

Abstract
Poly(N-isopropyl acrylamide) or pNIPAM is a thermoresponsive polymer that is widely studied for use in bioengineering applications. The interest in this polymer lies in the polymer’s unique capability to undergo a sharp property change near physiological temperatures, which aids in the spontaneous release of biological cells from substrates. Currently, there are many methods to deposit pNIPAM onto substrates, including atom transfer radical polymerization (ATRP) and electron beam ionization. Each method yields pNIPAM coated substrates with different surface characteristics which can influence cell behavior. In this work, we compare two methods of pNIPAM deposition: plasma deposition and co-deposition with a sol gel. The resulting pNIPAM films were analyzed for use as substrates for mammalian cell culture based on surface characterization (XPS, ToF-SIMS, AFM, contact angles), cell attachment/detachment studies, and an analysis of exocytosis function using carbon-fiber microelectrode amperometry (CFMA). We find that, although both methods are useful for the deposition of functional pNIPAM films, plasma deposition is much preferred for cell-sheet engineering applications due to the films’ thermoresponse, minimal change in cell density, and maintenance of supported cell exocytosis function.
INTRODUCTION
Stimuli-responsive polymers (SRP) are exploited by the biomedical community in tissue engineering,1 drug delivery,2 and biosensing.3 Specifically, poly (N-isopropyl acrylamide) (pNIPAM) is an SRP commonly used for biomedical applications and was the topic of the recent review by Cooperstein, et al.4 pNIPAM has the unique characteristic of being thermoresponsive near physiologically relevant temperatures. At 37°C, the polymer is relatively hydrophobic, and biological cells will readily grow on a substrate coated with pNIPAM. By decreasing the temperature beyond the lower critical solution temperature (LCST) to room temperature, the polymer becomes relatively hydrophilic. The polymer swells below the LCST, and adhered cells detach spontaneously in a confluent cell sheet.5 This polymer response has previously been used for a wide range of bioengineering applications, including tissue engineering6, 7 and protein separation.8, 9
Depending on the application, the method used to deposit the pNIPAM film may be altered to achieve the desired properties, as reviewed by Da Silva.10 For instance, to study interactions of bacteria with pNIPAM, many groups have used self-assembled monolayers (SAMs) of NIPAM.11 For microfiltration applications, pNIPAM has been deposited using atom transfer radical polymerization (ATRP).12 Although these methods produce uniform films, they are most efficient and predictable on a flat surface, such as a glass slide, and are dependent on surface chemistry to initiate deposition. Other groups have used electron beam ionization to deposit pNIPAM for tissue engineering applications.13 This method also requires a flat substrate for deposition. In addition, the film thickness of electron beam ionization deposition of pNIPAM must be controlled very carefully. When a film deposited using this method is too thick (>20 nm), the cells do not attach to the coated substrate, but if the film is too thin (<10 nm), cells do not detach.14
Although a variety of methods have been used to deposit pNIPAM, to our knowledge few direct comparisons have been made between these methods to determine which method is most ideal for use with mammalian cells. In this work, we compare two methods of deposition: plasma polymerization (ppNIPAM)15 and deposition of pNIPAM with a sol gel (spNIPAM).16 Previously, these techniques were optimized separately15, 16 to ensure that the ideal conditions for both were used to perform the comparisons made in the current work. We performed additional surface characterization including time-of-flight secondary ion mass spectroscopy (ToF-SIMS) and atomic force microscopy (AFM) to determine the source of the differences in these two deposition methods. Furthermore, we have also examined cell response to these surfaces using carbon-fiber microelectrode amperometry (CFMA) to assess exocytosis function. From surface characterization of the resulting substrates, we found that successful deposition of pNIPAM may be achieved using either deposition method, but AFM revealed a difference in topography that could explain why the cells responded differently depending on the deposition method. Amperometry studies indicate that mammalian cells grown on ppNIPAM behave more similarly to cells grown on uncoated glass, suggesting that ppNIPAM is more applicable for mammalian cell studies. From these results, we conclude that deposition of pNIPAM using plasma polymerization yields films that have the best thermoresponsive and mechanical properties, as well as minimal impact on cellular viability and function.
EXPERIMENTAL METHODS
Chemicals and supplies
Round glass cover slips were purchased from Ted Pella, Inc. (Redding, CA). The silicon chips were obtained from Silitec (Salem, OR). The 200 proof ethanol, HPLC grade methanol, HPLC grade dichloromethane and hydrochloric acid were purchased from Honeywell Burdick & Jackson (Deer Park, TX). The acetone was purchased from Fisher Scientific (Pittsburgh, PA). pNIPAM (molecular weight of ~40,000) for spNIPAM deposition was purchased from Polysciences, Inc (Warrington, PA). The tetraethyl orthosilicate (TEOS) for spNIPAM deposition was purchased from Sigma-Aldrich (St. Louis, MO). N-isopropyl acrylamide monomer (99%) for ppNIPAM deposition was purchased from Acros Organics (Geel, Belgium).
Surface preparation
Glass cover slips used for cell culture were cleaned with an acid wash (1:1 vol HCl:MeOH), rinsed with deionized water, and dried with nitrogen. Silicon chips used for surface analysis were washed with 10-minute intervals in dichloromethane, acetone, and then methanol in an ultrasonic cleaner from VWR International (Brisbane, CA).
spNIPAM preparation and deposition
Deposition of the spNIPAM films follows a previously described method.16 Briefly, 35 mg of pNIPAM, 5mL of distilled water, and 200 μL of 1 Normal HCl were mixed, and a weight percentage of pNIPAM was determined. In a separate container 250 μL TEOS sol (1 TEOS:3.8 ethanol:1.1 water:0.0005 HCl), 43 μL distilled water, 600 μL ethanol were mixed and weighed. To obtain 0.35wt% pNIPAM in sol, the appropriate amount of the pNIPAM solution was added to the TEOS solution in order to achieve the desired weight percentage (ex. 3.5g pNIPAM solution added to 996.5g TEOS sol).
100–200 μL of the prepared solution was evenly distributed on clean surfaces placed on a spin coater, model 100 spinner from Brewer Science, Inc. (Rolla, MO). These surfaces were then spun at 2000 rpm for 60s. The surfaces were stored under nitrogen in a Parafilm-covered Petri dish until either used for a) surface analysis or b) cell culture tests.
ppNIPAM preparation and deposition
Plasma polymerization was performed in a reactor chamber fabricated to our design specifications by Scientific Glass (Albuquerque, NM) following a method previously described.15 To spark a plasma in the chamber, two 2.5 cm copper electrodes were connected to a Dressler (Stolberg, Germany) matching network and Cesar radio frequency (RF) power generator from Advanced Energy (Fort Collins, CO). Ar and CH4 adhesion promoting layers were deposited before pNIPAM deposition. During pNIPAM deposition, the power setting of the RF generator was slowly decreased from 100W to 0W over 35 minutes.
After the samples were removed from the reactor, they were rinsed with cold DI water in order to remove any uncrosslinked NIPAM from the surface, and dried with nitrogen. The surfaces were placed in a Petri dish, backfilled with nitrogen, and sealed with Parafilm. The samples were then stored in a desiccator until surface analysis or cell culture was performed.
Contact angle
An Advanced Goniometer model 300-UPG (ramé-hart instrument co., Mountain Lakes, NJ) with an environmental chamber and DROPimage Standard program was used to measure inverted bubble contact angles in Ultrapure water (18 M-Ohm-cm). Contact angles were taken at room temperature and 37°C using the Temp Controller model 100–500 connected to the environmental chamber. Contact angles were obtained at room temperature (vs. 4°C, which was the temperature used for cell detachment), as it is experimentally simpler to perform the experiments at room temperature; furthermore, we previously demonstrated that the degree of thermoresponse was unchanged between these two temperatures < the LCST.1 Statistically relevant data was obtained by repeating all procedures three times, with each replicate using three surfaces and each surface analyzed at three different sites along the surface.
X-ray photoelectron spectroscopy (XPS)
All XPS were obtained using a Kratos Axis-Ultra DLD spectrometer. This instrument has a monochromatized Al Kα X-ray and a low energy electron flood gun for charge neutralization. X-ray spot size for these acquisitions was on the order of 300×700μm (Kratos ‘Hybrid’ mode). Pressure in the analytical chamber during spectral acquisition was ~5 × 10−9 Torr. Pass energy for survey spectra was 80 eV and the pass energy was 20 eV for high-resolution spectra (carbon).
Data treatment was performed on CasaXPS software (Manchester, UK). Core-level spectra were peak fit using the minimum number of peaks possible to obtain random residuals. A 70% Gaussian/30% Lorentzian line shape was used to fit the peaks, and a linear function was used to model the background.
Time-of-flight secondary ion mass spectroscopy (ToF-SIMS)
ToF-SIMS spectra were acquired on an IonTof ToF-SIMS 5 spectrometer using a 25 keV Bi+ ion source in the pulsed mode. Spectra were acquired for positive secondary ions over a mass range of m/z = 0 to 500. The ion source was operated with a current of 0.35 pA. Secondary ions of a given polarity were extracted and detected using a reflectron time-of-flight mass analyzer. Spectra were acquired using an analysis area of 0.01 mm2. Positive ion spectra were calibrated using the CH3+, C2H3+, C3H5+, and C7H7+ peaks. Calibration errors were kept below 10 ppm. Mass resolution (m/Δm) for a typical spectrum was between 8000 to 10,000 for m/z = 27 (pos).
Principle Component Analysis (PCA)
PCA was performed by used PLS Toolbox version 2.0 (Eigenvector Research, Manson, WA) for MATLAB (Math-Works, Inc., Natick, MA). All spectra were mean-centered before running PCA. Although a detailed description of PCA is not warranted here, the interested reader is referred to the more complete discussion of PCA by Jackson.17 Briefly, a “complete” peak set was constructed by using all of the major peaks. Selected peaks were then normalized to the total ion intensity to account for the fluctuations in secondary ion yield between different spectra. PCA was then used to capture the linear combination of peaks that described most of the variation within the data set. From this, an output of both a “scores” and a “loadings” plot was obtained.
Atomic Force Microscopy (AFM)
Force measurements and imaging were performed with a Veeco Nanoscope IIIa controller (Plainview, NY) and J type scanner. An O-ring and fluid cell containing the AFM cantilever was then set on top of the sample. Degassed nanopure water was injected into the fluid cell and the film was allowed to equilibrate with the water for 30 minutes. The temperature was controlled with infrared heat lamps directed at the AFM. The entire apparatus was then placed in a Plexiglas enclosure on an isolation setup. There was a minimum of thirty minutes between temperature changes. Data was collected with Veeco software version 6.1 (Plainview, NY). A silicon nitride cantilever, MSCT-UNM Veeco Probes, with a spring constant of 0.02 N/m was used for all force-distance and imaging results.
Mammalian cell culture
Immortal bovine aortic endothelial cells (BAECs) were purchased from Genlantis (San Diego, CA) and cultured in Dulbecco’s modified eagle medium (MEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 4.5 g/L glucose, 0.1 mM MEM nonessential amino acids, and 1 mM MEM sodium pyruvate. The cell culture media was from Hyclone (Logan, UT) and cells were cultured in 6 well tissue culture polystyrene (TCPS) plates from Greiner Bio-one (Monroe, NC). The DPBS without calcium-chloride or magnesium-chloride was purchased from HyClone (Logan, UT). The 0.25% trypsin-EDTA was from Gibco (Carlsbad, CA). Cells were incubated at 37°C in a humid atmosphere with 5% CO2. Cells were washed with Dulbecco’s phosphate buffered saline and lifted from culture flasks with 0.25% trypsin to seed 6 well plates with inserted pNIPAM deposited and blank control cover glass.
Primary culuture murine adrenal medullary chromaffin cells (MAMCs) were also used in this work. All mouse handling was done in accordance with the University of Minnesota Institutional Animal Care and Use Committee practices established under approved protocol #0509A75006. Primary culture MAMCs were harvested from wild-type brown male mice (C57BL/6J, Jackson Laboratories, Bar Harbor, ME) less then 4 months of age, as previously described.18 Briefly, following euthanasia both adrenal glands were located and excised; then, cortical tissue was removed to reveal the medullary tissue. The medullary tissue was digested with neutral protease (Worthington Biochemical Corporation, Lakewood, NJ) for 30 minutes, rinsed with DMEM/F12 media (Hyclone, Logan, UT), triturated to create a cell suspension and then plated on coated or uncoated coverslips in 35mm petri dishes. Cells were incubated at 37°C in a 5% CO2 atmosphere for 24 hours. At the time of CFMA examination, media was replaced with warm Tris buffer, kept at 37°C using a temperature controller, and plates were examined within two hours of removal from the incubator.
Cell attachment/detachment
Studies of BAECs adhesion to and release from the pNIPAM films follows a previously described method. Briefly, BAECs were cultured until confluence (approximately 4 days for these substrates).19 The medium was removed, and 4°C serum-free media was added to each well, since a previous investigation indicated this facilitated the fastest release from pNIPAM. The culture plate was then placed on a shaker platform for 2 hours and observed via light microscopy (Nikon F100, Melville, NY) with a 20x objective to determine the percentage of cells detached. Images were captured using Spot Advanced software (Sterling Heights, MI).
Carbon-fiber Microelectrode Amperometry (CFMA)
Carbon-fiber microelectrodes and stimulating pipettes were fabricated in-house, following previously published methods.20 Briefly, for microelectrodes, a single carbon fiber (Amoco, Greensville, SC) was aspirated into a glass capillary (AM Systems, Carlsberg, WA). It was then pulled, trimmed, epoxied (Epoxy Technology, Billerica, MA) and cured. Electrodes were beveled at 45 degrees using a BV-10 microelectrode beveler (Sutter Instruments, Novato, CA) and soaked in isopropyl alcohol saturated with activated carbon for at least 10 minutes prior to use.
Experiments were performed, as described previously,18, 21–23 on a Nikon® Eclipse TE2000U inverted microscope (Nikon USA, Melville, NY) equipped with Burleigh PCS500 piezoelectric micromanipulators (EXFO Photonics Solutions Inc, Mississauga, Onterio). Cell media was removed from the petri dish, cells were washed and media was replaced with warmed Tris buffer (300 mM sodium chloride, 12.5 mM trishydroxymethylaminomethane, 8.4 mM potassium chloride, 5.6 mM R-D-glucose, 4.5mM calcium chloride, and 4.2 mM magnesium chloride). The temperature of the dish was maintained at 37°C with a TC-324B single channel temperature controller (Warner Inst, Hamden, CT) throughout experiments. Micromanipulators were used to position the microelectrode on the surface of the cell with visual confirmation that the cell swelled slightly. The stimulating pipette was positioned approximately 30 to 50 micrometers from the cell being examined, and exocytosis was stimulated using a 3-second-bolus of 60 mM K+, delivered using a 1.5 psi nitrogen pulse (as depicted in Figure 1). The microelectrode was held at +700 mV vs Ag/AgCl reference electrode to oxidize only secreted epinephrine/norepinephrine while current was monitored. Recording began 3 seconds prior to stimulation of exocytosis and data was collected for a total of 30 seconds. The current recording was obtained using an Axopatch™ 200B potentiostat (Molecular Devices, Inc., Sunnyvale, CA) where oxidation current was low-pass Bessel filtered (5 kHz) and amplified (20 mV/pA). This was collected using a home-built breakout box in combination with Tarheel Electrochemistry software run in LabVIEW™ (National Electrochemistry Suite software module in LabVIEW, National Instruments, Austin, TX). Captured data was then filtered digitally (500 Hz) and every current spike with an area greater than 60 fC within each trace was analyzed using MiniAnalysis software (Synaptosoft Inc., Fort Lee, NJ). Exocytosis release (shown in Figure 1) was monitored from multiple cells in a dish within two hours of removal from incubation conditions, which typically resulted in traces from 5 to 15 cells per culture dish.
Figure 1.
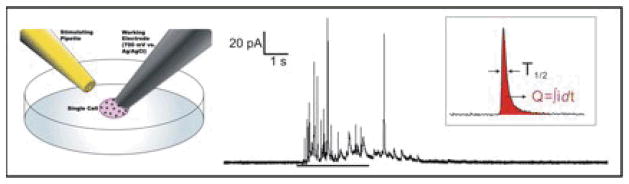
Schematic of a carbon-fiber microelectrode amperometry setup (left), a typical CFMA trace (right) and two CFMA spike parameters (inset). CFMA was used to examine functional changes in exocytosis after culture on pNIPAM surfaces. A simplified CFMA setup is presented at left, (not to scale; reference electrode not shown) with a stimulating pipette (gold) placed near the cell of interest and a carbon fiber microelectrode (grey) placed on the surface of a single cell. A typical CFMA trace is presented at right for a cell from the glass only control. The small bar is the period of stimulation (using 3 second K+ bolus) to induce exocytosis. Individual spikes parameters shown inset, including charge (Q) and full-width half-maximum (T1/2), are calculated, pooled and averaged for each condition. Comparison of these parameters revealed changes in exocytosis in a deposition dependent manner.
The average release characteristics for each trace were determined, focusing on the spike area (Q), spike full-width at half maximum (T1/2) and spike frequency. For each trace, all individual spikes were evaluated for these parameters and then each trace average was pooled to determine the experimental condition average, which is reported for Q, T1/2 and frequency. Average spike parameters for traces from individual cells (n ≥ 28) were assembled for a given experimental condition to determine the experimental average and the standard error of the mean (SEM). For experiments conducted on different days (done in triplicate), data was combined for pooled conditions where no significant difference was found between control conditions on separate days. Statistical significance was examined in Microsoft Excel (Microsoft Inc., Seattle, WA) using the 2-tailed student’s t-test (assuming equal variance, α= 0.05). All data was converted to log values and outliers were removed if they were found to be more than two log standard deviations from the log mean for that condition. The average cell release parameters for each experimental condition were then evaluated for significant differences using the 2-tailed student’s t-test as described above.
CFMA reveals a wealth of information about cellular exocytosis from individual spike characteristics. These spike characteristics can be understood in terms of biophysical properties of exocytosis, from the years of study with this method.24–31 In order to compare the effects of surface deposition methods, these types of exocytosis parameters and the physical properties behind them can reveal surface-related changes in cellular function. While numerous spike characteristics can be examined, this research focuses on the spike frequency, spike half-width or full-width at half-maximum (T1/2) and overall spike charge (Q). These three characteristics describe the rate of successful exocytosis events, chemical messenger exchange rate between the vesicle and extracellular space, and the number of molecules released from a vesicle (as calculated from Faraday’s law, shown in equation 1, where Q is the charge, n is the number of electrons in the oxidation (2 for epinephrine), N is the number of moles of epinephrine secreted and F is Faraday’s constant).
| Equation 1 |
By examining these three specific characteristics, average Q, T1/2, and frequency, one can determine if there are alterations to normal cell signaling via exocytosis, after exposure to coated surfaces fabricated using various deposition methods. This sensitive measurement of a critical, and highly conserved, cell function in primary culture cells will reveal a nuanced view of how mammalian cells interact with and are influenced by the pNIPAM coatings.
RESULTS AND DISCUSSION
pNIPAM has many potential applications, thus there are many methods of deposition used to tailor the coating to each use. For example, SAMs and ATRP deposition methods are used for bacterial studies, while electron beam ionization is used for tissue engineering. Each of the aforementioned techniques results in different surface characteristics. For instance, the change in wettability of these pNIPAM surfaces yield contact angles between 5°-65°.32–34 Since differences in surface characteristics can influence cell attachment, proliferation, and release,35 a multiple surface analysis approach was used to compare the two deposition methods of interest, followed by cellular response studies, to verify that these surfaces are biocompatible and support the maintenance of normal cellular functions.
Previous work has investigated two promising pNIPAM deposition methods, including plasma polymerization (ppNIPAM)15 and deposition of pNIPAM with a sol gel (spNIPAM).16 Using these two methods, we have seen that there are distinct differences in the cell response to the substrate coatings, and hypothesize that pNIPAM deposition must not only be altered to fit the substrates and surface chemistries necessary for the desired application, but also to obtain the least altered cell response. To test this hypothesis, this work compared the two methods currently used to deposit pNIPAM for applications using mammalian cells.
Surface characterization
XPS was used to ensure that pNIPAM was successfully deposited, regardless of deposition method. The presence of nitrogen in the elemental XPS data (see Figure 2) demonstrates that both ppNIPAM and spNIPAM deposition methods resulted in pNIPAM substrates. The ppNIPAM substrates more closely mimicked the theoretical relative atomic percentages that were determined from the monomer structure. However, this is mainly due to the large presence of silicon in the spNIPAM surfaces, since this method requires the use of a silicon-based sol gel (TEOS). The amide (288 eV) and amine (286 eV) peaks on the XPS high-resolution C1s spectra also indicated pNIPAM deposition (see Figure 3). Unlike in the elemental XPS data, the high-resolution C1s spectra of spNIPAM and ppNIPAM were very similar, thus supporting the fact that the relative atomic percentages for spNIPAM in Figure 2 were different from ppNIPAM and theoretical values due to the Si peak from the sol gel.
Figure 2.
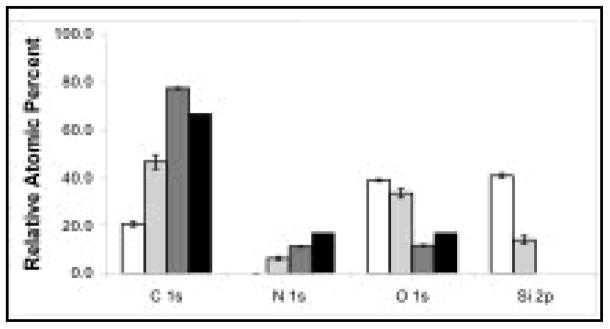
XPS elemental analysis of ppNIPAM (dark grey), spNIPAM (light grey), and blank Si controls (white). The presence of N1s on the ppNIPAM and spNIPAM surfaces indicates successful deposition of pNIPAM. When compared to the theoretical composition determined from the NIPAM monomer (black), ppNIPAM is most similar.
Figure 3.

XPS high resolution C1s spectra of a blank Si control (top), spNIPAM (middle), and ppNIPAM (bottom) films. Presence of amide and amine peaks confirm pNIPAM depostion using both ppNIPAM and spNIPAM deposition methods.
ToF-SIMS was used to further characterize the surfaces. While XPS can determine the molecular bonding environments present and the elemental make up of a substrate, ToF-SIMS is useful for looking at the molecular fragments present. However, due to the complex data sets that ToF-SIMS returns, it is common to use PCA to analyze the data (see Figure 4). The PC2 scores plot shows that there is a distinct difference between the two deposition methods. However, as was shown with XPS, the difference is primarily due to the TEOS fragments. There are pNIPAM fragments in both the ppNIPAM and spNIPAM substrates. There is more intact monomer in the spNIPAM surfaces (MW=114), while the ppNIPAM substrates tend to have more fragments with high molecular weight. The plasma used to deposit the ppNIPAM is high energy, thus resulting in larger fragments than those from the spNIPAM substrates where a pre-polymerized pNIPAM was used.
Figure 4.

PCA scores (top) and loadings (bottom) of ToF-SIMS data collected from ppNIPAM (triangles) and spNIPAM (asterisks) films. There is a distinct separation between the data obtained from samples prepared by the two deposition methods, which is due to the silicon based sol gel (TEOS) used for spNIPAM deposition.
Thermoresponse
Contact angles were used to determine if there was a change in wettability above and below the LCST (see Table 1). Both spNIPAM and ppNIPAM surfaces demonstrated a change in hydrophobicity when the temperature was shifted from 37°C to 25°C. There was a larger shift in the ppNIPAM substrates (19 ± 10°) than in the spNIPAM substrates (13 ± 8°), which suggests more efficient detachment when using the ppNIPAM substrates, a property that may be beneficial when working with mammalian cells.
Table 1.
Contact Angle Measurements
Contact angles for ppNIPAM, spNIPAM, and blank Si control surfaces, obtained above (right) and below (left) the LCST. Both ppNIPAM and spNIPAM demonstrated thermoresponse. There was no observable change on the Si controls.
| 25 °C | 37 °C | |
|---|---|---|
| Blank Si control | 43±2 | 44±2 |
| spNIPAM | 49±6 | 62±5 |
| ppNIPAM | 24±4 | 43±9 |
Further investigation of thermoresponse showed a difference in the topography of the surfaces above and below the LCST as revealed by AFM analysis (see Figure 5). Above the LCST, at cell culture temperature, both types of surfaces are relatively smooth. Obviously, the spNIPAM surfaces that were spun cast onto substrates are rougher than the plasma deposited surfaces due to the manner of deposition. However, when the polymer swells below the LCST, there are significant differences in the topography depending on the deposition method. The spNIPAM surfaces appear to have large islands of aggregated and swollen pNIPAM that create ~250 nm features, with an RMS of 6.8 ± 1.8. The ppNIPAM surfaces remain relatively flat, with only ~12 nm features, and an RMS of 1.1 ± 0.1. This is due to the fact that the ppNIPAM surfaces are entirely pNIPAM that is tethered to the substrate, and the spNIPAM has a copolymer that the pNIPAM is separating from upon the change in temperature.
Figure 5.
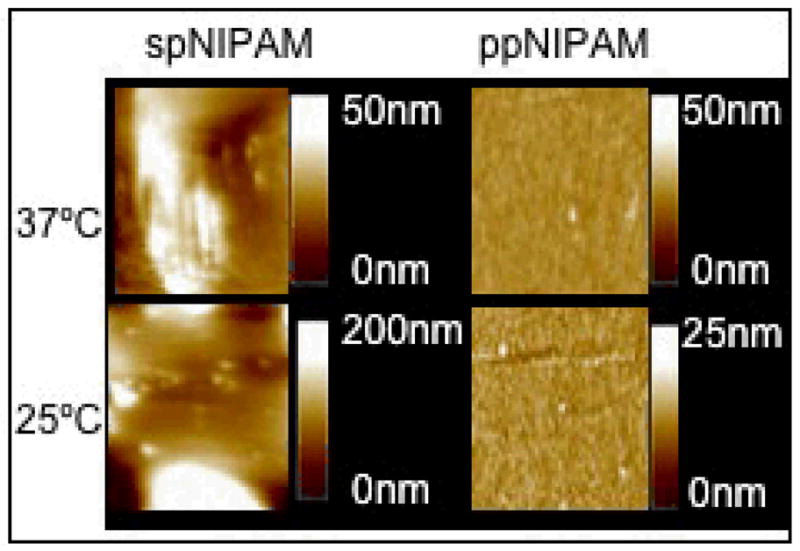
AFM images of ppNIPAM (left) and spNIPAM (right) substrates imaged in water above (top) and below (bottom) the LCST. At 37°C, the surfaces are relatively smooth. However, when the temperature is shifted below the LCST, spNIPAM surfaces are extremely rough.
Mammalian Cell Culture
BAECs were cultured and proliferated on bare glass, spNIPAM and ppNIPAM substrates. MAMCs were unable to be used for cell detachment studies, as they are primary culture cells that do not proliferate in culture. Previous work suggests that cell-cell interactions have been shown to assist in cell release from pNIPAM.36 Upon changing the temperature to below the LCST, the cells from the pNIPAM coated surfaces did begin to detach from the surfaces; however, the form of detachment varied depending on the deposition method (see Figure 6). The spNIPAM surfaces released in small aggregates of cells, consistent with the topographical changes seen using AFM where only large islands on the surface swelled. The ppNIPAM surfaces release as full cell sheets, with only a few cells remaining on the surfaces. The uniformity in surface topography along with the larger shift in contact angles likely determined the success in cell detachment seen using this deposition method.
Figure 6.

Bright field microscopy images of BAECs cultured on spNIPAM (bottom), ppNIPAM (middle), and blank glass control (top) surfaces. The cells attached and proliferated above the LCST (left) on all surfaces. After the temperature was shifted to 4°C (right), as expected there was no cell release from the control surfaces. There was complete cell detachment from ppNIPAM surfaces as a sheet, but aggregated clumps of cells detached from spNIPAM films. (Black scale bars = 100μm)
CFMA exocytosis response
CFMA was used to assess the influence of pNIPAM-coated surfaces on normal critical cell function in primary culture cells. This method gives both quantitative and biophysical mechanistic insight into any deposition-related changes in real-time delivery of chemical messengers. Three main amperometric spike characteristics were used to examine the effects of surface coating on the maintenance of cellular exocytosis function. The average Q, related to the number of molecules released, informs us about the magnitude of signaling response while the average T1/2, or full-width at half-maximum, informs us about the kinetics of exchange between the vesicle and extracellular space (shown in Figure 1). These two features provide insight into specific components of exocytosis, while the average frequency reveals insight into the overall number of successful exocytosis events. Any perturbations in the spike frequency would suggest that pNIPAM exposure impacts internal components of the exocytosis machinery, such as the trafficking of vesicles via the actin/myosin cytoskeleton network. Herein, the results indicate that these surface coatings do not have an impact in this fashion, as there were no perturbations in the average spike frequency for any of the coating conditions (p>0.05, data not shown). Despite this result, cells were found to have altered exocytosis function after 24 hours of culture on both types of pNIPAM surfaces, as shown in Figure 7.
Figure 7.
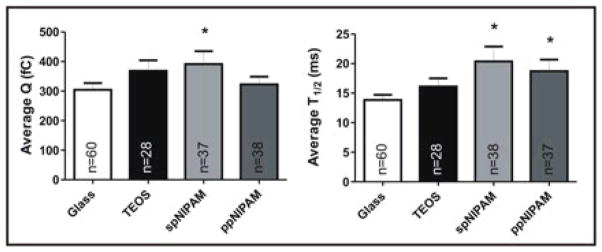
CFMA results from cells cultured on glass (white) and TEOS (black) control substrates, as well as spNIPAM (light grey) and ppNIPAM (dark grey) substrates. Perturbations in exocytosis for average charge (Q) and spike half-width (T1/2) were revealed to change with surface deposition method, with more perturbations for the spNIPAM deposition. Statistical significance is denoted with * as calculated using a students t-test, where p was < 0.05.
In both conditions where pNIPAM surfaces were used, there was a corresponding increase in the spike T1/2, 35 and 47% increase for plasma or spin coated surfaces, respectively, indicating that exchange between the vesicle and extracellular space was slowed (p<0.05). As this process gives insight into the release kinetics of the matrix unfolding within the average vesicle, it appears that matrix expansion is inhibited by the presence of the pNIPAM coating, yielding slower release of vesicular content into the extracellular space. As the exchange between vesicles and extracellular space is driven by a variety of forces, including an osmolarity gradient, the presence of a surface coating is likely to perturb this local environment and thus the driving forces of exchange. This was compared to a TEOS only-coated control, where an increase in T1/2 was not seen, indicating that this alteration was specifically a result of the presence of a pNIPAM coating (p>0.05).
Additionally, in the case of cells incubated on spNIPAM, there was also an impact on the average spike area, which is related to the total number of molecules released (see Average Q in Figure 7). The presence of the spNIPAM coating lead to an increase in the number of molecules secreted from the average vesicle, going from 960,000 to 1.3 × 106 molecules, a 35% increase (p<0.05). Considering that there are approximately 22,000 vesicles within a single chromaffin cell, if they all released at this augmented level, a single cell would be releasing 4.5 × 109 more chemical messenger molecules. For tissue engineering applications, this type of hyper-activated state and exaggerated release could lead to dire consequences for the resultant tissues. For example, if instead of MAMCs the engineered tissue contained cells secreting a neurotransmitter like histamine, hundreds to thousands of cells releasing 35% more molecules of histamine would certainly be detrimental to normal growth in this pro-inflammatory state.
These CFMA results suggest that pNIPAM coatings do have effects on the maintenance of normal cell functions during mammalian cell culture in a deposition-dependant manner. During CFMA studies, it was also clear that there were qualitatively fewer cells in the TEOS only (control) and spNIPAM conditions. This suggests that while both deposition methods can support cell culture, ppNIPAM surfaces allow more normal cells to grow while having a smaller impact on each cell’s function.
CONCLUSIONS
In this work, we investigated the differences in surface properties and cellular response of two pNIPAM deposition methods. Using a multi-technique approach, including XPS and ToF-SIMS, we analyzed the surface chemistry of films deposited using both deposition methods, demonstrating successful deposition of pNIPAM. In addition, using AFM and contact angle measurements, we demonstrated that thermoresponse was maintained. Topographical differences in the surfaces showed that, although both spNIPAM and ppNIPAM-coated substrates were relatively smooth above the LCST, there was significant roughness on spNIPAM substrates below the LCST. Finally, as these surfaces were primarily fabricated for mammalian cell studies, we analyzed cell attachment, proliferation, detachment, and critical cell exocytosis function. We found that cells did proliferate on surfaces coated using both methods of deposition, with the most cell detachment from the ppNIPAM surfaces. Additionally, although cells grow on pNIPAM-coated surfaces obtained from both methods, there were significant changes in the cell densities and perturbations in cellular communications (as measured using CFMA) on spNIPAM surfaces. Overall, cells cultured on ppNIPAM surfaces provided cellular responses, including both cell survival and function, most similar to cells cultured on uncoated glass.
From these results, it is clear that although pNIPAM can be successfully deposited using different techniques and maintain thermoresponse, the deposition method influences coating uniformity and behavior which, in turn, determine which deposition method is appropriate for the desired application. For instance, although both spNIPAM and ppNIPAM substrates both successfully release mammalian cells, for applications such as cell sheet engineering, cell culture using ppNIPAM substrates would be preferable, because maintenance of normal cellular function is more successful using ppNIPAM surfaces. In contrast, spNIPAM is a simple, inexpensive method of deposition that may be more appropriate for applications not requiring confluent (and unperturbed) cell sheets, such as protein separation. In summary, the work reported herein demonstrates that plasma deposition of pNIPAM is most useful for any application requiring an even topography, similar response across the substrate, and/or cells with minimal functional perturbations.
Acknowledgments
The authors would like to thank Brett Andrzejewski, Kateryna Artyushkova, and Steven Candelaria from the University of New Mexico as well as Lara Gamble and Jim Hull from the National ESCA and Surface Analysis Center for Biomedical Problems (NESAC/BIO) for data acquisition, contributions, and helpful discussions.
Financial support of this work was generously provided by separate 3M Non-Tenured Faculty Grants to C.L.H. at UMN and H.E.C. at UNM; grants from the National Science Foundation (No. CHE-0645041 at UMN, and DMR-0611616 at UNM); a grant from the National Institutes of Health to UW (NIBIB grant EB-002027), as well as the UNM Center for Biomedical Engineering. J.A.R. was funded by the NSF Graduate Research Fellowship Program.
References
- 1.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 2.Lopez VC, Raghavan SL, Snowden MJ. Reactive Func Poly. 2004;58:175–185. [Google Scholar]
- 3.Gerlach G, Guenther M, Suchaneck G, Sorber J, Arndt KF, Richter A. Macromol Symp. 2004;210:403–410. [Google Scholar]
- 4.Cooperstein MA, Canavan HE. 2010;26:7695–7707. doi: 10.1021/la902587p. [DOI] [PubMed] [Google Scholar]
- 5.Canavan HE, Cheng XH, Graham DJ, Ratner BD, Castner DG. J Biomed Mater Res Part A. 2005;75A:1–13. doi: 10.1002/jbm.a.30297. [DOI] [PubMed] [Google Scholar]
- 6.Okano T, Yamada N, Sakai H, Sakurai Y. 1993;27:1243–1251. doi: 10.1002/jbm.820271005. [DOI] [PubMed] [Google Scholar]
- 7.Canavan HE, Cheng XH, Graham DJ, Ratner BD, Castner DG. Plasma Proc Poly. 2006;3:516–523. [Google Scholar]
- 8.Turan E, Ozcetin G, Caykara T. Macromole Biosci. 2009;9:421–428. doi: 10.1002/mabi.200800273. [DOI] [PubMed] [Google Scholar]
- 9.Lin P, Feng JJ, Wang YL, Xie FJ, Wang WG, Chen Y, Huang LQ. Acta Chim Sinica. 2009;67:2703–2708. [Google Scholar]
- 10.Da Silva RMP, Mano JF, Reis RL. Trends Biotechnol. 2007;25:577–583. doi: 10.1016/j.tibtech.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Ista LK, Mendez S, Lopez GP. 2010;26:111–118. doi: 10.1080/08927010903383455. [DOI] [PubMed] [Google Scholar]
- 12.Lin F, Li Q, Jiang DB, Yu XH, Li L. Iranian Poly J. 2009;18:561–568. [Google Scholar]
- 13.Idota N, Tsukahara T, Sato K, Okano T, Kitamori T. Biomater. 2009;30:2095–2101. doi: 10.1016/j.biomaterials.2008.12.058. [DOI] [PubMed] [Google Scholar]
- 14.Akiyama Y, Kikuchi A, Yamato M, Okano T. 2004;20:5506–5511. doi: 10.1021/la036139f. [DOI] [PubMed] [Google Scholar]
- 15.Lucero AE, Reed JA, Wu X, Canavan HE. Plasma Proc Poly. 2010;7:992–1000. doi: 10.1002/ppap.201000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reed JA, Lucero AE, Hu S, Ista LK, Bore MT, Lopez GP, Canavan HE. ACS Appli Mater Interfaces. 2010;2:1048–51. doi: 10.1021/am900821t. [DOI] [PubMed] [Google Scholar]
- 17.Jackson JE. J Qual Technol. 1980;12:201–213. [Google Scholar]
- 18.Love SA, Haynes CL. Anal Bioanal Chem. 2010 doi: 10.1007/s00216-010-3735-3. accepted. [DOI] [PubMed] [Google Scholar]
- 19.Reed JA, Lucero AE, Cooperstein MA, Canavan HE. J Appl Biomater Biomech. 2008;6:81–88. [PMC free article] [PubMed] [Google Scholar]
- 20.Kawagoe K, Zimmerman J, Wightman R. J Neurosci Meth. 1993;48:225–240. doi: 10.1016/0165-0270(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 21.Marquis B, Haynes CL. Biophys Chem. 2008;137:63–69. doi: 10.1016/j.bpc.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Marquis B, Maurer-Jones M, Braun K, Haynes C. Analyst. 2009 doi: 10.1039/b913967b. accepted. [DOI] [PubMed] [Google Scholar]
- 23.Marquis B, McFarland A, Braun K, Haynes C. Anal Chem. 2008;80:3431–3437. doi: 10.1021/ac800006y. [DOI] [PubMed] [Google Scholar]
- 24.Clark R, Ewing A. Mol Neurobiol. 1997;15:1–16. doi: 10.1007/BF02740612. [DOI] [PubMed] [Google Scholar]
- 25.Grabner C, Fox A. J Neurophysiol. 2006;96:3082–3087. doi: 10.1152/jn.00017.2006. [DOI] [PubMed] [Google Scholar]
- 26.Amatore C, Arbault S, Bonifas I, Bouret Y, Erard M, Ewing A, Sombers L. 2005;88:4411–20. doi: 10.1529/biophysj.104.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amatore C, Arbault S, Bonifas I, Guille M. 2009;143:124–31. doi: 10.1016/j.bpc.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Amatore C, Arbault S, Bonifas I, Guille M, Lamaitre F, Verchier Y. 2007;129:181–9. doi: 10.1016/j.bpc.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Schroeder TJ, Jankowski JA, Kawagoe K, Wightman R, Lefrou C, Amatore C. Anal Chem. 1992;64:3077–3083. doi: 10.1021/ac00048a003. [DOI] [PubMed] [Google Scholar]
- 30.Sombers L, Maxson M, Ewing A. J Neurochem. 2005;93:1122–1131. doi: 10.1111/j.1471-4159.2005.03087.x. [DOI] [PubMed] [Google Scholar]
- 31.Travis E, Wightman R. Annu Rev Biophys Biomol Struct. 1998;27:77–103. doi: 10.1146/annurev.biophys.27.1.77. [DOI] [PubMed] [Google Scholar]
- 32.Cheng XH, Canavan HE, Stein MJ, Hull JR, Kweskin SJ, Wagner MS, Somorjai GA, Castner DG, Ratner BD. 2005;21:7833–7841. doi: 10.1021/la050417o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamirisa PA, Koskinen J, Hess DW. 2006;515:2618–2624. [Google Scholar]
- 34.Teare DOH, Barwick DC, Schofield WCE, Garrod RP, Beeby A, Badyal JPS. J Phys Chem B. 2005;109:22407–22412. doi: 10.1021/jp052869f. [DOI] [PubMed] [Google Scholar]
- 35.Boontheekul T, Hill EE, Kong HJ, Mooney DJ. Tissue Eng. 2007;13:1431–1442. doi: 10.1089/ten.2006.0356. [DOI] [PubMed] [Google Scholar]
- 36.Okano T, Yamada N, Okuhara M, Sakai H, Sakurai Y. Biomater. 1995;16:297–303. doi: 10.1016/0142-9612(95)93257-e. [DOI] [PubMed] [Google Scholar]