

Abstract
Inappropriate production of the iron-regulatory hormone hepcidin contributes to the pathogenesis of common iron disorders. Absolute or relative deficiency of hepcidin causes iron overload in hereditary hemochromatosis and iron-loading anemias. Elevated hepcidin causes iron restriction in inflammatory conditions including autoimmune disease, critical illness, some cancers and chronic kidney disease. Multiple agents targeting hepcidin and its regulators are under development as novel therapeutics for iron disorders. This review summarizes hepcidin biology and discusses the current landscape of hepcidin-targeting therapeutic strategies.
Keywords: Iron disorders, anemia, iron overload, ferroportin
Hepcidin regulates systemic iron homeostasis
The peptide hormone hepcidin is primarily produced in hepatocytes, and it regulates plasma iron concentrations [1]. The molecular target of hepcidin is the cellular iron exporter ferroportin [2]. Ferroportin supplies iron into plasma from duodenal enterocytes engaged in dietary iron absorption, from macrophages of the spleen and liver that recycle old red blood cells, and from hepatocytes involved in iron storage [3] (Figure 1). Hepcidin is the ligand of ferroportin, and their interaction results in rapid ubiquitination of ferroportin, and endocytosis and degradation of the ligand-receptor complex [4]. The loss of ferroportin from cell membranes decreases the delivery of iron into plasma. If hepcidin is chronically elevated, persistent hypoferremia can lead to the development of iron-restricted anemia. Conversely, chronic hepcidin deficiency results in excessive iron absorption, increased levels of non-transferrin bound iron in circulation and the development of iron overload. Because of its critical role in iron homeostasis and the pathogenesis of iron disorders, hepcidin has emerged as a promising drug target. Here we review the biology of hepcidin and highlight various pharmacological strategies focused on antagonizing or agonizing hepcidin.
Figure 1. The role of hepcidin in iron metabolism.

Hepcidin-ferroportin interaction determines the flow of iron into plasma. Hepcidin concentration is in turn regulated by iron, erythropoietic activity, and inflammation. Republished with permission from Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010 Dec 2;116(23):4754–61. © the American Society of Hematology.
Hepcidin is synthesized as a preprohormone which is cleaved intracellularly and secreted as a mature 25 amino acid peptide [5]. The production of hepcidin appears to be regulated predominantly at the transcriptional level. The major stimuli regulating hepcidin production include its substrate, iron, and the signals reflecting erythropoietic demands for iron (Figure 1). Hepcidin is also an acute phase reactant and is increased during inflammation. A number of other hepcidin regulators have also been described including the growth hormones (HGF/EGF) [6], steroid hormones (estrogen [7], testosterone [8,9]), and metabolic pathways (starvation/gluconeogenesis [10]), but their role in hepcidin and iron homeostasis and pathobiology is less well understood.
Hepcidin regulation by iron
Hepcidin production increases in response to iron loading [11,12], and this prevents further absorption of dietary iron and the development of iron overload. Both plasma iron and liver iron stores regulate hepcidin transcription through distinct but overlapping pathways [12,13]. These converge onto the bone morphogenetic protein (BMP) pathway to increase hepcidin transcription (Figure 2). Both serum iron and liver iron accumulation activate the BMP receptor and its Smad1/5/8 pathway [13], and increase hepcidin mRNA concentration in hepatocytes. Additionally, the BMP co-receptor hemojuvelin (HJV) is required for this response [12].
Figure 2. Regulation of hepcidin transcription.
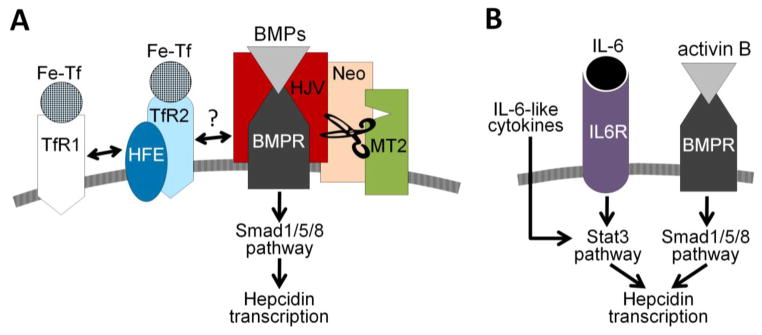
(A) Hepcidin regulation by extracellular iron. The BMP/Smad pathway is central to the transcriptional regulation of hepcidin expression. The BMP pathway signaling is further modulated by hemojuvelin (HJV), a BMP co-receptor. The sensing of holotransferrin is proposed to occur through the following mechanism: binding of holo-transferrin (Fe-Tf) to TfR1 displaces HFE from the complex with TfR1 and promotes its interaction with TfR2. TfR2 protein is itself stabilized by the binding of Fe-Tf. The HFE/TfR2 may promote BMP signaling by forming a complex with HJV, or even acting independently of HJV or each other. Additional proteins (TMPRSS6/matriptase-2 (MT2) and neogenin) modulate the cleavage of membrane HJV and thus alter hepcidin transcription. (B) Hepcidin regulation by inflammation. IL-6 and other cytokines (e.g. oncostatin M, IL-22) were shown to regulate hepcidin expression by activating the Stat3 pathway. Activin B acting via BMP receptors and the Smad1/5/8 pathway was also proposed to stimulate hepcidin expression during inflammation.
It is not yet known how intracellular iron concentrations are sensed. One of the mediators may be BMP6, whose expression increases with liver iron loading in mice [14]. For extracellular iron, transferrin receptor 1 and 2 (TfR1 and TfR2) are the likely sensors of holo-transferrin concentrations, but multiple hypotheses have been proposed as to how this signal is transmitted. One line of evidence suggests that HFE protein, an MHC-class I molecule, shuttles between the two TfR receptors depending on holo-Tf concentrations. At higher holo-Tf concentrations, HFE is displaced from TfR1 and associates with TfR2 [15]. HFE and TfR2 in turn may interact with HJV [16], thus potentiating BMP signaling. Other evidence suggests that HFE and TfR2 regulate hepcidin independently [17], but the details of this proposed mechanism are not yet known. Holo-Tf binding to TfR2 by itself stabilizes the receptor as TfR2 is redirected to a recycling pathway [18].
Though the details of the iron sensing circuitry remain to be determined, mutations in Hfe, TfR2, Hjv, Bmp6, BMP receptors Alk2 and Alk3, or Smad4 were all shown to impair hepcidin regulation by iron in mice [12,19,20]. HFE, TfR2 and HJV mutations were also described in humans and result in hepcidin levels that are inappropriately low for the degree of iron overload observed in these patients [21–23]. Hepcidin production is further modulated by transmembrane serine protease TMPRSS6, also known as matriptase-2 [24,25], and by neogenin, a multifunctional transmembrane receptor [26]. Although the mechanism is still controversial, these proteins have been proposed to act by posttranslationally regulating the levels of membrane-associated HJV [27,28]. The specific involvement of these proteins in iron sensing is also uncertain.
Hepcidin regulation by erythropoietic signals
Hepcidin is suppressed in conditions associated with increased erythropoietic activity, presumably to make more iron available for hemoglobin synthesis. Hemorrhage, hemolysis or injections of erythropoietin, a hormone that promotes red blood cell production, all result in a rapid decrease in hepcidin [29]. In anemias with ineffective erythropoiesis, hepcidin levels are chronically suppressed, and this is thought to be the cause of iron overload in nontransfused patients [30]. It is not known whether the same pathways mediate both physiological and pathological suppression of hepcidin in response to increased erythropoietic activity, but these likely involve secretion of a hepcidin suppressor from the bone marrow erythroid precursors [31].
Hepcidin regulation by inflammation
Hepcidin is rapidly increased by inflammatory and infectious stimuli via the IL-6 pathway [32], although other pathways, including the BMP pathway may also contribute [33,34] (Figure 2). Inflammatory regulation of hepcidin may have evolved as a host defense mechanism to slow the growth of microorganisms by sequestering iron from microbes. Although the role of hepcidin in infections remains to be demonstrated, hepcidin increase in inflammatory conditions is thought to contribute to the development of iron-restricted anemia [1].
Hepcidin and iron disorders
Hepcidin deficiency in iron overload disorders
Hepcidin deficiency is the pathogenic cause of iron overload in most forms of hereditary hemochromatosis. Hepcidin insufficiency results from the deleterious mutations in the genes encoding hepcidin regulators (HFE, TfR2 and HJV) or hepcidin itself [1]. In all of these cases, dietary iron is hyperabsorbed, resulting in the deposition of excess iron in the liver and other parenchyma. The degree of hepcidin deficiency correlates with the severity of iron overload: mutations in HJV or hepcidin, which are associated with absolute hepcidin deficiency, cause juvenile hemochromatosis, whereas mutations in HFE and TfR2 in which hepcidin response to iron loading is partially preserved [12,21] result in the less severe adult form of the disease. A rare form of hemochromatosis is also caused by mutations in the hepcidin receptor ferroportin which lead to the resistance of ferroportin to hepcidin-induced endocytosis [35,36]. Hereditary hemochromatosis patients are currently treated by bleeding. Each 1 ml removal of blood eliminates 1 mg of iron from the body. As new red blood cells are made, excess iron from other organs is mobilized and used for erythropoiesis. Although this treatment is effective, repeated phlebotomies may be a significant inconvenience for many patients, and may be difficult for those with poor venous access or coexisting medical conditions. Furthermore, once iron-depleted, hereditary hemochromatosis patients lower their hepcidin dramatically [21], further accelerating iron absorption, and the need for more phlebotomy.
As mentioned before, hepcidin deficiency also contributes to iron loading in non-transfused patients suffering from anemias with ineffective erythropoiesis, including β-thalassemia and congenital dyserythropoietic anemias. In these patients, despite iron loading, hepcidin remains insufficient because of the suppressive effect of the excess erythroid activity [30]. In transfused patients, hepcidin levels decrease toward the end of the transfusion cycle [37] suggesting that even in those patients relative hepcidin deficiency could contribute to iron loading. Currently, iron overload in thalassemia is treated by iron chelation, which can have significant side effects [38].
Hepcidin excess in iron-restrictive disorders
Increased hepcidin and associated hypoferremia are thought to contribute to the development of anemia in diverse human disorders [1]. The causes of hepcidin increase include high levels of inflammatory cytokines, decreased clearance of hepcidin, or mutations in the negative regulators of hepcidin. IL-6 and other cytokines cause high hepcidin levels in autoimmune disorders, infections and some cancers. In chronic kidney disease (CKD), apart from the common presence of inflammation, decreased clearance of hepcidin in the kidneys may also contribute to the development of anemia in CKD [39]. Finally, mutations in protease TMPRSS6, a hepcidin suppressor, lead to the development of iron-refractory iron deficiency anemia (IRIDA) [25].
How much hepcidin excess contributes to anemia of inflammation remains to be determined. Although hepcidin knockout mice develop milder anemia in mouse models of inflammation [40], hemoglobin decrease is not entirely prevented suggesting that other mechanisms also contribute. These may include direct effects of cytokines such as interferon-γ and IL-6 on hematopoietic precursor differentiation and erythrocyte maturation [41,42].
Manipulation of the hepcidin pathway for therapeutic purposes
As hepcidin deficiency or excess play important roles in pathogenesis of various iron disorders, hepcidin agonists and antagonists may be potentially useful in clinical practice. Genetic studies in animal models have provided the initial proof of principle that hepcidin could be an effective therapeutic target. For example, overexpression of hepcidin in Hfe-/- mice, a model of the most common form of human hereditary hemochromatosis, prevented liver iron overload normally seen in these mutants [43]. Similarly, in β-thalassemia intermedia mouse models, moderate transgenic hepcidin expression decreased iron loading of the liver, diminished splenomegaly and improved anemia [44]. In case of anemia of inflammation, hepcidin knockout mice demonstrated milder anemia with faster recovery than wild-type mice [40].
Hepcidin agonists
Compounds that can mimic hepcidin’s function or potentiate its endogenous synthesis may be able to prevent systemic accumulation of iron. The development of both types of therapeutics is in progress (Table 1). Such compounds may provide additional treatment options for patients who do not respond well to standard treatment regimens, like hereditary hemochromatosis patients with limited tolerance to phlebotomy or β-thalassemia patients undergoing iron chelation therapy and suffering from its side effects.
Table 1.
Principles of the hepcidin-targeting therapeutic approaches
| Therapeutic approach | Targeted disease | Mode of action | Agents |
|---|---|---|---|
| Hepcidin agonists | Iron overload (hereditary hemochromatosis and iron-loading anemias) | Hepcidin mimics | Minihepcidins [47] |
| Stimulators of hepcidin production | Gene silencing of TMPRSS6 [50,51] | ||
| BMP pathway agonists [52] | |||
| Hepcidin antagonists | Iron-restricted anemias (anemia of inflammation, anemia of chronic kidney disease, anemia of cancer, IRIDA) | Suppressors of hepcidin production | BMP pathway inhibitors [54,56,76] |
| Anti-inflammatory agents [60–62] | |||
| Erythropoiesis-stimulating agents [65] | |||
| Gene silencing of hepcidin and its regulators [66,67] | |||
| Hepcidin peptide neutralizing binders | Anti-hepcidin antibodies [68,69] | ||
| Anticalins [70] | |||
| Spiegelmers [71] | |||
| Agents interfering with hepcidin-ferroportin interaction | Anti-ferroportin antibodies [73] | ||
| Thiol modifiers [74] |
Hepcidin mimics
The hepcidin peptide itself does not have desirable pharmacological properties. The hormone has a short half-life in circulation (several minutes) [45]. Moreover, the synthesis of bioactive peptide is difficult and expensive, mostly due to complicated disulfide bond connectivity (4 -S-S- bridges) that diminishes the yield of the correctly folded product. In addition, recently published studies aimed at the development of more stable cyclic full-length hepcidin analogs proved unsuccessful, as cyclic analogs showed no biological activity despite increased stability in the human serum [46]. Hepcidin mimetics may thus be more viable therapeutic candidates.
Minihepcidins are peptide-based hepcidin agonists which were rationally designed based on the region of hepcidin that interacts with ferroportin [47]. Mutagenesis and truncation studies established that 9 amino acids long N-terminal fragment of hepcidin (DTHFPICIF) is crucial for hormone’s activity [47]. This particular fragment was further engineered: unnatural amino acids (N-substituted and β–homo-amino acids) were introduced to increase resistance to proteolysis, and fatty acids were conjugated to prolong the half-life in circulation, yielding analogs that were at least as potent as the full-length hepcidin and had a longer duration of action [48]. One of such analogs, a minihepcidin PR65, was tested in hepcidin knockout mice, a model of severe hemochromatosis. A two-week treatment prevented the development of iron overload when given to non-overloaded hepcidin knockout mice [48]. The treatment of mice with pre-existing iron overload was less effective but still caused a partial redistribution of iron from the liver to the spleen within two weeks [48]. At high doses, PR65 caused profound iron restriction and anemia indicating that minihepcidin therapy will likely require titration to effect to avoid excessive hypoferremia and iron restriction. Collectively, these data strongly suggest that minihepcidins may be useful for the prevention of iron overload, or as an auxiliary to phlebotomy or chelation for the treatment of existing iron overload. The development of the next generation of minihepcidins is underway (Ruchala unpublished).
Stimulators of hepcidin production
One approach to increase hepcidin production is to antagonize TMPRSS6, a negative regulator of hepcidin. Homozygous inactivation of Tmprss6 in thalassemic th3/+ mice increased hepcidin levels, ameliorated iron overload, and improved ineffective erythropoiesis [49]. Subsequently, targeting of Tmprss6 with RNA-based therapeutics proved to be a promising approach. Both antisense oligonucleotides (ASOs) and small interfering RNAs (siRNA) against Tmprss6 were successfully used in mouse models of iron overload [50,51]: Hfe-/- mice treated for 6 weeks increased their hepcidin mRNA and reduced their serum and liver iron concentrations. A small reduction in hemoglobin was also noted indicating mild iron restriction. Treatment of thalassemic th3/+ mice with ASOs and siRNA against Tmprss6 reproduced the effect of transgenic hepcidin overexpression in th3+/- mouse model [44] and improved anemia while decreasing iron overload [50,51].
Other approaches for increasing hepcidin production may include administration of BMP6 and its agonists [52]. However, clinical application of these agents in iron disorders will require an extensive further research considering that BMP pathway plays a role in bone morphogenesis, cell growth, differentiation, apoptosis, angiogenesis and cancer.
Hepcidin antagonists
Elevated hepcidin concentrations are associated with various pathologies: anemia of inflammation, chronic kidney disease, some cancers and iron-refractory iron deficiency anemia. These conditions are usually treated with erythropoiesis-stimulating agents (ESAs) with or without high dose intravenous iron. However, the effectiveness of these therapies is thought to be impaired by high hepcidin. Hepcidin-mediated iron restriction likely contributes to erythropoietin resistance, and high hepcidin may decrease mobilization of iron from macrophages which process IV iron preparations. Considering reported side effects of ESAs when used at high doses [53], introduction of hepcidin antagonists may offer a therapeutic advantage.
Antagonism of hepcidin activity can be achieved by decreasing hepcidin production, neutralizing hepcidin peptide, blocking hepcidin’s binding to ferroportin, preventing ferroportin endocytosis or promoting ferroportin synthesis (Table 1).
Suppressing hepcidin production by targeting the pathways regulating hepcidin transcription
BMP pathway. As the BMP-SMAD pathway is crucial for regulation of hepcidin transcription (Figure 2), it is not surprising that agents interfering with the BMP pathway are effective at decreasing hepcidin production. However, questions remain about what other processes may be affected in vivo by these agents, given the pleiotropic role of the BMP pathway in various biological responses.
Sequestration of BMPs by heparin was recently reported to decrease hepcidin expression in hepatic cell lines and in mice [54]. Patients undergoing low molecular weight heparin therapy to prevent deep vein thrombosis also decreased serum hepcidin concentration by ~80% within 2–5 days after the start of the treatment. This was associated with increased serum iron and transferrin saturation. Because heparin is a known anticoagulant, novel non-anticoagulant heparins were also developed and tested [55] showing potent activity in vitro and in vivo in mice, even in the presence of an inflammatory stimulus. Notably, heparin itself is an anti-inflammatory agent [54] which may be a contributory factor to its anti-hepcidin activity.
HJV, a BMP co-receptor essential for hepcidin expression, is another molecular target that can be exploited to interfere with hepcidin production. Membrane-linked HJV and its soluble form (sHJV) have opposing effects on hepcidin expression with sHJV decreasing Smad signaling and hepcidin levels [56]. Soluble HJV-Fc fusion protein (sHJV.Fc) was shown to ameliorate anemia of inflammation (AI) in a rat model [57] where AI was induced with Group A Streptococcal Peptidoglycan-Polysaccharide (PG-APS). Four-week therapy resulted in increased hemoglobin and serum iron, although by this point the hepcidin mRNA was not significantly decreased. Notably, in vitro studies suggested that sHJV.Fc acts as a broad spectrum BMP antagonist [58], and it remains to be determined whether this therapy will have significant off-target effects in vivo.
Small molecule inhibitor of the BMP type I receptor was another effective suppressor of hepcidin. LDN-193189, a derivative of dorsomorphin that specifically antagonizes the kinase activity of BMP receptor isotypes ALK2, ALK3 and ALK6, effectively reversed anemia in the rat model of AI caused by PG-APS [57]. In a rat model of anemia of chronic kidney disease caused by adenine treatment, 5 weeks of LDN-193189 injections decreased hepcidin mRNA, increased serum iron and hemoglobin content of reticulocytes, although no change in hemoglobin was noted [59].
Inflammatory pathway
Inflammation strongly induces hepcidin expression via IL-6/Stat3 and possibly other pathways (Figure 2). As expected, neutralizing monoclonal antibodies against IL-6 or IL-6 receptors decreased hepcidin synthesis in animal models and humans with inflammatory conditions. For example, anti-IL6 mAb decreased hepcidin and improved anemia in renal cell carcinoma, multiple myeloma and Castleman disease [60,61]. Similarly, administration of anti-TNFα antibody decreased hepcidin levels in patients with rheumatoid arthritis [62]. Drawbacks of the anti-cytokine regimens are usually related to impaired host defense immunoresponse [63] that translates to an increased risk of infections.
Erythropoietic pathway
Although the mechanism is poorly understood, increased erythropoietic activity suppresses hepcidin, and the potentiation of this pathway could be used in iron-restricted conditions associated with elevated hepcidin. High doses of erythropoietin (EPO) can overcome the resistance to erythropoietin seen in AI [64], ad this may be partially due to hepcidin suppression. Similarly, prolyl hydroxylase inhibitors which stabilize hypoxia-inducible factor and increase EPO synthesis, were also shown to decrease hepcidin and increase hemoglobin in CKD patients [65]. However, administration of large doses of ESAs in patients with CKD or cancer have been associated with rare but severe adverse effects [53]. Thus, agents that specifically target hepcidin production would be needed to alleviate safety concerns.
Gene silencing of hepcidin and its regulators. Gene silencing agents are known to preferentially accumulate in the liver, thus hepcidin and its regulators may be feasible targets for these approaches. Antisense oligonucleotides and siRNAs for hepcidin, TfR2 and HJV have been developed [66,67]. Experiments in PG-APS rat model of AI showed that TfR2 siRNA administration decreased hepcidin mRNA and reversed anemia [67]. Considering that human mutations in hepcidin, HJV and TfR2 are so far exclusively associated with iron overload disorders, off-target effects could be minimal.
Neutralizing hepcidin peptide
Neutralization of hepcidin bioactivity may be achieved by direct binding/sequestration of the hormone in circulation. To this end (i) monoclonal antibodies (mAbs), (ii) engineered proteins (anticalins) and (iii) RNA-based binders (spiegelmers) were investigated.
Fully human anti-hepcidin antibody (12B9m) was evaluated in transgenic mice expressing human hepcidin, using a model of AI caused by the heat-killed Brucella abortus. MAb-mediated neutralization of hepcidin by itself increased hemoglobin, and also reversed erythropoietin resistance observed in this model [68]. Furthermore, the MAb suppressed hepcidin in cynomolgus monkeys and caused a prolonged dose-dependent increase in serum iron. Another fully humanized mAb against hepcidin (LY2787106) is currently in Phase I human trials for cancer-related anemia [69].
Anticalins are small proteins (~20 kDa) that were derived from lipocalins, naturally occurring proteins which can bind diverse biological ligands. Target-specific anticalins with high specificities and picomolar affinities may be engineered via site-directed random mutagenesis in combination with selection via phage display. One of such compounds, anticalin PRS-080 binds human hepcidin with sub-nanomolar affinity. In nonhuman primates (cynomolgus monkeys), PRS-080 administration caused effective mobilization of iron and hyperferremia [70].
Spiegelmers are L-enantiomeric oligonucleotides that were engineered to bind pharmacologically relevant targets with high affinity. Therefore, conceptually they are a form of aptamers. Due to their non-natural structure that utilizes L-ribose instead of D-ribose in nucleotide units, they are highly resistant to degradation by nucleases, stable in circulation, immunologically passive and generally well tolerated. Spiegelmers for specific targets are selected by screening of large combinatorial libraries. NOX-H94 is a spiegelmer that neutralizes human hepcidin. In a Phase I human trial, NOX-H94 dose-dependently increased serum iron and transferrin saturation [71]. NOX-H94 is currently undergoing the Phase II clinical trials to examine its efficacy in patients with anemia of cancer.
The high specificity of hepcidin binders for their target is the main advantage of these approaches. The main challenge for their efficacy is likely the high rate of hepcidin production, estimated at ~12 mg/day per 75 kg for humans [72] and even higher during inflammation. Considering the difference in molecular weight between hepcidin (only 2.7 kDa) and its neutralizing binders, massive doses of intravenously-delivered therapeutic agents may be required. Moreover, the neutralization of hepcidin with large molecular weight binders will most likely impede its natural clearance rate resulting in further hepcidin accumulation. However, even a partial or intermittent neutralization of hepcidin activity may be sufficient to reach desired therapeutic effects.
Blocking hepcidin-ferroportin interaction
A humanized anti-ferroportin monoclonal antibody (LY2928057) was developed against an extracellular loop of ferroportin adjacent to the hepcidin-binding region [73]. LY2928057 prevents hepcidin binding to the transporter without interfering with the iron efflux through ferroportin. LY2928057 increased serum iron in a dose-dependent manner in cynomolgus monkeys, and clinical evaluation is underway.
Mechanistically, hepcidin binding to ferroportin relies on the extracellular loop of ferroportin that contains sulfhydryl Cys326 residue and surrounding hydrophobic residues [47]. Thiol-reactive compound fursultiamine was shown to prevent hepcidin binding to ferroportin in vitro [74], although the short half-life of fursultiamine in circulation prevented any consistent effects in mice in vivo. Similar agents may be effective in preventing hepcidin-ferroportin interaction, provided that the non-specific effects on other proteins containing an active thiol are minimized.
Targeting ferroportin endocytosis or synthesis
Although no agents have been tested in preclinical models yet, any approaches that prevent ferroportin endocytosis or stimulate ferroportin synthesis would be expected to increase iron delivery to plasma, thus circumventing the effect of elevated hepcidin. Ferroportin endocytosis is dependent on the ubiquitination of several intracellular lysine residues [4]. Because of the fairly generic nature of endocytic pathways, this process may be difficult to target unless E3 ubiquitin ligases and/or other endocytic components are identified that have high ferroportin substrate specificity.
Unrelated to the effect on the systemic iron metabolism, agents that increase ferroportin synthesis may also be beneficial in the treatment of some cancers. Ferroportin expression was a strong and independent predictor of prognosis in breast cancer, with higher ferroportin being associated with a favorable prognosis [75].
Concluding remarks
The hepcidin-ferroportin axis plays an important role in the pathogenesis of iron disorders including iron overload diseases and iron-restricted anemias. It is therefore not surprising that within only a dozen years since the first publications on hepcidin, multiple hepcidin-targeting strategies have been developed. Although most agents have only been evaluated in preclinical studies, several have reached the stage of human clinical trials. It is still too early to try to assess which of the many approaches have the best chance of success. While the clinical effectiveness of hepcidin-targeted therapies remains to be established, the hope is that hepcidin agonists and antagonists will improve the treatment of patients with iron disorders, either alone or in combination with existing therapies.
Highlights for “The pathophysiology and pharmacology of hepcidin”.
The hormone hepcidin regulates systemic iron homeostasis
Hepcidin deficiency or excess contribute to the pathogenesis of iron disorders
Hepcidin agonists and antagonists are being developed as novel therapeutics
Footnotes
Disclosures
Elizabeta Nemeth is a stockholder and consultant for Intrinsic LifeSciences, a biotech company developing hepcidin diagnostics, and Merganser Biotech, a biotech company developing hepcidin therapeutics. Piotr Ruchala is a stockholder and consultant for Merganser Biotech.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med. 2011;62:347–360. doi: 10.1146/annurev-med-050109-142444. [DOI] [PubMed] [Google Scholar]
- 2.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 3.Donovan A, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Qiao B, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15:918–924. doi: 10.1016/j.cmet.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis. 2008;40:132–138. doi: 10.1016/j.bcmd.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodnough JB, et al. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56:291–299. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou Y, et al. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene. 2012;511:398–403. doi: 10.1016/j.gene.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 8.Latour C, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology. 2013 doi: 10.1002/hep.26648. [DOI] [PubMed] [Google Scholar]
- 9.Guo W, et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell. 2013;12:280–291. doi: 10.1111/acel.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vecchi C, et al. Gluconeogenic Signals Regulate Iron Homeostasis Via Hepcidin in Mice. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- 12.Ramos E, et al. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology. 2011;53:1333–1341. doi: 10.1002/hep.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corradini E, et al. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology. 2011;54:273–284. doi: 10.1002/hep.24359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kautz L, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112:1503–1509. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- 15.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 16.D’Alessio F, et al. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012;57:1052–1060. doi: 10.1016/j.jhep.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt PJ, Fleming MD. Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am J Hematol. 2012;87:588–595. doi: 10.1002/ajh.23173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson MB, et al. Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol Biol Cell. 2007;18:743–754. doi: 10.1091/mbc.E06-09-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinbicker AU, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118:4224–4230. doi: 10.1182/blood-2011-03-339952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang RH, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Piperno A, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110:4096–4100. doi: 10.1182/blood-2007-06-096503. [DOI] [PubMed] [Google Scholar]
- 22.Nemeth E, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–1806. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 23.Papanikolaou G, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 24.Du X, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088–1092. doi: 10.1126/science.1157121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finberg KE, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA) Nat Genet. 2008;40:569–571. doi: 10.1038/ng.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee DH, et al. Neogenin inhibits HJV secretion and regulates BMP induced hepcidin expression and iron homeostasis. Blood. 2010 doi: 10.1182/blood-2009-11-251199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silvestri L, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–511. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enns CA, et al. Neogenin interacts with matriptase-2 to facilitate hemojuvelin cleavage. J Biol Chem. 2012;287:35104–35117. doi: 10.1074/jbc.M112.363937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicolas G, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Origa R, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92:583–588. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 31.Pak M, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez R, et al. Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infect Immun. 2013 doi: 10.1128/IAI.00983-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Besson-Fournier C, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120:431–439. doi: 10.1182/blood-2012-02-411470. [DOI] [PubMed] [Google Scholar]
- 34.Armitage AE, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118:4129–4139. doi: 10.1182/blood-2011-04-351957. [DOI] [PubMed] [Google Scholar]
- 35.Fernandes A, et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009;114:437–443. doi: 10.1182/blood-2008-03-146134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sham RL, et al. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol Dis. 2005;34:157–161. doi: 10.1016/j.bcmd.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Pasricha SR, et al. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: a longitudinal study. Blood. 2013;122:124–133. doi: 10.1182/blood-2012-12-471441. [DOI] [PubMed] [Google Scholar]
- 38.Porter JB. Optimizing iron chelation strategies in beta-thalassaemia major. Blood Rev. 2009;23(Suppl 1):S3–S7. doi: 10.1016/S0268-960X(09)70003-7. [DOI] [PubMed] [Google Scholar]
- 39.Troutt JS, et al. Hepcidin-25 concentrations are markedly increased in patients with chronic kidney disease and are inversely correlated with estimated glomerular filtration rates. J Clin Lab Anal. 2013;27:504–510. doi: 10.1002/jcla.21634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim A, et al. Mouse model of anemia of inflammation: complex pathogenesis with partial dependence on hepcidin. Blood. 2013 doi: 10.1182/blood-2013-08-521419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Libregts SF, et al. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU. 1 axis. Blood. 2011;118:2578–2588. doi: 10.1182/blood-2010-10-315218. [DOI] [PubMed] [Google Scholar]
- 42.Prince OD, et al. Late stage erythroid precursor production is impaired in mice with chronic inflammation. Haematologica. 2012;97:1648–1656. doi: 10.3324/haematol.2011.053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicolas G, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34:97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 44.Gardenghi S, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120:4466–4477. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao JJ, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J. 2010;12:646–657. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark RJ, et al. Design, synthesis, and characterization of cyclic analogues of the iron regulatory peptide hormone hepcidin. Biopolymers. 2013;100:519–526. doi: 10.1002/bip.22350. [DOI] [PubMed] [Google Scholar]
- 47.Preza GC, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121:4880–4888. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nai A, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119:5021–5029. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123:1531–1541. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt PJ, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121:1200–1208. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corradini E, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology. 2010;139:1721–1729. doi: 10.1053/j.gastro.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett CL, et al. A review of safety, efficacy, and utilization of erythropoietin, darbepoetin, and peginesatide for patients with cancer or chronic kidney disease: a report from the Southern Network on Adverse Reactions (SONAR) Semin Thromb Hemost. 2012;38:783–796. doi: 10.1055/s-0032-1328884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poli M, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011;117:997–1004. doi: 10.1182/blood-2010-06-289082. [DOI] [PubMed] [Google Scholar]
- 55.Poli M, Girelli D, Naggi A, Campostrini N, Asperti M, Finazzi D, Maccarinelli F, Castagna A, Arosio P. Identification of heparins without anticoagulant activity which inhibit hepcidin in vivo. American Journal of Hematology. 2013 May 1;88(5):E40. Ref Type: Abstract. [Google Scholar]
- 56.Babitt JL, et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–1939. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Theurl I, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118:4977–4984. doi: 10.1182/blood-2011-03-345066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nili M, et al. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem. 2010;285:24783–24792. doi: 10.1074/jbc.M110.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun CC, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant. 2013;28:1733–1743. doi: 10.1093/ndt/gfs584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schipperus M, Rijnbeek B, Reddy M, Qin X, Cornfield MJ. CNTO328 (Anti-IL-6 mAb) Treatment Is Associated with An Increase in Hemoglobin (Hb) and Decrease in Hepcidin Levels in Renal Cell Carcinoma (RCC) Blood. 2009 Nov 20;114(22):4045. Ref Type: Abstract. [Google Scholar]
- 61.Kurzrock R, et al. A Phase I, Open-Label Study of Siltuximab, an Anti-IL-6 Monoclonal Antibody, in Patients with B-cell Non-Hodgkin Lymphoma, Multiple Myeloma, or Castleman Disease. Clin Cancer Res. 2013;19:3659–3670. doi: 10.1158/1078-0432.CCR-12-3349. [DOI] [PubMed] [Google Scholar]
- 62.Doyle MK, et al. Effects of subcutaneous and intravenous golimumab on inflammatory biomarkers in patients with rheumatoid arthritis: results of a phase 1, randomized, open-label trial. Rheumatology (Oxford) 2013;52:1214–1219. doi: 10.1093/rheumatology/kes381. [DOI] [PubMed] [Google Scholar]
- 63.van de Vosse E, van Agtmael MA. Targets of anticytokine therapy and the risk of infections in humans and mice. Curr Opin Rheumatol. 2007;19:626–635. doi: 10.1097/BOR.0b013e3282f05c6d. [DOI] [PubMed] [Google Scholar]
- 64.Elliott J, et al. Hyporesponsiveness to erythropoietin: causes and management. Adv Chronic Kidney Dis. 2009;16:94–100. doi: 10.1053/j.ackd.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 65.Besarab A, Chernyavskaya EN, Motylev I, Evgeny S, Yampolskiy AF, Kumbar LM, Gurevich K, Chan DTM, Leong R, Zhong M, Franco M, Yu KHP, Neff TB. FG-4592, an Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor, Corrects Anemia without Iron Supplementation in Incident Dialysis Patients. J Am Soc Nephrol. 2012;23:21A. Ref Type: Abstract. [Google Scholar]
- 66.Isis Pharmaceuticals. Xenon licenses antisense drug XEN701 from Isis and initiates preclinical toxicology studies. 2013 http://ir.isispharm.com/phoenix.zhtml?c=222170&p=irol-newsArticle&ID=1828284&highlight=
- 67.Akinc Akin, Chan-Daniels Amy, Sehgal Alfica, Foster Don, Bettencourt Brian R, Hettinger Julia, Racie Tim, Aubin Justin, Kuchimanchi Satya, Epstein-Barash Hila, Nakayama Tomoko. Targeting the Hepcidin Pathway with RNAi Therapeutics for the Treatment of Anemia. Blood. 2011 Nov 18;118(21):688. Ref Type: Abstract. [Google Scholar]
- 68.Cooke KS, et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood. 2013;122:3054–3061. doi: 10.1182/blood-2013-06-505792. [DOI] [PubMed] [Google Scholar]
- 69.Eli Lilly and Company. A Phase 1 Study of LY2787106 in Cancer and Anemia. 2013 http://www.clinicaltrials.gov/ct2/show/NCT01340976.
- 70.Hohlbaum A, Gille H, Christian J, Allersdorfer A, Jaworski J, Burrows J, Rattenstetter B, Kolodziejczyk M, Olwill S, Audoly L. Iron mobilization and pharmacodynic marker measurements in non-human primates following administration of PRS-080, a novel and highly specific anti-hepcidin therapeutic. American Journal of Hematology. 2013 May 1;88(5):E41. Ref Type: Abstract. [Google Scholar]
- 71.Riecke K, Zollner S, Boyce M, vanHecke B, Vauleon S, Summo L, Laarakkers CM, Swinkels DW, Schwoebel F. Single and repeated dose first-in-human study with the anti-hepcidin spiegelmer NOX-H94. American Journal of Hematology. 2013 May 1;88(5):E42. Ref Type: Abstract. [Google Scholar]
- 72.Fung E, Nemeth E. Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica. 2013;98:1667–1676. doi: 10.3324/haematol.2013.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leung Donmienne, Hill Karen A, De Rosa David C, Xu Jianghuai, Manetta Joseph, Wroblewski Victor J, Benschop Robert J. LY2928057, An Antibody Targeting Ferroportin, Is a Potent Inhibitor Of Hepcidin Activity and Increases Iron Mobilization In Normal Cynomolgus Monkeys. Blood. 2013 Oct 21;122(21):3433. Ref Type: Conference Proceeding. [Google Scholar]
- 74.Fung E, et al. High-throughput screening of small molecules identifies hepcidin antagonists. Mol Pharmacol. 2013;83:681–690. doi: 10.1124/mol.112.083428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pinnix ZK, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu PB, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]