricts 40S ribosomal subunit production, a known response to the depletion of available nutrients. Therefore, methylation of RPS2 may play a role in regulating export of rRNA to the cytosol.22, 23
Our observation of RPS2 R10 dimethylation increasing 10-fold in stationary phase yeast provides new evidence for the role of RPS2 methylation in 20S rRNA processing and export. To our knowledge, the observation of RPS1B K243 methylation decreasing in stationary phase yeast has not been reported, suggesting a novel role for RPS1B methylation that invites further investigation. This work exhibits the utility of mass spectrometry for quantifying dynamic changes in PTMs without prior knowledge of target enzymes and validates mass spectrometry as a valuable tool for investigating the biological role of PTMs.
Supplementary Material
Highlights.
Ribosomal protein modifications were quantified for yeast in different phases of growth.
Of 27 confidently identified methylated and acetylated peptides, two change significantly.
Methylated ribosomal protein (RP) S1B decreases 1.9-fold, inviting further investigation.
Dimethylated RP S2 increases 10-fold, suggesting a role in regulating ribosome synthesis.
Mass spectrometry is useful for investigating the role of post-translational modifications.
Acknowledgments
This work was supported by the National Institutes of Health: NIGMS Program Project P01GM081629 and NHGRI Center of Excellence in Genomic Science 1P50HG004952.
We thank Toshifumi Inada for his generous donation of FLAG-tagged yeast strain YIT613.
Abbreviations
- PTM
post-translational modification
- RPS1B
ribosomal protein S1B
- RPS2
ribosomal protein S2
- BCA
bicinchoninic acid
- HPLC
high-performance liquid chromatography
- ESI
electrospray ionization
- m/z
mass-to-charge ratio
- HCD
higher-energy collisional dissociation
- ETD
electron transfer dissociation
- PSM
peptide spectral match
- XIC
extracted ion chromatogram
- TIC
total ion chromatogram
Footnotes
Supplementary Material. Protein lists for representative log phase and stationary phase ribosome data are shown in the supplementary material.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces Responds to Nutrients. Annu Rev Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
- 2.Warner JR, Vilardell J, Sohn JH. Economics of Ribosome Biosynthesis. Cold Spring Harb Symp Quant Biol. 2001;66:567–574. doi: 10.1101/sqb.2001.66.567. [DOI] [PubMed] [Google Scholar]
- 3.Zwietering MH, Jongenburger I, Rombouts FM, Vantriet K. Modeling of the Bacterial-Growth Curve. Appl Environ Microbiol. 1990;56:1875–1881. doi: 10.1128/aem.56.6.1875-1881.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. “Sleeping beauty”: Quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2004;68:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis PP, Nomura M. Stringent Control of Ribosomal-Protein Gene-Expression in Escherichia-Coli. Proc Natl Acad Sci U S A. 1974;71:3819–3823. doi: 10.1073/pnas.71.10.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic Expression Programs in the Response of Yeast Cells to Environmental Changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen DR, Toone WM, Mata J, Lyne R, Burns G, Kivinen K, Brazma A, Jones N, Bahler J. Global Transcriptional Responses of Fission Yeast to Environmental Stress. Mol Biol Cell. 2003;14:214–229. doi: 10.1091/mbc.E02-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of Yeast Genome Expression in Response to Environmental Changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geyer PK, Meyuhas O, Perry RP, Johnson LF. Regulation of Ribosomal-Protein Messenger-RNA Content and Translation in Growth-Stimulated Mouse Fibroblasts. Mol Cell Biol. 1982;2:685–693. doi: 10.1128/mcb.2.6.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH. Dissection of the Mechanism for the Stringent Factor ReIA. Mol Cell. 2002;10:779–788. doi: 10.1016/s1097-2765(02)00656-1. [DOI] [PubMed] [Google Scholar]
- 11.Ueta M, Yoshida H, Wada C, Baba T, Mori H, Wada A. Ribosome Binding Proteins YhbH and YfiA have Opposite Functions During 100S Formation in the Stationary Phase of Escherichia coli. Genes Cells. 2005;10:1103–1112. doi: 10.1111/j.1365-2443.2005.00903.x. [DOI] [PubMed] [Google Scholar]
- 12.Thomas G, Siegmann M, Gordon J. Multiple Phosphorylation of Ribosomal Protein-S6 during Transition of Quiescent 3t3 Cells into Early G1, and Cellular Compartmentalization of the Phosphate Donor. Proc Natl Acad Sci U S A. 1979;76:3952–3956. doi: 10.1073/pnas.76.8.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cameron DM, Gregory ST, Thompson J, Suh MJ, Limbach PA, Dahlberg AE. Thermus thermophilus L11 Methyltransferase, PrmA, is Dispensable for Growth and Preferentially Modifies Free Ribosomal Protein L11 Prior to Ribosome Assembly. J Bacteriol. 2004;186:5819–5825. doi: 10.1128/JB.186.17.5819-5825.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL. Regulated Release of L13a from the 60S Ribosomal Subunit as a Mechanism of Transcript-Specific Translational Control. Cell. 2003;115:187–198. doi: 10.1016/s0092-8674(03)00773-6. [DOI] [PubMed] [Google Scholar]
- 15.Porras-Yakushi TR, Whitelegge JP, Clarke S. Yeast Ribosomal/Cytochrome C SET Domain Methyltransferase Subfamily - Identification of Rpl23ab Methylation Sites and Recognition Motifs. J Biol Chem. 2007;282:12368–12376. doi: 10.1074/jbc.M611896200. [DOI] [PubMed] [Google Scholar]
- 16.Sadaie M, Shinmyozu K, Nakayama JI. A Conserved SET Domain Methyltransferase, Set11, Modifies Ribosomal Protein Rpl12 in Fission Yeast. J Biol Chem. 2008;283:7185–7195. doi: 10.1074/jbc.M709429200. [DOI] [PubMed] [Google Scholar]
- 17.Kamita M, Kimura Y, Ino Y, Kamp RM, Polevoda B, Sherman F, Hirano H. N-alpha-Acetylation of Yeast Ribosomal Proteins and its Effect on Protein Synthesis. J Proteomics. 2011;74:431–441. doi: 10.1016/j.jprot.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Shin HS, Jang CY, Kim HD, Kim TS, Kim S, Kim J. Arginine Methylation of Ribosomal Protein S3 affects Ribosome Assembly. Biochem Biophys Res Commun. 2009;385:273–278. doi: 10.1016/j.bbrc.2009.05.055. [DOI] [PubMed] [Google Scholar]
- 19.Ren JQ, Wang YQ, Liang YH, Zhang YQ, Bao SL, Xu ZH. Methylation of Ribosomal Protein S10 by Protein-arginine Methyltransferase 5 Regulates Ribosome Biogenesis. J Biol Chem. 2010;285:12695–12705. doi: 10.1074/jbc.M110.103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachand F, Silver PA. PRMT3 is a Ribosomal Protein Methyltransferase that affects the Cellular Levels of Ribosomal Subunits. EMBO J. 2004;23:2641–2650. doi: 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swiercz R, Cheng DH, Kim D, Bedford MT. Ribosomal Protein RPS2 is Hypomethylated in PRMT3-Deficient Mice. J Biol Chem. 2007;282:16917–16923. doi: 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- 22.Lipson RS, Webb KJ, Clarke SG. Rmt1 Catalyzes Zinc-Finger Independent Arginine Methylation of Ribosomal Protein Rps2 in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2010;391:1658–1662. doi: 10.1016/j.bbrc.2009.12.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young BD, Weiss DI, Zurita-Lopez CI, Webb KJ, Clarke SG, McBride AE. Identification of Methylated Proteins in the Yeast Small Ribosomal Subunit: A Role for SPOUT Methyltransferases in Protein Arginine Methylation. Biochem. 2012;51:5091–5104. doi: 10.1021/bi300186g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith LM, Kelleher NL. Proteoform: a Single Term Describing Protein Complexity. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyder MS, Willison KR, Klug DR, deMello AJ, Ces O. Affinity Chromatography and Capillary Electrophoresis for Analysis of the Yeast Ribosomal Proteins. Biochem Mol Biol Rep. 2012;45:233–238. doi: 10.5483/bmbrep.2012.45.4.233. [DOI] [PubMed] [Google Scholar]
- 26.Lee SW, Berger SJ, Martinovic S, Pasa-Tolic L, Anderson GA, Shen YF, Zhao R, Smith RD. Direct Mass Spectrometric Analysis of Intact Proteins of the Yeast Large Ribosomal Subunit Using Capillary LC/FTICR. Proc Natl Acad Sci USA. 2002;99:5942–5947. doi: 10.1073/pnas.082119899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold RJ, Reilly JP. Observation of Escherichia coli Ribosomal Proteins and their Posttranslational Modifications by Mass Spectrometry. Anal Biochem. 1999;269:105–112. doi: 10.1006/abio.1998.3077. [DOI] [PubMed] [Google Scholar]
- 28.Running WE, Ravipaty S, Karty JA, Reilly JP. A Top-Down/Bottom-Up Study of the Ribosomal Proteins of Caulobacter crescentus. J Proteome Res. 2007;6:337–347. doi: 10.1021/pr060306q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strader MB, VerBerkmoes NC, Tabb DL, Connelly HM, Barton JW, Bruce BD, Pelletier DA, Davison BH, Hettich RL, Larimer FW, Hurst GB. Characterization of the 70S Ribosome from Rhodopseudomonas palustris Using an Integrated “Top-Down” and “Bottom-Up” Mass Spectrometric Approach. J Proteome Res. 2004;3:965–978. doi: 10.1021/pr049940z. [DOI] [PubMed] [Google Scholar]
- 30.Lauber MA, Running WE, Reilly JP. B. subtilis Ribosomal Proteins: Structural Homology and Post-Translational Modifications. J Proteome Res. 2009;8:4193–4206. doi: 10.1021/pr801114k. [DOI] [PubMed] [Google Scholar]
- 31.Carroll AJ, Heazlewood JL, Ito J, Millar AH. Analysis of the Arabidopsis Cytosolic Ribosome Proteome Provides Detailed Insights into its Components and their Post-Translational Modification. Mol Cell Proteomics. 2008;7:347–369. doi: 10.1074/mcp.M700052-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Forbes AJ, Patrie SM, Taylor GK, Kim YB, Jiang LH, Kelleher NL. Targeted Analysis and Discovery of Posttranslational Modifications in Proteins from Methanogenic Archaea by Top-Down MS. Proc Natl Acad Sci USA. 2004;101:2678–2683. doi: 10.1073/pnas.0306575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louie DF, Resing KA, Lewis TS, Ahn NG. Mass Spectrometric Analysis of 40 S Ribosomal Proteins from Rat-1 Fibroblasts. J Biol Chem. 1996;271:28189–28198. doi: 10.1074/jbc.271.45.28189. [DOI] [PubMed] [Google Scholar]
- 34.Inada T, Winstall E, Tarun SZ, Yates JR, Schieltz D, Sachs AB. One-Step Affinity Purification of the Yeast Ribosome and its Associated Proteins and mRNAs. RNA. 2002;8:948–958. doi: 10.1017/s1355838202026018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simons SP, McLellan TJ, Aeed PA, Zaniewski RP, Desbonnet CR, Wondrack LM, Marr ES, Subashi TA, Dougherty TJ, Xu ZY, Wang IK, LeMotte PK, Maguire BA. Purification of the large ribosomal subunit via its association with the small subunit. Anal Biochem. 2009;395:77–85. doi: 10.1016/j.ab.2009.07.042. [DOI] [PubMed] [Google Scholar]
- 36.Hardy SJS, Kurland CG, Voynow P, Mora G. Ribosomal Proteins of Escherichia Coli .I. Purification of 30s Ribosomal Proteins. Biochem. 1969;8:2897–2905. doi: 10.1021/bi00835a031. [DOI] [PubMed] [Google Scholar]
- 37.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B: Stat Methodol. 1995;57:289–300. [Google Scholar]
- 38.Shen YF, Tolic N, Xie F, Zhao R, Purvine SO, Schepmoes AA, Ronald JM, Anderson GA, Smith RD. Effectiveness of CID, HCD, and ETD with FT MS/MS for Degradomic-Peptidomic Analysis: Comparison of Peptide Identification Methods. J Proteome Res. 2011;10:3929–3943. doi: 10.1021/pr200052c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yishai L, Schwarz E, Wang L, Leweke FM, Bahn S. Label-Free LC-MS/MS Quantitative Proteomics for Large-Scale Biomarker Discovery in Complex Samples. J Sep Sci. 2007;30:2198–2203. doi: 10.1002/jssc.200700189. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. Detecting Differential and Correlated Protein Expression in Label-Free Shotgun Proteomics. J Proteome Res. 2006;5:2909–2918. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
- 41.Bachand F, Lackner DH, Bahler J, Silver PA. Autoregulation of Ribosome Biosynthesis by a Translational Response in Fission Yeast. Mol Cell Biol. 2006;26:1731–1742. doi: 10.1128/MCB.26.5.1731-1742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perreault A, Bellemer C, Bachand F. Nuclear Export Competence of pre-40S Subunits in Fission Yeast Requires the Ribosomal Protein Rps2. Nucleic Acids Res. 2008;36:6132–6142. doi: 10.1093/nar/gkn625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perreault A, Lemieux C, Bachand F. Regulation of the Nuclear Poly(A)-Binding Protein by Arginine Methylation in Fission Yeast. J Biol Chem. 2007;282:7552–7562. doi: 10.1074/jbc.M610512200. [DOI] [PubMed] [Google Scholar]
- 44.Choi S, Jung CR, Kim JY, Im DS. PRMT3 Inhibits Ubiquitination of Ribosomal Protein S2 and Together Forms an Active Enzyme Complex. Biochim Biophys Acta Gen Subj. 2008;1780:1062–1069. doi: 10.1016/j.bbagen.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Ferreira-Cerca S, Pöll G, Gleizes PE, Tschochner H, Milkereit P. Roles of Eukaryotic Ribosomal Proteins in Maturation and Transport of Pre-18S rRNA and Ribosome Function. Mol Cell. 2005;20:263–275. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Shen EC, Henry MF, Weiss VH, Valentini SR, Silver PA, Lee MS. Arginine Methylation Facilitates the Nuclear Export of hnRNP Proteins. Genes Dev. 1998;12:679–691. doi: 10.1101/gad.12.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green DM, Marfatia KA, Crafton EB, Zhang X, Cheng XD, Corbett AH. Nab2p is Required for Poly(A) RNA Export in Saccharomyces cerevisiae and is Regulated by Arginine Methylation via Hmt1p. J Biol Chem. 2002;277:7752–7760. doi: 10.1074/jbc.M110053200. [DOI] [PubMed] [Google Scholar]
- 48.McBride AE, Cook JT, Stemmler EA, Rutledge KL, McGrath KA, Rubens JA. Arginine Methylation of Yeast mRNA-Binding Protein Npl3 Directly Affects its Function, Nuclear Export, and Intranuclear Protein Interactions. J Biol Chem. 2005;280:30888–30898. doi: 10.1074/jbc.M505831200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.