

Abstract
Immunofluorescent imaging has been a powerful technique in helping to identify intracellular nuclear and cytoplasmic molecules which are target antigens of autoantibodies in systemic autoimmune disorders. Patterns of staining can be correlated with molecules engaged in specific cellular functions and distributed in distinct cellular domains. Different autoimmune disorders have different profiles of autoantibodies, and immunodiagnostics has become an important adjunct in differential diagnosis. An important finding that has eluded explanation is the presence of autoantibodies to many different antigens, manifested strikingly in systemic lupus erythematosus. In cancer, the occurrence of autoantibodies to tumor-associated antigens is not uncommon and a characteristic feature is also the presence of multiple autoantibodies. The targeted tumor-associated antigens are either oncogene or tumor suppressor gene products or their coactivators, which are altered or mutated and driving the autoimmune response. Most cancer cells have between two and eight mutated genes before oncogenic transformation occurs, initiating a process called synthetic lethality in tumorigenesis pathways. These observations beg the question of whether there are similar mechanisms in systemic lupus erythematosus and other disorders driving autoimmunity pathways. Targeting molecules that are synthetic lethal to each other is in the forefront of the search for anticancer therapy, and this could also be an objective in systemic autoimmune disorders.
Introduction
Antinuclear antibodies (ANAs) have been used for several decades as diagnostic biomarkers and are involved in autoantibody-mediated immune complex inflammation in the kidney, lung, brain, skin, joints and many other organs [1,2]. Many of the major nuclear and cytoplasmic components which are the target antigens of these autoantibodies have been identified, but the reasons why these cellular components acquired immunogenicity and induced autoantibody formation are largely unknown. Elucidation of this enigma is arising from studies of autoantibodies to tumor-associated antigens (TAAs) in cancer, suggesting that, in addition to their known roles in diagnosis and pathogenesis, ANAs might be revealing the cellular components involved in autoimmunity pathways in the way that autoantibodies to TAAs inform on partners in tumorigenesis pathways.
Some unique features of antinuclear antibodies in systemic autoimmune disorders
The immunofluorescent imaging technique, using tissue culture cells such as HEp2 (an epithelial tumor cell line) as the substrate for reaction with autoimmune sera, has been an important technique for detecting ANAs. Figure 1 shows autoantibodies in systemic lupus erythematosus (SLE) binding to particles in the nucleoplasm of HEp2 cells, but no binding to any component in the cytoplasm. Subsequent studies have shown that this is a staining pattern characteristic of autoantibodies reacting with Sm antigen, a complex of small nuclear RNA and proteins called snRNPs, which are involved in the processing of precursor mRNAs to mature mRNAs [2]. Autoantibodies to Sm are unique to SLE. Figure 2 shows the reaction of another SLE autoantibody reacting with proliferating cell nuclear antigen in nuclei of cells that are in the S (DNA synthesis) phase of the cell cycle. The different sizes and densities of speckled staining relate to early or late stages of DNA synthesis [3]. There was at first some skepticism concerning the importance and significance of ANA staining patterns, but this was in large part due to lack of appreciation of the structure, function and location of intracellular micro-organelles.
Figure 1.
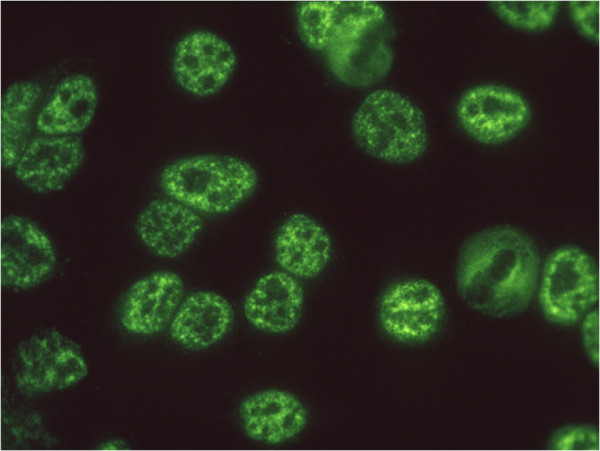
Immunofluorescence histochemistry depicting the Sm staining pattern on HEp2 cells using autoantibodies to Sm in the sera of patients with systemic lupus erythematosus. Sm antigen has been identified as a component of mRNA splicing particles distributed in the nucleoplasm in the size and location shown here. This pattern has been called nuclear speckles. The cytoplasm is nonreactive with the antibody and the nucleolar domains contain a few small speckles.
Figure 2.
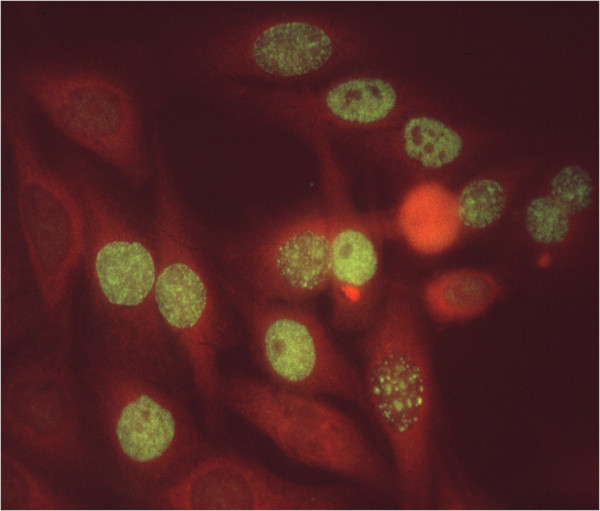
Immunofluorescence histochemistry depicting the proliferating cell nuclear antigen staining pattern. The antibody in this systemic lupus erythematosus serum reacts with proliferating cell nuclear antigen (PCNA), identified as an auxiliary protein of DNA polymerase delta that is involved in DNA synthesis. PCNA is seen to localize in nuclei that are in the S phase of the cell cycle, and the variation in size and density of speckled staining is related to early or late stages of the S phase. The nuclei of cells in interphase which are not in DNA synthesis do not react with the antibody. The cytoplasm was counterstained (orange) to delineate cytoplasm from the yellow–green speckled staining of PCNA particles in the nucleus.
Studies on ANAs spread quickly to other rheumatic autoimmune disorders and it became clear that some ANAs were highly specific and associated predominantly with one disease, such as autoantibodies to double-strand DNA and to Sm antigen in SLE, anti-DNA topoisomerase 1 and anti-centromere in scleroderma and the CREST syndrome, and anti-transfer RNA synthetases in dermato/polymyositis. Other ANAs such as anti-histones are present in several diseases, including SLE and rheumatoid arthritis. Nevertheless, combinations of ANAs with high specificity and others with lower specificity produced different ANA profiles that were useful in differential diagnosis of clinical disorders. Testing for ANAs is now a widely used tool in the diagnostic armamentarium of the rheumatologist.
In almost every patient with systemic autoimmune disease, there is multiplicity of autoantibodies present at the same time. In SLE, antibody to double-stranded DNA, anti-Sm and anti-histones might occur concurrently. In scleroderma, antibody to DNA topoisomerase 1 and anti-nucleolar antibodies are often present together. In many instances, autoantibodies of three or more specificities might be present. This phenomenon is an enigma that has not been elucidated.
Tumorigenesis pathways and autoimmunity pathways
One-third of patients with chronic hepatitis and liver cirrhosis eventually develop hepatocellular carcinoma. We examined serial serum samples and showed that autoantibodies were detectable in the pre-cancer period, but novel autoantibodies appeared with transformation to hepatocellular carcinoma. The molecular targets of these novel autoantibodies were identified as insulin-like growth factors [4,5], coregulators of oncogenes [6,7], or tumor suppressor genes [8,9]. Extension of such studies to other types of solid tumors showed frequent occurrence of autoantibodies to a number of cellular antigens that have been called TAAs. Other features include autoantibodies to multiple TAAs, with the majority of patients having two or more antibodies, a characteristic that recapitulates observations in systemic autoimmune diseases.
Recent studies on genetic abnormalities in cancer have helped in elucidating the immunological findings in cancer that might contribute to clarifying similar questions in systemic autoimmunity. In breast and colorectal cancers, a typical tumor contains two to eight driver-gene mutations that modulate or alter signaling pathways [10,11]. These driver genes include c-myc (an oncogene) and p53 (a tumor suppressor gene), and both gene products are TAAs and major targets of anti-TAA autoantibodies. To date, as many as 140 genes have been identified as driver genes, and mutations in several genes combine to confer growth advantage to cancer cells.
The concept of synthetic lethality in cancer is based on early observations in yeast and Drosophila that have been extended to higher organisms [12,13]. When two genes are synthetic lethal, mutation in one gene alone is nonlethal but simultaneous mutation in both genes is lethal. This concept has been expanded to include the situation called synthetic sickness/lethality. One example of the synthetic sickness/lethality concept is where dysfunction of the breast tumor suppressor gene BRCA1/2 is synthetically lethal with inhibition of the DNA repair enzyme poly(ADP-ribose) polymerase 1, and this combination of events leads to oncogenesis [13]. The implications of this concept explain why multiple genetic mutations are found in tumors because one mutation alone may not be lethal, and some multiple mutations may be synthetically lethal but others may not be synthetically lethal until the right combination occurs. Autoimmune response to the altered gene products would result in the production of autoantibodies.
Genetic studies in cancer have been highly productive partly because the target cell for analysis can be readily accessed. In systemic autoimmunity, such as SLE, the target cell that drives autoimmune responses is unknown. However, by making antibodies, the immune system is also reporting the identity of players in the pathway to autoimmunity [14]. There are many murine models of autoimmunity that would be useful in exploring potential autoimmunity pathways. In addition to advancing our knowledge of basic mechanisms in autoimmunity, there is the likelihood of developing an approach to therapy by targeting molecules that are synthetic sickness/lethal to each other. The latter approach is at the forefront of the search for anticancer therapy [15,16].
Note
This article is part of the collection Research through the eyes of pioneers. Other articles in this series can be found at http://arthritis-research.com/series/pioneers.
Box 1. About Eng M Tan
Eng M Tan is Professor Emeritus at the Scripps Research Institute in La Jolla, CA, USA. He received his MD degree from the Johns Hopkins University School of Medicine in Baltimore, MD and completed training in internal medicine at Duke University Hospital in Durham, NC, and Case Western University Hospitals in Cleveland, OH. His postdoctoral fellowship was in immunology at the Rockefeller University in New York, NY, under the mentorship of Dr Henry Kunkel. He has been on the faculty of Washington University School of Medicine in St Louis, MO, of University of Colorado in Denver, CO, and of the Scripps Research Institute in La Jolla, CA. His research interest has been in the field of autoimmunity with special focus on the nature of cellular antigens that are the targets of autoantibodies and on the use of autoantibodies as diagnostic biomarkers in clinical medicine. This interest has taken him from the study of autoantibodies in rheumatic autoimmune diseases such as systemic lupus erythematosus to autoantibodies in cancer and has shown how information from two different clinical fields mutually contribute to each other in further understanding of important pathogenic pathways in disease.
Abbreviations
ANA: Antinuclear antibody; SLE: Systemic lupus erythematosus; TAA: Tumor-associated antigen.
Competing interests
The author declares that he has no competing interests.
References
- Tan EM. Autoantibodies, autoimmune disease and the birth of immune diagnostics. J Clin Invest. 2012;122:3835–3836. doi: 10.1172/JCI66510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- Miyachi K, Fritzler MJ, Tan EM. Autoantibody to a nuclear antigen in proliferating cells. J Immunol. 1978;121:2228–2234. [PubMed] [Google Scholar]
- Zhang J-Y, Chan EKL, Peng X-X, Tan EM. A novel cytoplasmic protein with RNA-binding motifs is an autoantigen in human hepatocellular carcinoma. J Exp Med. 1999;189:1101–1110. doi: 10.1084/jem.189.7.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Weaver UM, Nielsen FC. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol Cell Biol. 1999;19:1261–1270. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Chan EKL, Kiyosawa K, Fu X-D, Tan EM. Novel nuclear autoantigen with splicing factor motifs identified with antibody from hepatocellular carcinoma. J Clin Invest. 1993;92:2419–2426. doi: 10.1172/JCI116848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta J, Fan G, Gelinas C. CAPER alpha is a novel Rel-TAD interacting factor that inhibits lymphocyte transformation by the potent Rel/NF-kB oncoprotein v-Rel. J Virol. 2008;82:10792–10802. doi: 10.1128/JVI.00903-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo Hoo L, Zhang JY, Chan EKL. Characterization of a novel 90 kDa ‘companion’ autoantigen of p62 overexpressed in cancer. Oncogene. 2002;21:5006–5015. doi: 10.1038/sj.onc.1205625. [DOI] [PubMed] [Google Scholar]
- Juntilla MR, Puustinen P, Niemala M, Ahola R, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, Lu SL, Lin S, Chan EK, Wang XJ, Grenman R, Kast J, Kallunki T, Sears R, Kahari VM, Westermarck J. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Sillman N, Szabo S, Dezso Z, Ustyansky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshae R, Willis J, Dawson D, Shipitsin M, Wilson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Brough R, Frankum JR, Costa-Cabral S, Lord CJ, Ashworth A. Searching for synthetic lethality in cancer. Curr Opin Gen Dev. 2011;21:34–41. doi: 10.1016/j.gde.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Tan EM, Zhang J-Y. Autoantibodies to tumor-associated antigens. Reporters from the immune system. Immunol Rev. 2008;222:328–340. doi: 10.1111/j.1600-065X.2008.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Jiang H, Hemann MT, Jaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112–3119. doi: 10.4161/cc.8.19.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]