

Abstract
Attempts to target mutant KRAS have been unsuccessful. Here, we report the identification of Smad ubiquitination regulatory factor 2 (SMURF2) and UBCH5 as a critical E3:E2 complex maintaining KRAS protein stability. Loss of SMURF2 either by small interfering RNA/short hairpin RNA (siRNA/shRNA) or by overexpression of a catalytically inactive mutant causes KRAS degradation, whereas overexpression of wild-type SMURF2 enhances KRAS stability. Importantly, mutant KRAS is more susceptible to SMURF2 loss where protein half-life decreases from >12 hours in control siRNA-treated cells to <3 hours on Smurf2 silencing, whereas only marginal differences were noted for wild-type protein. This loss of mutant KRAS could be rescued by overexpressing a siRNA-resistant wild-type SMURF2. Our data further show that SMURF2 monoubiquitinates UBCH5 at lysine 144 to form an active complex required for efficient degradation of a RAS-family E3, β-transducing repeat containing protein 1 (β-TrCP1). Conversely, β-TrCP1 is accumulated on SMURF2 loss, leading to increased KRAS degradation. Therefore, as expected, β-TrCP1 knockdown following Smurf2 siRNA treatment rescues mutant KRAS loss. Further, we identify two conserved proline (P) residues in UBCH5 critical for SMURF2 interaction; mutant of either of these P to alanine also destabilizes KRAS. As a proof of principle, we demonstrate that Smurf2 silencing reduces the clonogenic survival in vitro and prolongs tumor latency in vivo in cancer cells including mutant KRAS-driven tumors. Taken together, we show that SMURF2:UBCH5 complex is critical in maintaining KRAS protein stability and propose that targeting such complex may be a unique strategy to degrade mutant KRAS to kill cancer cells.
Introduction
KRAS is the most frequently mutated oncogenic driver, reported in approximately 15% to 30% of all human malignancies, and is more prevalent in pancreatic (90%), colon (50%), and lung (30%) cancers [1–3]. Patients with tumors carrying a KRAS mutation show resistance to anti-epidermal growth factor receptor (EGFR) therapies [4–6], and attempts to target mutant KRAS have been unsuccessful [7,8]. As KRAS activity is known to be regulated by farnesylationmediated protein modifications [9], farnesyl transferase inhibitors have been developed. Although they were effective in preclinical models, they failed in the clinic [10,11]. Therapeutic approaches to inhibit KRAS downstream signaling have focused on the development of kinase inhibitors targeting rapidly accelerated fibrosarcoma (RAF), mitogen-activated protein kinase kinase (MEK), and extracellular signal-regulated kinase (ERK). Only a minority of these provided marginal survival advantage to patients carrying KRAS mutations, and they also resulted in significant adverse events [12]. Recently, various preclinical studies of synthetic lethality approaches targeting specific kinases were also reported to specifically induce cell death of mutant KRAS-driven tumors [13–17], but these approaches remain to be tested clinically. Hitherto, currently available therapeutic approaches of indirectly targeting mutant KRAS have had limited success [18–21] and there remains a great need to identify more effective therapeutic approaches for KRAS mutant cancers.
On the basis of our recent findings as well as several independent studies [22–25], we hypothesized that the physical loss or degradation of an oncoprotein provides a more robust and durable antitumor effect compared to inhibition of oncogene function, a commonly used strategy, which has so far provided only a transient antitumor response. On the basis of such a provocative hypothesis, we felt the need to better understand the regulators involved in maintaining oncogene protein stability, particularly mutant KRAS. Increasingly, it is becoming recognized that ubiquitin-mediated protein modifications of RAS family members (H-, N-, and KRAS) play critical roles in protein abundance, maintenance of their activity, and association with downstream signaling molecules. Particularly, in the case of mutant KRAS, mono-/bi-ubiquitination enhances its GTP binding and its association with downstream signaling molecules [26,27], whereas polyubiquitination mediated through an F-box family E3, β-transducing repeat containing protein 1 (β-TrCP1), induces RAS degradation [28,29].
While investigating the role of Smad ubiquitination regulatory factor 2 (SMURF2) in EGFR protein stability [22], we observed that SMURF2 loss had greater impact on the clonogenic survival of mutant KRAS-driven cancer cells compared to wild-type KRAS-containing cell lines. In this report, we have extended this observation to several mutant KRAS-driven lung and colorectal cancer cell lines and discovered that, although KRAS protein is stable (half-life > 12 hours) under normal physiological condition, on the loss of SMURF2, mutant KRAS protein is rapidly degraded (half-life < 3 hours). Subsequently, we found that targeting SMURF2 in mutant KRAS-driven cells significantly reduces their clonogenic survival and growth of tumor xenografts in nude mice. Furthermore, we have identified the molecular mechanism of SMURF2-mediated protection of KRAS protein, where SMURF2 in complex with UBCH5 polyubiquitinates and degrades RAS family E3, β-TrCP1, thus indirectly protects KRAS from degradation. Furthermore, we found that in various cancer cell lines as well as in a large cohort of human lung adenocarcinoma specimens Smurf2 and KRAS gene and protein expression are significantly correlated, which suggests that these interactions occur in patients.
Materials and Methods
Reagents
A monoclonal antibody against human SMURF2 has been described previously [30]. Rabbit polyclonal SMURF2 antibody was purchased from Upstate Biotechnology (Lake Placid, NY), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-TrCP1 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-KRAS (F234, Cat. No. SC-30) and anti-ubiquitin (P4D1) antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-KRAS (OP24) antibody was obtained from Calbiochem (EMD Chemicals, San Diego, CA). Anti-MYC (Cat. No. 46-0603) antibody was purchased from Invitrogen (Life Technologies, Grand Island, NY). Anti-UBCH5 (UBE2D1, Cat. No. ab66600) antibody was obtained from Abcam (Cambridge, MA). Anti-UBCH7 (Cat. No. AB3859) antibody was obtained fromMillipore (EMD Millipore, Billerica, MA). Sepharose-conjugated anti-FLAG (M2) antibody, protease inhibitor cocktail, and 3-methyladenine (3-MA; Cat. No. M9281) were obtained from Sigma (St Louis, MO). Agarose-conjugated anti-c-MYC monoclonal antibody (Cat. No. 631208) was obtained from Clontech (Mountain View, CA). Glutathione-sepharose beads were purchased from GE Healthcare (Pittsburgh, PA). Cycloheximide (Cat. No. 239763) was obtained from Calbiochem (EMD Chemicals). Control (Cat. No. D-001810), Smurf2 (Cat. No. D-007194), β-TrCP (Cat. No. D-003463), and UbcH5 (Cat. No. J-009387) small interfering RNA (siRNA) were purchased from Thermo Fisher Scientific (Lafayette, CO), whereas another Smurf2 siRNA pool (Cat. No. sc-41675), Smurf1 (Cat. No. sc-41673), and AIP4/ITCH (Cat. No. sc-40364) siRNAs were obtained from Santa Cruz Biotechnology.
Cell Cultures
Human embryonic kidney-293 (HEK293) cell line, human lung cancer cell lines (H2347, Hcc78, H358, H441, and A549), and cervical carcinoma cell line (HeLa) were purchased from the American Type Culture Collection (Manassas, VA) and were cultured either in Dulbecco's modified Eagle's medium (for HEK293 and HeLa) or RPMI-1640 (for rest of the cell lines) supplemented with 10% FBS. Isogenic colorectal cancer cell lines (HCT116) harboring either wild-type or mutant KRAS alleles were kind gifts from Dr Vogelstein (Johns Hopkins University, Baltimore, MD) and were grown in McCoy's medium supplemented with 10% FBS. For plasmid transfection, Lipofectamine 2000 (Invitrogen) and, for the siRNA transfection, Lipofectamine RNAi-max (Invitrogen) were used according to the manufacturer's instructions.
Protein Analyses
Immunoblot analysis and immunoprecipitation techniques were performed as described previously [22] with a minor modification of buffer, such that excess calcium was added [50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 1.3 mM CaCl2, 1 mM DTT, 10 mM β-glycerophosphate, 1 mM NaF, 0.1 mM sodium orthovanadate, 10% glycerol, 1% NP-40, and 1x protease inhibitor cocktail (Sigma; Cat. No. P8340)].
Glutathione S-Transferase Pull-Down Assay
Glutathione S-transferase (GST) pull-down assays were performed as described previously [22]. Briefly, GST-SMURF2 proteins [either wild-type or C716A (CA) mutants] were purified from bacteria by standard techniques using GST-Sepharose beads (GE Healthcare) and were equilibrated in 0.5x Superdex buffer [1x Superdex buffer: 25 mM Hepes (pH 7.5), 12.5 mM MgCl2, 10 µM ZnSO4, 150 mM KCl, 20% glycerol, 0.1% NP-40, and 1 mM EDTA] by washing three times with the buffer. The beads were then incubated with about 500 ng of UBCH5 protein (Cat. No. E2-616; Boston Biochemicals, Cambridge, MA) and incubated overnight at 4°C. The beads were then washed three times using 0.5 Superdex buffer and boiled in Laemmli buffer. The SMURF2:UBCH5 complex was immunodetected using UBCH5- and SMURF2-specific antibodies.
Clonogenic Cell Survival Assay
Clonogenic survival assays were performed using techniques described previously [31]. The effects of different siRNAs (e.g., Smurf2, β-TrCP1, and UbcH5) on clonogenic survival of different cancer cell lines were determined by normalizing the survival fraction of control siRNA-treated group as 1.
RNA Isolation and Quantitation
Total cellular RNA was isolated using Qiagen RNeasy mini kit (Qiagen, Inc, Valencia, CA) according to the manufacturer's instructions. For quantitation, RNA samples (1 µg) were reverse transcribed with random hexamers using the High-Capacity cDNA Reverse Transcription System (Applied Biosystems, Foster City, CA). Realtime polymerase chain reaction (PCR) was carried out with an ABI Prism 7700 sequence detector using Power SYBR GREEN PCR master mix (Applied Biosystems). The following human gene-specific primers were used for the PCR reaction: 1) KRAS (forward, 5′ TACAGTGCAATGAGGGACCA 3′ and reverse, 5′ CTGTTCTAGAAGGCAAATCACA 3′) and 2) GAPDH (forward, 5′ GAGTCAACGGATTTGGTCGT 3′ and reverse, 5− TTGATTTTGGAGGGATCTCG 3′).
Lentiviral shRNA-Mediated Gene Knockdown
Control and Smurf2 small hairpin RNA (shRNA) lentiviral constructs were obtained from Open Biosystems (Lafayette, CO), and viral particles were produced using a standardized procedure as described previously [32]. For efficient knockdown, target cells were transduced twice with the lentiviral particles with an interval of 24 hours, and 48 hours after the second transduction, cells were lysed for immunoblot analysis to check Smurf2 knockdown efficiency and also trypsinized and used for either clonogenic cell survival assays or tumor xenograft studies.
Tumor Xenograft Studies
All animal procedures were carried out in accordance with a University Committee on Use and Care of Animals of the University of Michigan-approved protocol. Briefly, either control or Smurf2 shRNA lentiviruses were transduced into H358 and HCT116 isogenic cell lines. To generate tumors, 300,000 cells were injected into 4- to 6-week-old female athymic nu/nu mice (Harlan Laboratories, Indianapolis, IN). Tumor onset was detected by palpation by examining the mice on a regular basis, and tumor latency was calculated on the basis of the time from injection to detection of palpable tumors. Throughout the experiment, mice were weighed every other day, and the length and width of the tumors were measured on a regular basis. The tumor volumes were calculated using the formula 0.5 x length x width2 [33]. Relative tumor volumes were calculated by normalizing to the tumor size when first detected.
In Vitro Ubiquitination Assay
The in vitro ubiquitination reaction was carried out in a 15-µl reaction volume containing reaction buffer [250 mM Tris-HCl (pH 7.5), 50 mM MgCl2, 50 µM DTT, and 20 mM ATP), 10 βg of Myc-tagged ubiquitin (Cat. No. U-115), 0.35 βg of UBE1 (Cat. No. E305), and 0.5 βg of UBCH5 (Cat. No. E2-616; all from Boston Biochemicals), and Flag-tagged β-TrCP1 was overexpressed in HEK293 cells and pulled down using affi-Flag (M2) beads. Human recombinant SMURF2 protein (Cat. 468H; Creative Biomart, New York, NY) was then added, and the reaction mixtures were incubated at 37°C for 2 hours. The reaction was terminated after boiling with 4x gel loading dye. The samples were then resolved and immunoblotted using indicated antibodies. For certain reactions where indicated, GST-SMURF2 (either wild type or C716A mutants) proteins were purified in the laboratory using a GST-Sepharose column and were used in the in vitro ubiquitination assay.
Mass Spectrometry
The in vitro ubiquitination reaction mix was separated on a polyacrylamide gel, and proteins were visualized with colloidal Coomassie stain. In-gel digestion followed by identification of ubiquitination site mapping was carried out essentially as described previously [34]. Briefly, on trypsin digestion, peptides were resolved on a nano-capillary reverse phase column and subjected to a high-resolution, linear ion-trap mass spectrometer (LTQ Orbitrap XL; Thermo Fisher Scientific). The full mass spectrometry (MS) scan was collected in Orbitrap (resolution 30,000 at 400 m/z), and data-dependent MS/MS spectra on the nine most intense ions from each full MS scan were acquired. Proteins and peptides were identified by searching the data against Swissprot human protein database, appended with decoy (reverse) sequences, using X!Tandem/Trans-Proteomic Pipeline (TPP) software suite. All proteins identified with a ProteinProphet probability of >0.9 (fdr < 1%) were accepted. Spectral matches to ubiquitinated peptides were manually verified.
Analytical Size Exclusion Chromatography
Analytical size exclusion chromatography (SEC) was performed with the purified GST-SMURF2 and UBCH5 using an S200pc.column and AKTAmicro (GE Healthcare) equilibrated with buffer [50 mM Hepes (pH 8.0) and 100 mM NaCl] at 4°C. In a 50-µl reaction volume, 50 µg each of affinity-purified GST-SMURF2 and UBCH5 were mixed either in the presence or absence of in vitro ubiquitination components (as mentioned above). Reactions were then incubated at 37°C for 2 hours. Each reaction mixture was then injected into the pre-equilibrated SEC column. Fractions (50 µl per fraction) were collected in a 96-well plate and A280 values were measured to identify protein fractions. A tenth of such protein fractions were then subjected to immunoblot analysis using SMURF2 and UBCH5 antibodies.
Statistics
Pearson correlation (r) was used to quantify the correlation between SMURF2 and KRAS. Unless noted otherwise, results are presented as mean ± standard error of the mean (SEM; estimate) of at least three experiments. To assess the correlation between KRAS and SMURF2, we first normalized their values by calculating the ratios KRAS/GAPDH and SMURF2/GAPDH. We then calculated Pearson correlation coefficients for the log of the normalized KRAS and SMURF2 values. Time to tumor detection was calculated as the time from injection of cells to detection of palpable tumors. The Kaplan-Meier method was used to summarize time to development of tumor in mice under various experimental conditions, and the log-rank test was used to compare groups with respect to time to tumor development. Mice without a tumor at the end of the study were censored at that time. A significance level threshold of two-sided P < .05 was used to determine statistical significance.
Results
SMURF2 Ligase Activity Maintains KRAS Steady-State Levels
To explore the involvement of SMURF2 on KRAS steady-state levels, we altered SMURF2 levels and catalytic activity either by using multiple siRNA/shRNA or by overexpressing wild-type and catalytically inactive mutant (Cys716Ala) of SMURF2, respectively. As shown in Figure 1A and quantified in Figure 1B, knockdown of Smurf2 in mutant KRAS-driven lung adenocarcinoma cell lines [H358 (KRASG12C) and H441 (KRASG12V)] caused ∼60% to 80% reduction in mutant KRAS steady-state levels compared to control siRNA-treated cells as early as 24 hours post-transfection (Figure 1A, compare lanes 1 and 2; lanes 3 and 4). In lung adenocarcinoma cell lines carrying wild-type KRAS (H2347), silencing of Smurf2 had minimal effects on wild-type KRAS protein steady-state level (Figure 1A, lanes 5 and 6). Using different siRNAs (S1 and S2) for Smurf2 and different cell lines (HeLa and A549), we found that loss of endogenous Smurf2 leads to reduced steady-state levels of KRAS (Figure W1A). Probing the membrane with different KRAS antibodies confirmed our observation (Figure W1A). To show the Smurf2 knockdown specificity on KRAS steady-state levels, we performed rescue experiments by overexpressing an siRNA-resistant (si-R) construct of wild-type SMURF2. Expression of SMURF2 si-R before siRNA transfection significantly rescued the KRAS down-regulation (Figure 1C). Furthermore, our study with several other homologous to E6AP carboxyl terminus (HECT)-type ubiquitin ligases (SMURF1 and AIP4/ITCH) showed no alteration in KRAS steady-state level (Figure W1B), suggesting SMURF2 specificity.
Figure 1.
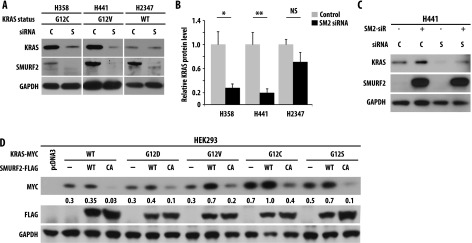
SMURF2 ubiquitin ligase activity controls mutant KRAS steady-state levels. (A) H358 (KRASG12C), H441 (KRASG12V), and H2347 (KRASWT) human lung adenocarcinoma cells were transfected with either control (C) or Smurf2 (S) siRNA. Forty-eight hours posttransfection, cell lysates were subjected to immunoblot analysis using specified antibodies. (B) Quantification of KRAS steady-state level in H358, H441, and H2347 cells on C or S siRNA. Immunoblot obtained from A were scanned and quantified using ImageJ software, and KRAS steady-state levels were normalized using GAPDH as loading control. Relative KRAS protein levels were obtained, and mean ± SEM values were calculated from three independent experiments; * denotes significant difference from control at P < .05; **, denotes significant difference from control at P < .005; NS, not significant. (C) H441 cells were transfected with Smurf2 si-R (SM2-siR) construct before siRNA-mediated Smurf2 knockdown. Cell lysates were prepared 24 hours post siRNA transfection and immunoblotted using indicated antibodies. (D) HEK293 cells were co-transfected with Myc-tagged KRAS [either wild type or various mutants (G12D, V, C, or S)] in the presence or absence of FLAG-tagged SMURF2 [either wild type or C716A (CA) mutants]. Twelve hours post-transfection, cell lysates were prepared and subjected to immunoblot analysis using indicated antibodies.
To test the importance of SMURF2 ubiquitin ligase activity in regulating KRAS steady-state levels, we co-overexpressed FLAG-tagged wild type or a ligase-dead Cys716Ala (CA) mutant of SMURF2 with Myc-tagged KRAS [either wild-type or different mutants (G12D/V/C/S)] in HEK293 cells. Overexpression of wild-type SMURF2 increased mutant (G12D, V, C or S) KRAS steady-state levels, whereas SMURF2 (CA) overexpression downregulated KRAS significantly (Figure 1D), indicating the importance of SMURF2 ubiquitin ligase activity in maintaining KRAS levels. As shown earlier, we also noted that SMURF2 (CA) expression was significantly higher than SMURF2 (wild type), because SMURF2 (CA) is unable to undergo auto-ubiquitination-mediated degradation [35]. Taken together, these results show the critical involvement of SMURF2 physical presence and ubiquitin ligase activity in maintaining steady-state levels of KRAS and most importantly mutant form of the protein.
SMURF2 Ubiquitin Ligase Activity Positively Regulates KRAS Protein Stability
To address whether the changes in KRAS steady-state levels resulting from SMURF2 depletion or overexpression are due to changes in KRAS protein stability or changes in gene transcription, we performed experiments using lysosomal or proteasomal inhibitors and protein half-life studies using the protein synthesis inhibitor cycloheximide and also quantitated KRAS gene transcription following SMURF2 alterations. As shown in Figure 2A, a lysosomal inhibitor, 3-MA, not only increased the steady-state basal level of KRAS (lane 5), indicative of constitutive lysosomal degradation of KRAS in cells, but it also protected against SMURF2 (CA)-mediated KRAS down-regulation (compare lanes 3 and 6). In this study, the proteasomal inhibitor MG132 had minimal protective effects on SMURF2 (CA)-mediated KRAS down-regulation (Figure 2A, lane 9), supporting the notion that KRAS degradation might occur in the lysosomes [36]. (This is in contrast to other RAS family members such as H- or N-RAS, which undergo proteasomal degradation.) [28,29] Furthermore, in HEK293 cells overexpressing KRAS-G12V, the protein half-life decreased from >12 hours to ∼3.5 hours following SMURF2 (CA) overexpression [Figure 2, B (right panel) and C], suggesting the importance of SMURF2 ligase activity on mutant KRAS protein stability. To determine the half-life of the endogenous KRAS protein on Smurf2 knockdown, we transfected lung cancer cells carrying either wild type (H2347) or KRAS-G12V (H441) with either control or Smurf2 siRNA. As shown in Figure 2D and quantified in Figure 2E, on Smurf2 loss, mutant KRAS half-life was reduced from >12 hours to ∼2.5 hours, whereas wild-type KRAS half-life changed marginally, indicating that KRAS-G12V protein was more labile on Smurf2 loss. In a similar experiment, KRAS-G12C mutant behaved similarly as KRAS-G12V protein. In contrast, KRAS gene transcription remained unaffected on Smurf2 knockdown (Figure W2). Taken together, these data support the involvement of SMURF2 ubiquitin ligase activity in maintaining KRAS protein stability including the mutant form.
Figure 2.
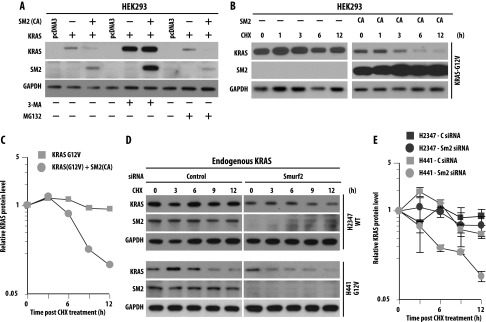
Ubiquitin ligase activity of SMURF2 positively regulates KRAS protein stability. (A) HEK293 cells were transfected with KRAS in the presence or absence of SMURF2 (SM2) CA. Twelve hours post-transfection, indicated cells were treated with either 5 mM 3-MA or 2 µM MG132 for 4 hours. Cell lysates were then subjected to immunoblot analysis with the indicated antibodies. (B) HEK293 cells were transfected either with G12V mutant KRAS in the presence or absence of SM2-CA. Twelve hours post-transfection, cells were treated with cycloheximide (50 µg/ml), and cell lysates were harvested at the indicated time points and analyzed by immunoblot analysis with the antibodies mentioned. (C) Graphical representation of the quantification of KRAS protein levels shown in B to determine protein half-life. Relative KRAS levels were determined by densitometric scanning of the representative immunoblot considering 0 hour band intensity as 1 (arbitrary units). (D) Endogenous KRAS half-life was determined for H2347 and H441 cells on Smurf2 knockdown. Indicated cells were transfected with Smurf2 siRNA, and 48 hours post-transfection, cells were treated with cycloheximide (50 µg/ml), harvested at indicated times, and analyzed by immunoblot analysis. (E) Protein half-lives were determined as explained in C. Each value represents protein intensity average ± SD from three independent experiments and plotted on a log-linear scale.
SMURF2 Downregulates β-TrCP1 to Indirectly Protect Mutant KRAS from Degradation
Recently, β-TrCP, an F-box ubiquitin ligase, has been implicated in RAS (including KRAS) ubiquitination and degradation [28,29]. As reported earlier, we also observed that siRNA-mediated downregulation of β-TrCP1 in mutant KRAS-driven H358 and H441 cells enhances KRAS steady-state level (Figure W3A); overexpression of β-TrCP1 causes degradation of KRAS (Figure W3B), and immunoprecipitation studies show interaction between KRAS and β-TrCP1 only when KRAS degradation was inhibited by a lysosomal inhibitor, 3-MA (Figure W3C). To determine the role of β-TrCP1 in degradation of KRAS on Smurf2 siRNA-mediated down-regulation, we knocked down β-TrCP1 by siRNA before altering SMURF2 expression. As shown in Figure 3A, prior down-regulation of β-TrCP1 protected Smurf2 siRNA-mediated KRAS down-regulation, pointing to its involvement in the process. Prior knockdown of other ubiquitin ligases including a β-TrCP1 homologue, β-TrCP2, did not rescue Smurf2 siRNA-mediated KRAS down-regulation (data not shown). Taken together, our data show that on loss of SMURF2, β-TrCP1 degrades mutant KRAS effectively. We also observed that with Smurf2 siRNA treatment, the steady-state level of β-TrCP1 was significantly higher (Figure 3, A and B). Furthermore, protein half-life studies indicated enhanced β-TrCP1 stabilization; t1/2 ∼ 1.5 hours for control siRNA-treated cells compared to ∼5 hours in Smurf2 siRNA-treated cells (Figure 3, C and D), thus supporting the role of SMURF2 in β-TrCP1 degradation. Similarly, overexpression of wild-type SMURF2 reduced β-TrCP1 levels, whereas overexpression of the SMURF2 (CA) mutant caused up-regulation of β-TrCP1 similar to Smurf2 siRNA treatment (Figure 3E). Using in vivo ubiquitination studies, we demonstrated that in H441 cells, overexpression ofwild-typeSMURF2 enhances β-TrCP1 polyubiquitination, whereas overexpression of catalytically inactive SMURF2 (CA) mutant inhibits it (Figure 3F). Furthermore, as SMURF2 is known to partner with different E2s including UBCH7 and UBCH5 [35,37], we examined the involvement of SMURF2 ubiquitination machinery components in β-TrCP1 polyubiquitination using in vitro ubiquitination assay. Results obtained from this study demonstrate enhanced polyubiquitination of β-TrCP1 in the presence of purified SMURF2 and UBCH5 (Figure 3G). Taken together, our data suggest that SMURF2:UBCH5 is the E3:E2 complex responsible for down-regulation of β-TrCP1, and in the absence of SMURF2, β-TrCP1 is stabilized, leading to an efficient degradation of KRAS.
Figure 3.

SMURF2 destabilizes β-TrCP1 to indirectly protect mutant KRAS from degradation. (A) H441 cells were transfected with either control or β-TrCP1 (βT1) siRNA before Smurf2 (S) knockdown. Twenty-four hours post-transfection, cell lysates were subjected to immunoblot analysis. (B) H358 and H441 cells were transfected with either C or S siRNA, and cell lysates were subjected to immunoblot analysis. (C) H441 cells were transfected with either control (C) or Smurf2 (S) siRNA. Twenty-four hours post-transfection, cells were treated with 50 µg/ml cycloheximide (CHX) for the indicated times. Cell lysates were prepared and subjected to immunoblot analysis. (D) Protein half-life of β-TrCP1 was determined as explained above. (E) HEK293 cells were transfected with either wild type or catalytically inactive (CA) mutants of SMURF2 along with β-TrCP1 as indicated. Twenty-four hours post-transfection, cell lysates were prepared and subjected to immunoblot analysis. (F) H441 cells were transfected with β-TrCP1 either alone or in combination with SMURF2 (either wild type or CA mutants). Forty-eight hours post-transfection, cells were treated with MG132, and 4 hours post-treatment, cell lysates were subjected to immunoprecipitation with β-TrCP1 antibodies followed by immunoblot analysis using indicated antibodies. (G) In vitro ubiquitination assays were performed using immunoprecipitated Flag-tagged β-TrCP1 in the presence or absence of purified recombinant SMURF2. UBCH5, a known E2 for SMURF2, was used in the reaction. Immunoblot analyses were performed using indicated antibodies.
Smurf2 Knockdown Reduces Clonogenic Survival and Increases Tumor Latency Including the Mutant KRAS-Driven Cancer Cells
To address the functional importance of SMURF2-mediated KRAS stabilization, we performed clonogenic survival assays on Smurf2 alteration using various lung adenocarcinoma cells carrying either wild-type or mutant KRAS alleles. Smurf2 loss reduced the clonogenic survival of mutant KRAS-driven H358 (KRASG12C) and H441 (KRASG12V) cells to 0.03 ± 0.002 and 0.05 ± 0.01, respectively, compared to 0.21 ± 0.03 in the case of wild-type KRAS containing H2347 cells (Figure 4A). Similarly, Smurf2 knockdown in HCT116 isogenic lines reduced the clonogenic survival to 0.25 ± 0.1 in wild-type KRAS HCT116 cells and to 0.08 ± 0.02 in mutant KRASG13D-HCT116 isogenic cells compared to control siRNA-treated cells (Figure 4B). In our previous studies, we have shown Smurf2 knockdown had minimal effects on clonogenic survival of various noncancerous cells (e.g., Het-1A and NIH-3T3 and CHO cells) [22]. As the effect of siRNA-mediated gene silencing is transient, we also used lentiviral vectors encoding Smurf2 shRNA (Table W1). As shown in Figure 4C, infection of cells with two different Smurf2 shRNA (shRNA#3 and 4) reduced SMURF2 expression and caused KRAS down-regulation (Figure W4, A and B). Like Smurf2 siRNA-treated cells, lentivirus-infected Smurf2 shRNA-expressing cells showed a significant decrease (0.41 ± 0.04 for H358 and 0.61 ± 0.05 for H441 cells) in clonogenic survival (Figure 4C), supporting the critical function of SMURF2 in regulating the clonogenic survival of cancer cells including mutant KRAS-driven cells. shRNA-mediated Smurf2 knockdown had a lesser effect on clonogenic survival (0.79 ± 0.2) of wild-type KRAS-driven H2347 cells.
Figure 4.
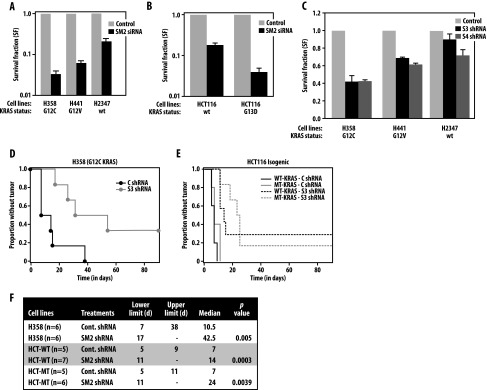
Smurf2 knockdown reduces clonogenic survival and increases tumor latency in KRAS-mutated cancer cells. (A) H358, H441, and H2347 cells were transfected with either control or Smurf2 siRNA. Forty-eight hours post-transfection, cells were plated for clonogenic survival assays as described in the Materials and Methods section. Survival fraction (SF) on Smurf2 knockdown was calculated relative to the control siRNA-treated group (set at 1) and presented as mean ± SEM from three independent experiments. (B) Isogenic colorectal cancer cell lines HCT-116 harboring either wild-type or mutant KRAS were transfected with either control or SM2 siRNA, and clonogenic survival fraction was calculated as described above. (C) H358, H441, and H2347 cells were transduced with either two different Smurf2 shRNA (S3 and S4) or a scrambled (control) shRNA as detailed in the Materials and Methods section. Forty-eight hours post-infection, cells were plated for clonogenic assays. Clonogenic survival of SMURF2 shRNA (S3 and S4) infected cells were calculated relative to the control shRNA-treated group (set at 1). Results are presented as mean ± SEM from three independent experiments. H358 (3 x 105; as in D) and HCT116 (either wild-type or KRASG13D; as in E) cells were infected with either control or SM2-shRNA and 24 hours post-infection injected into the specified number of nude mice, and tumor appearance was observed by palpation on a regular basis. Kaplan-Meier curves indicate the tumor-free survival in days. (F) Statistical analyses of the data obtained from tumor detection studies are shown in tabulated fashion showing the median time-to-tumor detection. The table also shows the P values from a log-rank test for any difference in the time-to-tumor development between two groups.
To test the in vivo relevance of such observations, we assessed the tumor forming potential of mutant KRAS (G12C) H358 and colorectal isogenic HCT116 cells carrying either wild-type or mutant KRAS, on transduction with control or Smurf2 shRNA. As shown in Figure 4D and tabulated in Figure 4F, control shRNA-treated H358 cells formed palpable tumors with a mean time of tumor detection of 10.5 days, which Smurf2 knockdown prolonged to 42.5 days (P < .005). For HCT116 isogenic cells, the mean time until tumor detection was calculated to be 7 days for both wild-type and mutant KRAS cells. Interestingly, Smurf2 down-regulation increased median time to tumor detection to 14 days post-injection for wild-type (P < .0003) and to 24 days for mutant (P < .004) KRAS, respectively (Figure 4, E and F). Such studies demonstrate the critical importance of SMURF2 in maintaining the tumorigenic potential of cancer cells including mutated KRAS-driven cells.
SMURF2 and KRAS Protein and Gene Expression Are Highly Correlated in Patients with Lung Adenocarcinoma
To address the physiological and clinical relevance of our observation, we started by assessing the relationship between protein expression of KRAS and SMURF2 using cell lysates isolated from a variety of cancer and normal cell lines. As shown in Figure 5A, SMURF2 and KRAS protein expression were positively correlated (r = 0.39, n 25, P = .05), suggesting the physiological relevance of our observation. Although it would be desirable to attempt to correlate KRAS and SMURF2 protein expression in patient samples, this is not possible because of the lack of availability of immunohistochemistry compatible KRAS-specific antibodies. To explore the clinical importance of our findings, we analyzed the gene expression data from 595 patients with lung adenocarcinoma and 130 patients with squamous lung cancer measured using oligonucleotide arrays. We found a highly significant correlation (r = 0.49, n = 595, P < .0001) between Smurf2 and KRAS gene expression among patients with lung adenocarcinoma [38,39] (Figure 5B). Among squamous lung cancers, the correlation was lower but still significant (r = 0.24, n = 130, and P < .005) [40] (Figure 5B). This was further supported by data available in the public domain, where a strong correlation between Smurf2 and KRAS gene expression was observed in 79 lung cancer cell lines (r = 0.31, n = 79, P = .005) [41] and among patients with nonhistologically defined non-small cell lung cancern (NSCLC; r = 0.46, n = 183, P < .001) [42,43] (Figure 5B). We determined the KRAS gene mutation status for 54 patients with lung adenocarcinoma in 100 tumors analyzed in the Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma [38]. In this cohort, 28 patients had KRAS mutations particularly at G12 and G13 amino acids, and interestingly, their Smurf2 gene expression was higher compared to the patients carrying wild-type KRAS (Figure W5). Interestingly, β-TrCP1 gene expression was found to be inversely correlated to both Smurf2 (-0.41, n = 595, P < .00001; Figure W6A) and KRAS (-0.52, n = 595, P < .0001; Figure W6B) gene expression in patients with lung adenocarcinoma, suggesting the clinical relevance of our findings. However, the mechanisms underlying these clinical correlations at the transcript levels are as yet undetermined.
Figure 5.
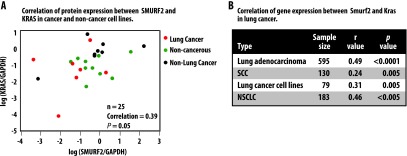
Smurf2 and KRAS gene and protein expression are highly correlated among lung adenocarcinoma patients and cell lines. (A) Scatter plot of KRAS and SMURF2 protein expression among different lung cancer, non-lung cancer, and noncancerous cell lines showing positive correlation (r 0.39, n = 25, P = .05). Cell lysates isolated from 25 different cell lines of lung cancer (H2347, Hcc78, H1793, H838, H441, H2009, SK-Lu1, H460, A549, and H1975), non-lung cancer (U2OS, HeLa, UMSCC-1, UMSCC-11B, UMSCC-74B, Panc1, SW620, and Lovo), and noncancerous cells (Het-1A, IMR-90, MH-S, MRC5, CHO, HDMEC, and skin fibroblasts) were subjected to immunoblot analysis using KRAS, SMURF2, and GAPDH antibodies. Band intensities (arbitrary units) were calculated as described in Figure 1B and normalized using GAPDH. Log-transformed values ofSMURF2/GAPDH and KRAS/GAPDHwere plotted and Pearson's correlation coefficient (r = 0.39, n = 25, P = .05) was calculated. (B) KRAS and Smurf2 matched expression obtained from a large cohort of gene expression microarray data show a strong correlation among lung adenocarcinoma patients (r = 0.49, n = 594, P < .001) and lung cancer cell lines (r = 0.57, n = 79, P < .005). However, the correlation was not as strong (r = 0.24, n = 130, P < .01) among patients with squamous lung cancer.
SMURF2 Monoubiquitinates UBCH5 at the K144 Position to Form a Stable Complex, Which Is Essential for Maintaining KRAS Stability
While performing SMURF2:UBCH5-mediated in vitro ubiquitination unexpectedly, we further identified monoubiquitinated species of UBCH5 (Figure 3G, lower panel). Experiments using either wildtype SMURF2 or ligase-dead SMURF2 (CA) demonstrated that SMURF2 ubiquitin ligase activity is essential for UBCH5 monoubiquitination (Figure 6A). Under similar reaction conditions, SMURF2 did not ubiquitinate the other E2 enzyme, UBCH7 (Figure 6B). MS/MS data further identified lysine (K) 144 as the site of monoubiquitination (Figure 6C). From these studies, we hypothesized that SMURF2 ligase activity may be essential in mediating UBCH5 monoubiquitination, which may be critical in polyubiquitinating β-TrCP1.
Figure 6.

SMURF2 monoubiquitinates UBCH5 at K144 position. (A) GST-SM2 (either wild type or CA mutant) was purified using GST-Sepharose beads, and in vitro ubiquitination reaction was performed and immunoblotted using indicated antibodies. (B) In vitro ubiquitination assays were performed as above using wild-type SMURF2 in the presence and absence of either UBCH5 or UBCH7 as E2. Immunoblot analyses were performed using indicated antibodies. (C) MS/MS spectrum of a peptide identifying ubiquitination of K144 in UBCH5. Peptides isolated on in-gel digestion were resolved on a reverse phase column, and collision-induced dissociation spectra were obtained using an Orbitrap XL mass spectrometer. An MS/MS corresponding to 140EWTQK*YAMox147 of UBCH5 (precursor peptide m/z = 593.76) is shown above. Observed b- and y-ions are indicated. Modified Lys (K) is denoted with *. Mox = oxidized methionine. (D) HEK293 cells were transfected with KRAS along with either wild type or mutants (C85A, K144R, P61A, and P95A) of Myc-tagged UBCH5. Twelve hours post-transfection, cell lysates were prepared and subjected to immunoblot analysis.
To address the importance of SMURF2-mediated UBCH5 monoubiquitination to KRAS stability, we tested the effects of wild-type UBCH5 and K144R mutants on KRAS steady-state levels. As shown in Figure 6D, lane 3, overexpression of wild-type UBCH5 caused a slight increase in KRAS steady-state level, whereas overexpression of UBCH5 (K144R) mutant significantly reduced the KRAS expression (Figure 6D, lane 5). Similar studies using catalytically inactive UBCH5 (C85A) mutant also caused KRAS destabilization (Figure 6D, lane 4). Taken together, our data indicate that SMURF2-mediated monoubiquitination of UBCH5 may be important for β-TrCP1 polyubiquitination and critical for maintaining KRAS levels and the loss of monoubiquitination or catalytic activity of UBCH5 leads to KRAS degradation.
To understand the functionality of SMURF2-mediated monoubiquitination of UBCH5, we performed analytical SEC. As shown in Figure 7, A and B, we detected SMURF2 and UBCH5 in the same complex but only when we performed the in vitro ubiquitination reaction before SEC analysis. From these observations, we hypothesized that SMURF2-mediated monoubiquitination ofUBCH5promotes the formation of a stable E3:E2 complex. Similarly, using HEK293 cells overexpressing SMURF2 and UBCH5 (Figure 7C) and also GST pull-down assays (Figure 7D), we confirmed SMURF2:UBCH5 interaction in the same immunocomplex as well as a direct interaction between them. On the basis of these findings, we conclude that SMURF2 mediates monoubiquitination of UBCH5 leading to autoactivation through an effective E3:E2 complex formation.
Figure 7.
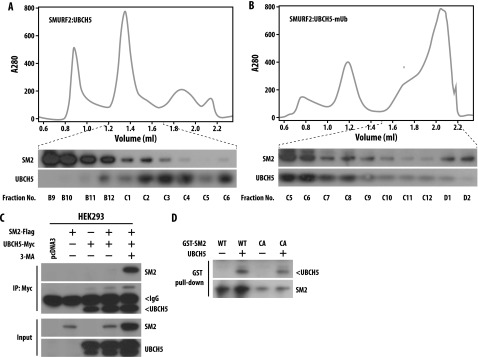
Monoubiquitinated UBCH5 forms a stable complex with SMURF2. (A and B) Purified GST-SMURF2 and recombinant UBCH5 (in A) and purified GST-SMURF2, UBCH5, and the ubiquitination machinery including E1, ubiquitin, and ATP (in B) were mixed, incubated, and subjected to analytical SEC. The top panel is a representative spectrum. In the bottom panel, indicated fractions were subjected to immunoblot analysis using indicated antibodies. The presence of both SM2 and UBCH5 in the same fraction indicates the formation of a stable complex. (C) HEK293 cells were transfected with Myc-tagged UBCH5 in the presence or absence of FLAG-tagged SM2 wild type. Twenty-four hours post-transfection, cells were treated with 3-MA in the indicated samples, and 4 hours post-treatment, lysates were prepared. Cell lysates were then subjected to immunoprecipitation using Myc-agarose beads and immunoblotted with the indicated antibodies. (D) Purified GST-SM2 (either wild type or CA) and purified UBCH5 was subjected to GST pull-down assay as described in the Materials and Methods section. Immunoblot analysis was performed using indicated antibodies.
Disruption of SMURF2:UBCH5 Interaction Reduces KRAS Steady-State Levels
SEC, immunoprecipitation analysis, and GST pull-down assays (Figure 7) identified a direct interaction between SMURF2 and UBCH5. Although the exact contact point(s) between SMURF2 and UBCH5 is currently unknown, the crystal structure data available between SMURF2 and UBCH7 revealed that 61PF62 and 95PA96 are the critical residues in UBCH7 that mediate the E3:E2 interaction [35]. Although, on the basis of primary amino acid alignment analysis, UBCH7 show only 32% sequence identity with UBCH5, the proline, phenylalanine (PF) and proline, alanine (PA) residues are conserved between the two ubiquitin-conjugating enzymes (Figure W7). To determine the importance of proline residues in UBCH5 on the maintenance of KRAS protein stability, we overexpressed the P61A and P95A mutants in the context of KRAS. As shown in Figure 6D, lanes 6 and 7, overexpression of UBCH5 (P61A and P95A) mutants reduced the KRAS steady-state levels similar to those produced by the UBCH5 (C85A) and UBCH5 (K144R) mutants. Taken together, our data show that any alteration of either catalytic activity or disruption of complex formation of SMURF2 and UBCH5 may be an effective strategy to degrade mutant KRAS.
Discussion
Oncogenic KRAS remains an undruggable target in spite of decades of effort attempting to target it either directly through controlling its physical presence or its catalytic activity or indirectly by targeting its downstream signaling particularly the RAF-MEK-ERK or phosphoinositide 3-kinase (PI3K)-AKT pathway [44–47]. The recent identification of ubiquitination-mediated regulation of KRAS protein stability, GTP loading, and association with downstream partners [26,28,29] has provided novel clues for developing efficient strategies to molecularly target mutant KRAS-driven tumors. However, a clear understanding of such ubiquitination-mediated regulation of KRAS still remained largely unclear. In this report, we have identified SMURF2:UBCH5 as a candidate E3:E2 complex that regulates the stability of KRAS; interestingly, Smurf2 knockdown effects were more profound on mutant KRAS as oppose to wild-type protein. Our data suggest that SMURF2 monoubiquitinates its cognate ubiquitin-conjugating enzyme (E2), UBCH5/UBE2D, leading to the active E3:E2 complex formation. Further, we show that this active SMURF2:UBCH5 complex is responsible for polyubiquitinating and degrading another F-box family E3 ligase, β-TrCP1, which is known to degrade RAS including KRAS. In addition, the loss of SMURF2 causes KRAS degradation leading to pronounced reductions in clonogenic survival and tumor-forming abilities of various cancer cells including mutant KRAS-driven ones. The strong correlation between SMURF2 and KRAS protein and transcript expression in human lung adenocarcinoma patients and cell lines further strengthens the clinical and physiological relevance of our observation. On the basis of these findings, in Figure 8, we have proposed a model explaining the critical role of SMURF2: UBCH5 in KRAS protein stability.
Figure 8.

Schematic representation of the hypothetical pathway involving SMURF2, UBCH5, and β-TrCP1 controlling KRAS protein stability. Steps 1 to 4 represent events mediated by active SMURF2:UBCH5 complex. SMURF2 monoubiquitinates UBCH5 at K144 to activate the complex (step 1), which is responsible for polyubiquitination and degradation of β-TrCP1 (step 2). Loss of β-TrCP1, an ubiquitin ligase to degrade RAS family members including KRAS, thus indirectly causes active KRAS accumulation (steps 3 and 4). Steps 5 to 8 represent events on SMURF2 loss (either siRNA/shRNA), where due to the absence of the active ligase complex (step 5), β-TrCP1 gets accumulated (step 6). This active β-TrCP1 then causes polyubiquitination (step 7) and degradation of KRAS (step 8). In this degradation process, glycogen synthase kinase-3 beta (GSK-3β)-mediated phosphorylation of RAS has been reported to prime its binding with β-TrCP1 [28].
An important finding of this work was the differential effects of SMURF2 loss on protein stability of wild-type versus mutant KRAS. We found that physical loss, or loss of SMURF2 catalytic activity, affected mutant KRAS protein's steady-state level and half-life to a greater degree than the wild-type protein. In many instances, protein activity tends to correlate inversely with protein stability. For example, the activated form of SRC kinase is less stable than its wild-type or a kinase-dead counterpart [48], and a constitutively active mutant RHEB GTPase has been reported to be unstable [49]. In case of mutant KRAS, as it is known to be highly active compared to the wild-type protein, we hypothesized and demonstrated here that mutant KRAS protein is particularly labile specifically in the absence of SMURF2 ligase activity and such observation is providing rationale to target SMURF2 to specifically kill mutant KRAS-dependent cells. We would like to point out that although Smurf2 targeting is found to be most effective against mutant KRAS-driven cells, it has substantial effects on the clonogenic survival of wild-type KRAS-driven cells as well. We believe that such effects are at least partially attributable to Smurf2 targeting effects on EGFR [22].
Here, we also establish a direct inhibitory role of a HECT-type ubiquitin ligase SMURF2 on a RING-type E3, β-TrCP1 protein stability. Previously, we and others have reported that SMURF2 is a unique E3 ligase, which can cause stabilization of its substrates including mitotic arrest deficient 2 (MAD2) [50], human enhancer of filamentation 1 (HEF1) [51], EGFR [22], mothers against decapentaplegic homolog 3 (SMAD3) [52], and mouse double minute 2 homolog (MDM2) [53] in a catalytic activity-dependent or -independent manner. In the case of MDM2, SMURF2 promotes MDM2 stabilization by enhancing its heterodimerization with MDMX [53]. In contrast, β-TrCP1 degrades MDM2 and downregulates its activity [54], thus counteracting SMURF2's protective function. Similarly, β-TrCP1 polyubiquitinates and degrades RAS family members including KRAS, whereas our data suggest that SMURF2 is responsible for downregulating β-TrCP1 steady-state levels, thus indirectly protecting KRAS from β-TrCP1-mediated degradation and promoting oncogenesis. Furthermore, we show that SMURF2 is polyubiquitinating and degrading β-TrCP1, suggesting the critical balance of ubiquitin ligase activity in themaintenance of KRAS protein stability. Similarly, whether β-TrCP1 has any counteractive effects on SMURF2 activity remains an open question. Furthermore, inverse correlations between β-TrCP1 gene expression with either Smurf2 or KRAS expression in patients with lung adenocarcinoma also suggest the existence of certain transcriptional/post-transcriptional regulation in the process, an area of research that is beyond the scope of this work.
In this study, although we could recapitulate a previous report of β-TrCP1 involvement in KRAS degradation [28], however, the mode of degradation remains controversial. In 2009, Lu et al. reported that the KRAS is the only RAS isoform, which undergoes lysosomal degradation [36]. In contrast, Jeong et al. reported that β-TrCP-induced RAS degradation is a proteasomal event [28]. In this study, we have obtained data that a lysosomal inhibitor, 3-MA, can protect KRAS from SMURF2 (CA)-induced degradation, whereas a proteasomal inhibitor, MG132, was largely ineffective. Recently, in another study, 4-hydroxy-tamoxifen was reported to induce lysosome-mediated KRAS degradation [55]. In the light of such controversy, we hypothesize that such differences in the mode of KRAS degradation may be attributed to different inducers of KRAS degradation and a more detailed analysis may be necessary to resolve the issue.
While the present data suggest that SMURF2 could be an attractive molecular target to develop anti-KRAS therapy, our previous observations of the role of SMURF2 as a major mitotic regulator in spindle assembly checkpoint [50] and that forced alterations cause chromosomal instability and aneuploidy and promote enhanced tumor initiation in Smurf2-null mice [56] suggest that the inhibition of SMURF2 catalytic activity or siRNA/shRNA-mediated targeting of Smurf2may not be an ideal strategy. However, our study also identified a novel regulation of an E2 through an E3-mediated monoubiquitination that may provide a locus for a different strategy. Currently, there are no reports of covalent ubiquitination of an E2 by its partner E3. We identified SMURF2-mediated monoubiquitination of UBCH5 at the lysine (K) 144 position, which may be responsible for its efficient association to form the active E3:E2 complex required to downregulate β-TrCP1. Furthermore, we also identified two proline residues located at 61 and 95 positions in UBCH5, which may be critical for its association with SMURF2 to form an active complex. Interestingly, although the above-mentioned proline residues are conserved between UBCH5 and UBCH7, the surrounding 95PA96 amino acids are less conserved between the two, and we hypothesize that such differences might permit specific targeting of the SMURF2:UBCH5 interaction without affecting SMURF2:UBCH7 complex required for other cellular functions of SMURF2. Thus, it seems possible that disruption of specific SMURF2: UBCH5 interaction through either a peptidomimetic approach or by small molecule inhibitors may offer a strategy for inducing mutant KRAS degradation.
Supplementary Material
Acknowledgments
We thank Bert Vogelstein for providing isogenic HCT-116 and DLD-1 cell lines harboring either wild-type or mutant KRAS. The authors thank Yi Sun for valuable discussion during the course of the work, Mary Davis for assistance in manuscript preparation, and Steven Kronenberg for graphic assistance.
Abbreviations
- E3
ubiquitin ligase
- E2
ubiquitin-conjugating enzyme
Footnotes
This work was supported by grants 5R01CA160981 (to D.R.), 5R01CA131290 (to M.K.N.), and R01 CA154365 (to A.M.C. and D.G.B.) and by the University of Michigan's Cancer Center Support grant (5P30CA46592).
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 to W7 and are available online at www.neoplasia.com.
References
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010;29:49–60. doi: 10.1007/s10555-010-9209-4. [DOI] [PubMed] [Google Scholar]
- 3.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 4.Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, Haller A, Lothaire P, Meert AP, Noel S, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martini M, Vecchione L, Siena S, Tejpar S, Bardelli A. Targeted therapies: how personal should we go? Nat Rev Clin Oncol. 2012;9:87–97. doi: 10.1038/nrclinonc.2011.164. [DOI] [PubMed] [Google Scholar]
- 7.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 9.Prior IA, Hancock JF. Ras trafficking, localization and compartmentalized signalling. Semin Cell Dev Biol. 2012;23:145–153. doi: 10.1016/j.semcdb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009;5:105–116. doi: 10.2217/14796694.5.1.105. [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg S, Henis YI. Interactions of Ras proteins with the plasma membrane and their roles in signaling. Cell Signal. 2008;20:31–39. doi: 10.1016/j.cellsig.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 13.Singh A, Settleman J. Oncogenic KRAS “addictiony” and synthetic lethality. Cell Cycle. 2009;8:2676–2677. doi: 10.4161/cc.8.17.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, Barbacid M. A synthetic lethal interaction between K-RAS oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 18.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 19.Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T, Dostalova J, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–3957. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 20.Harousseau JL, Martinelli G, Jedrzejczak WW, Brandwein JM, Bordessoule D, Masszi T, Ossenkoppele GJ, Alexeeva JA, Beutel G, Maertens J, et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood. 2009;114:1166–1173. doi: 10.1182/blood-2009-01-198093. [DOI] [PubMed] [Google Scholar]
- 21.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray D, Ahsan A, Helman A, Chen G, Hegde A, Gurjar SR, Zhao L, Kiyokawa H, Beer DG, Lawrence TS, et al. Regulation of EGFR protein stability by the HECT-type ubiquitin ligase SMURF2. Neoplasia. 2011;13:570–578. doi: 10.1593/neo.11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Argiris A, Duffy AG, Kummar S, Simone NL, Arai Y, Kim SW, Rudy SF, Kannabiran VR, Yang X, Jang M, et al. Early tumor progression associated with enhanced EGFR signaling with bortezomib, cetuximab, and radiotherapy for head and neck cancer. Clin Cancer Res. 2011;17:5755–5764. doi: 10.1158/1078-0432.CCR-11-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng FY, Varambally S, Tomlins SA, Chun PY, Lopez CA, Li X, Davis MA, Chinnaiyan AM, Lawrence TS, Nyati MK. Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene. 2007;26:3431–3439. doi: 10.1038/sj.onc.1210129. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, Cantley LC. Ubiquitination of K-RAS enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker R, Lewis SM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol. 2013;20:46–52. doi: 10.1038/nsmb.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, Kim H, Yoon JB, Choi KY. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5:ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 29.Kim SE, Yoon JY, Jeong WJ, Jeon SH, Park Y, Yoon JB, Park YN, Kim H, Choi KY. H-Ras is degraded by Wnt/β-catenin signaling via β-TrCP-mediated polyubiquitylation. J Cell Sci. 2009;122:842–848. doi: 10.1242/jcs.040493. [DOI] [PubMed] [Google Scholar]
- 30.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 31.Nyati MK, Maheshwari D, Hanasoge S, Sreekumar A, Rynkiewicz SD, Chinnaiyan AM, Leopold WR, Ethier SP, Lawrence TS. Radiosensitization by pan ErbB inhibitor CI-1033 in vitro and in vivo. Clin Cancer Res. 2004;10:691–700. doi: 10.1158/1078-0432.ccr-1041-03. [DOI] [PubMed] [Google Scholar]
- 32.Ray D, Terao Y, Nimbalkar D, Hirai H, Osmundson EC, Zou X, Franks R, Christov K, Kiyokawa H. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 2007;67:6605–6611. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- 33.Hahnfeldt P, Panigrahy D, Folkman J, Hlatky L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999;59:4770–4775. [PubMed] [Google Scholar]
- 34.Maine GN, Li H, Zaidi IW, Basrur V, Elenitoba-Johnson KS, Burstein E. A bimolecular affinity purification method under denaturing conditions for rapid isolation of a ubiquitinated protein for mass spectrometry analysis. Nat Protoc. 2010;5:1447–1459. doi: 10.1038/nprot.2010.109. [DOI] [PubMed] [Google Scholar]
- 35.Ogunjimi AA, Briant DJ, Pece-Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F, Wrana JL. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell. 2005;19:297–308. doi: 10.1016/j.molcel.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 36.Lu A, Tebar F, Alvarez-Moya B, López-Alcalá C, Calvo M, Enrich C, Agell N, Nakamura T, Matsuda M, Bachs O. A clathrin-dependent pathway leads to KRAS signaling on late endosomes en route to lysosomes. J Cell Biol. 2009;184:863–879. doi: 10.1083/jcb.200807186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwamborn JC, Muller M, Becker AH, Püschel AW. Ubiquitination of the GTPase Rap1B by the ubiquitin ligase Smurf2 is required for the establishment of neuronal polarity. EMBO J. 2007;26:1410–1422. doi: 10.1038/sj.emboj.7601580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma, author. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–4424. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006;66:7466–7472. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 41.Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E, Lo Y, Baribaud F, Mikami I, Reguart N, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 43.Bröet P, Camilleri-Broet S, Zhang S, Alifano M, Bangarusamy D, Battistella M, Wu Y, Tuefferd M, Régnard JF, Lim E, et al. Prediction of clinical outcome in multiple lung cancer cohorts by integrative genomics: implications for chemotherapy selection. Cancer Res. 2009;69:1055–1062. doi: 10.1158/0008-5472.CAN-08-1116. [DOI] [PubMed] [Google Scholar]
- 44.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002;1:599–606. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- 45.Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 46.Adjei AA. K-ras as a target for lung cancer therapy. J Thorac Oncol. 2008;3:S160–S163. doi: 10.1097/JTO.0b013e318174dbf9. [DOI] [PubMed] [Google Scholar]
- 47.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 48.Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci USA. 1999;96:13738–13743. doi: 10.1073/pnas.96.24.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan L, Findlay GM, Jones R, Procter J, Cao Y, Lamb RF. Hyperactivation of mammalian target of rapamycin (mTOR) signaling by a gain-of-function mutant of the Rheb GTPase. J Biol Chem. 2006;281:19793–19797. doi: 10.1074/jbc.C600028200. [DOI] [PubMed] [Google Scholar]
- 50.Osmundson EC, Ray D, Moore FE, Gao Q, Thomsen GH, Kiyokawa H. The HECT E3 ligase Smurf2 is required for Mad2-dependent spindle assembly checkpoint. J Cell Biol. 2008;183:267–277. doi: 10.1083/jcb.200801049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore FE, Osmundson EC, Koblinski J, Pugacheva E, Golemis EA, Ray D, Kiyokawa H. The WW-HECT protein Smurf2 interacts with the Docking Protein NEDD9/HEF1 for Aurora A activation. Cell Div. 2010;5:22. doi: 10.1186/1747-1028-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, Deng CX, Cheng SY, Zhang YE. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011;30:4777–4789. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nie J, Xie P, Liu L, Xing G, Chang Z, Yin Y, Tian C, He F, Zhang L. Smad ubiquitylation regulatory factor 1/2 (Smurf1/2) promotes p53 degradation by stabilizing the E3 ligase MDM2. J Biol Chem. 2010;285:22818–22830. doi: 10.1074/jbc.M110.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCFβ-TRCP ubiquitin ligase. Cancer Cell. 2010;18:147–159. doi: 10.1016/j.ccr.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohli L, Kaza N, Coric T, Byer SJ, Brossier NM, Klocke BJ, Bjornsti MA, Carroll SL, Roth KA. 4-Hydroxytamoxifen induces autophagic death through K-RAS degradation. Cancer Res. 2013;73:4395–4405. doi: 10.1158/0008-5472.CAN-12-3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med. 2012;18:227–234. doi: 10.1038/nm.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.