

Abstract
Pathway analysis is important in interpreting the functional implications of high-throughput experimental results, but robust comparison across platforms and species is problematic. A new approach, Pathprinting, provides a cross-platform, cross-species comparative analysis of pathway expression signatures. This method calculates pathway-level statistics from gene expression across nearly 180,000 microarrays in the Gene Expression Omnibus. Pathprinting can accurately retrieve phenotypically similar samples and identify sets of human and mouse genes that are prognostic in cancer.
See related Research paper, http://genomemedicine.com/content/5/7/68
Pathway analysis methods in transcriptomics
Over the past two decades, microarray technologies have been used to characterize gene expression in various contexts, notably complex human disease and corresponding animal models. Many, perhaps most, analyses could be enriched by comparison with other experiments across species and platforms. However, experimental and platform biases tend to drown out changes in biological signal [1], and comparison across species presents a further challenge: it is difficult to accurately identify orthologs. Aggregating gene sets based on function is known to improve consistency [2], but fewer than half of human genes are represented in pathway databases. In a recent article published in Genome Medicine [3], Winston Hide and colleagues describe Pathprinting, a statistics-based approach to map gene expression to function in humans and the main animal models of disease (mouse, rat, zebrafish, fruit fly and nematode). By standardizing pathway analysis, basing it more globally across functional interactions and controlling for biases, Pathprinting will enable researchers and clinicians to use data from multiple platforms, experiments and animal models to explore complex disease.
The Pathprinting analysis pipeline can be classified as a second-generation method according to the criteria of Atul Butte and colleagues [4], who recognized three generations of these methods. First-generation methods take a list of genes over-, under- or differentially expressed in a study, compute the proportion of pathway members therein compared with the proportion in a background dataset, and statistically test for enrichment. Second-generation methods improve on this by using information from the entire experiment (all genes, ranked according to a gene-level statistic) to generate pathway-level statistics that capture coordinated changes in the expression of genes in a pathway or gene set. Third-generation methods move beyond treating pathways as lists of genes, adding information about the connectivity and directionality of interactions. In this sense, Pathprinting is a second-generation method, but in moving beyond canonical pathways and including information from nearly the entire corpus of microarray data to generate pathway statistics (fingerprints), it captures crucial information on conserved and divergent co-expression that is absent from other methods.
Expression-based pathway signatures across platforms and species
Hide and colleagues' approach [3] was to retrieve normalized data (176,971 arrays) for six species, spanning 31 single-channel array platforms, from Gene Expression Omnibus (GEO) and to map probes to Entrez Gene identifiers. They computed a mean expression level for each gene by combining values for multiple probes representing single Entrez genes. They sourced pathway gene sets from KEGG, Reactome, Wikipathways and Netpath. To avoid introducing a bias towards well-annotated pathways, the authors used interactions derived from gene co-expression, protein-protein and protein-domain databases, Gene Ontology annotations and text mining to generate a 'functional interaction network' covering 181,706 interactions involving 9,452 human genes. They then applied Markov clustering to decompose the connected portion of this graph into 144 functional interaction clusters (static modules) covering 6,458 genes, 1,542 of which are not found in these pathway databases. This process yields 633 human pathways and static modules. Using NCBI Homologene, they then mapped the corresponding gene sets in the other five species.
Figure 1 illustrates a key part of the workflow. The authors ranked genes by expression level and computed the mean squared rank. The null hypothesis was generated by sample permutation of all arrays of the same platform type, thereby preserving gene-expression correlation structure within gene sets, particularly within pathways. As the expected distribution is unknown, background distributions were fitted to a two-component mixture model, with the normal component corresponding to the core distribution of pathway scores and the uniform distribution corresponding to expression outliers. From these they calculated a probability of expression, that is, the probability that a pathway expression score belongs to the uniform component, and assigned it a score of +1, zero or -1. These ternary scores formed components of the Pathprinting vector. Within a group of fingerprints (such as for a tissue type) the mean score of each extended pathway was then binarized (+1 if above the threshold, -1 if below) and summarized in a vector (consensus fingerprint) that represents the set of functional modules significantly over- and under-expressed in a cell type or condition. This associated a set of pathway activities with a phenotype.
Figure 1.
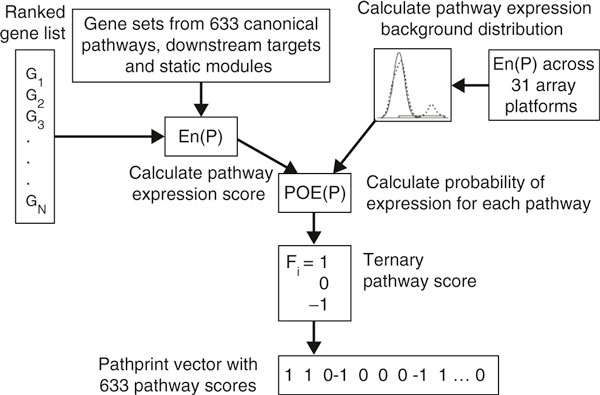
The core workflow for calculating Pathprints for a microarray expression dataset. The Pathprinting vector for an experiment summarizes the expression of genes across 633 pathways and functional modules. It can be combined or compared with other such fingerprints in various ways as described by Hide and colleagues [3]. En(P) indicates the pathway expression score; POE(P) indicates the probability of expression for a pathway; Fi indicates the pathway score (see text for further details).
It is straightforward to calculate a 'functional distance' between fingerprint vectors. This distance is necessarily threshold-dependent, and the authors [3] considered at some length how thresholds might be optimized for the problem at hand. By seeding a consensus fingerprint profile, phenotypes can be matched into an expression database (here, GEO). The question of threshold significance is also relevant at this point, and the authors present a simple but appealing approach that assumes that the database contains a few highly matched but many non-matched samples.
Code and data were implemented in the R package Pathprint. As few research groups are likely to have the necessary resources to implement this independently, the authors [3] helpfully provide pre-computed Pathprinting scores for these six species in a searchable database.
Applications and remaining challenges
The authors [3] briefly describe computational experiments illustrating three applications of Pathprinting. Using 127 human and mouse expression datasets, the authors derived an embryonic stem cell fingerprint indicating pluripotency and matched it to GEO. Of the top 1,000 matches, 90% are induced pluripotent stem cells from 140 human and mouse studies over 13 platforms; the others are cancer cell lines known to express embryonic stem cell functions. In another experiment, they used Pathprinting to jointly analyze human and mouse hematopoietic lineages; parsimony analysis of the individual Pathprinting states resolved the major myeloid and lymphoid lineages, irrespective of species. They also used Pathprinting to recognize four stemness-associated self-renewal pathways shared between human and mouse. The authors demonstrated the clinical relevance of these four pathways by computing Pathprints for four independent clinical studies of gene expression in patients with acute myeloid leukemia; high scores for these pathways were significantly associated with poor patient outcomes, and together these pathways had greater prognostic value than did the human or mouse pathways on their own.
Scope remains for further development of the Pathprinting framework. Not unreasonably, the authors [3] did not re-normalize historical array data using modern approaches. They averaged probe expression at the gene level, although this flattens out the signal from alternative splicing. Alternative approaches are available for orthology assignment. Their phenotype-matching threshold ignores potential multimodal distributions in tissue datasets; for example, datasets annotated as 'kidney' include not only normal kidney but also disease states including cancer, which can have different gene copy numbers and transcriptional programs. One could imagine (as do the authors) a feature-selection approach to identify genes that contribute most toward performance. Finally, individual variation and environmental effects remain largely outside this paradigm.
Like other second-generation pathway analysis techniques [4], this approach [3] ignores topology once the pathway- or module-specific gene sets have been defined. Unlike other pathway analysis tools, however, Pathprinting is designed to enable integrated comparative pathway analysis. Although popular platforms, including GenMAPP [5] and DAVID [6], support pathway analyses for key model organisms, applying them cross-species requires the initial individual analyses to be followed by post hoc meta-analysis. OSCAR [7] enables integrated cross-species co-expression analysis and clustering, but for only a few datasets and without built-in functional analysis. PlaNet [8] performs co-expression and network analysis between Arabidopsis and six crop species, but only for Affymetrix GeneChip data. Pathprinting moves well beyond all these approaches by supporting the large-scale comparative functional analysis of clinical expression data across experiments, species and platforms within a computational framework.
Pathways and modules as computational units of cellular function
Waddington [9] famously depicted cellular phenotype as a canalized landscape, the topography of which is actively shaped by underpinning cables tethered to genetic loci. Individual cables are connected not only to the landscape but often to each other as well, forming a web of epistatic interactions. From a twenty-first century 'omic' perspective, it is difficult not to reinterpret this substructure as genes linked to their expression products through a network of physical interactions, with cellular phenotypes, both structural and functional, emerging from this network. In this way, functional phenotype in its diverse contexts arises from definable subsets of the cellular network, such as local protein interactions or signaling reactions. To a first approximation, then, modules of molecular interaction are computationally relevant units of functional phenotype.
Moving from the identification of characteristic gene expression profiles to delineating the pathways and networks that mechanistically underlie cellular function and disease has been, and remains, a major focus of molecular systems biology and systems medicine. Hide and colleagues [3] now provide the most comprehensive collection of modules so far, and a robust, principled approach to quantifying and comparing their effects along developmental trajectories, across species and in different patient groups. Rhodes and Chinnaiyan [10] envisaged an integrative analysis for molecular cancer research that allows experimental results to be analyzed in the context of existing data and compared on the basis of biological similarity. The achievement of Hide and colleagues brings this vision to reality.
Abbreviations
GEO: Gene Expression Omnibus.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Melissa J Davis, Email: m.davis@imb.uq.edu.au.
Mark A Ragan, Email: m.ragan@uq.edu.au.
Acknowledgements
The authors acknowledge Australian Research Council CE0348221 and strategic funding from The University of Queensland.
References
- Leek JT, Scharpf RB, Corrada Bravo H, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;5:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Su Z, Wen Z, Shi L, Chen T. Microarray platform consistency is revealed by biologically functional analysis of gene expression profiles. BMC Bioinformatics. 2009;5(Suppl 11):S12. doi: 10.1186/1471-2105-10-S11-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler GM, Hofmann O, Kalatskaya I, Payne R, Ho Sui SJ, Saxena U, Krivtsov AV, Armstrong SA, Cai T, Stein L, Hide WA. Pathway Fingerprinting: an integrative approach to understand the functional basis of disease. Genome Med. 2013;5:68. doi: 10.1186/gm472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;5:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Hanspers K, Zambon AC, Vranizan K, Lawlor SC, Dahlquist KD, Doniger SW, Stuart J, Conklin BR, Pico AR. GenMAPP 2: new features and resources for pathway analysis. BMC Bioinformatics. 2007;5:217. doi: 10.1186/1471-2105-8-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;5:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Lu Y, He X, Zhong S. Cross-species microarray analysis with the OSCAR system suggests an INSR->Pax6->NQO1 neuro-protective pathway in aging and Alzheimer's disease. Nucleic Acids Res. 2007;5:W105–W114. doi: 10.1093/nar/gkm408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M, Klie S, Tohge T, Giorgi FM, Wilkins O, Campbell MM, Fernie AR, Usadel B, Nikoloski Z, Persson S. PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell. 2011;5:895–910. doi: 10.1105/tpc.111.083667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The Strategy of the Genes. London: George Allen & Unwin; 1957. [Google Scholar]
- Rhodes DR, Chinnaiyan AM. Integrative analysis of the cancer transcriptome. Nat Genet. 2005;5:S31–S37. doi: 10.1038/ng1570. [DOI] [PubMed] [Google Scholar]