

Abstract
Introduction
Although a substantial proportion of male breast cancers (MBCs) are hereditary, the molecular pathways that are activated are unknown. We therefore examined the frequency and clinicopathological associations of the PIK3CA/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) pathways and their regulatory genes in familial MBC.
Methods
High resolution melting analysis and confirmatory sequencing was used to determine the presence of somatic mutations in PIK3CA (exon 9 and 20), AKT1 (exon 4), KRAS (exon 2) and BRAF (exon 15) genes in 57 familial MBCs. Further analysis of the PIK3CA/mTOR pathway was performed using immunohistochemistry for the pAKT1, pS6 and p4EBP1 biomarkers.
Results
PIK3CA somatic mutations were identified in 10.5% (6 of 57) of cases; there were no AKT1, KRAS or BRAF somatic mutations. PIK3CA mutations were significantly more frequent in cancers from BRCAX patients (17.2%, 5/29) than BRCA2 (0%, 0/25) carriers (P = 0.030). Two BRCAX patients had an E547K mutation which has only been reported in one female breast cancer previously. PIK3CA mutation was significantly correlated with positive pS6 (83.3% vs. 32.0%, P = 0.024) and negative p4EBP1 (100% vs. 38.0%, P = 0.006) expression, but not pAKT expression. Expression of nuclear p4EBP1 correlated with BRCA2 mutation carrier status (68.0% vs. 38.7%, P = 0.035).
Conclusions
Somatic PIK3CA mutation is present in familial male breast cancer but absent in BRCA2 carriers. The presence of two of the extremely rare E547K PIK3CA mutations in our cohort may have specific relevance in MBCs. Further study of PIK3CA in MBCs, and in particular BRCAX patients, may contribute to further establishing the relevance of specific PIK3CA mutations in MBC aetiology and in the identification of particular patient groups most likely to benefit from therapeutic targeting with the novel PIK3CA inhibitors that are currently in development.
Keywords: PIK3CA, E547K, mTOR, familial, male breast cancer, BRCA2, BRCAX
Introduction
Recent studies characterising male breast cancer (MBC) show that these rare tumours are very different to their female counterparts [1,2]. In particular, there are notable distinctions between familial female and MBC with a different pattern of penetrance and genotypic phenotypic correlation in BRCA1, BRCA2 and BRCAX subsets [1]. While it is likely that hormonal influence is a significant contributor, as yet, the characterisation of oncogenic drivers by mutation analysis of even the most common gene mutations in MBCs has not been undertaken.
Several significant targetable oncogenes are known and relatively well described in female breast cancer (FBC). The most frequent gain of function mutations is seen in phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha 9 (PIK3CA) which forms one of the catalytic subunits of the phosphatidylinositol 3-kinase (PI3K) holoenzyme [3,4]. Mutations of the helical or kinase domain lead to activation of the p110a kinase with subsequent downstream triggering of the mammalian target of rapamycin (mTOR) leading to cell proliferation, angiogenesis and promotion of the metastatic process [5,6]. Additional regulators of the PIK3CA/mTOR pathway include AKT1 and the RAS/RAF/mitogen-activated protein kinase (MAPK) pathway that intersect at multiple points [7-13].
Within FBC, the prevalence and prognostic significance of tumours with these driving mutations are unclear and may be dependent on both tumour histological type and estrogen receptor (ERα) status [14-17]. Notably, in vitro studies propose that activation of the PIK3CA/mTOR pathway may be important in tumours with deficient homologous recombination [18], suggesting a possible role in gaining resistance to poly ADP ribose polymerase (PARP) inhibitors in BRCA1/2 deficient tumours. However, although there are limited data (n = 22), an association between BRCA1/2 loss and activation of the PIK3CA/mTOR pathway in human tumours has not been confirmed [15].
Despite accruing data in FBC as to the significance of these oncogenes, there are few studies examining somatic mutation in sporadic MBC only [19-23], with the majority of studies focused on gene expression profiling [24-26] and germ-line mutational analysis [27-32].
Since the PIK3CA/mTOR pathway is more frequently associated with ERα positive FBC, and MBC is largely characterised by ERα positive disease, we have examined the frequency of activation of the PIK3CA/mTOR pathway and its regulators in a cohort of 57 familial MBCs. While the reported frequency of KRAS and BRAF mutations in female breast cancer is generally low (<5%) reference [33,34], a single sporadic MBC study showing a markedly high percentage of KRAS mutations (12%) also encouraged investigation of the mitogen-activated protein kinase (MAPK) pathway, which also interacts with the PIK3CA/mTOR pathway. Our aims were to; (1) identify PIK3CA, AKT1, KRAS and BRAF mutations in familial male breast cancer, (2) assess the relationship between such somatic gene mutations and clinicopathological factors, including BRCA1/2 mutation carrier status, and (3) identify and characterise the PIK3CA/mTOR and MAPK pathway and correlate with any clinicopathological factors and survival.
Materials and methods
Patient samples
Only primary breast cancers were examined in this study. Cases were obtained from the kConFab repository [35]. Prerequisites for cases to be included into kConFab are a strong family history of breast and ovarian cancer (Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA) scores [36] generated from family pedigree and stratified by BRCA1/2 mutation carrier status included as Additional file 1: Supplementary figure 1) with criteria for admission to the kConFab study as outlined previously [37]. Cases were from Australia and New Zealand and diagnosed between 1980 and 2009.
The flow of patients through the study according to the REMARK criteria [38] is listed in Additional file 2: Supplementary table 1. Of the 118 cases within the kConFab registry, 58 cases were excluded due to unavailability of tissue. Of the 60 cases where tissue was available, 2 cases had insufficient tumour tissue for DNA extraction or for a core to be taken for assembly of a tissue microarray (TMA) and a further single case had an extremely low DNA yield and insufficient material for tissue microarray. Fifty seven cases had sufficient material at an appropriate DNA concentration for somatic mutation testing and one case did not have adequate tissue for TMA construction with all tissue committed to DNA extraction. Clinical parameters, including disease specific mortality were obtained from referring clinical centres, kConFab questionnaires and state death registries. Information on pedigree, mutational status and testing were available from the kConFab central registry. Histological classification was based on criteria set by the World Health Organization 2012 [39] and all slides and pathological records from all cases were reviewed for tumour size, tumour grade, lymphovascular and perineural invasion. Immunohistochemistry for ERα, progesterone receptor (PgR), basal markers (cytokeratin (CK) 5, epidermal growth factor receptor (EGFR)) and HER2 silver in situ hybridisation (SISH) had been performed as previously reported [1]. Using stratification of intrinsic phenotypes based on Nielsen et al. [40], tumours were placed into luminal (ERα positive, HER2 negative, CK5 and/or EGFR negative or positive), basal (HER2 and ERα negative; CK5 and/or EGFR positive), HER2 (HER2 positive, ERα, CK5 and EGFR negative or positive) and null/negative (HER2, ERα, CK5 and EGFR negative) phenotypes. This work was carried out with approval from the Peter MacCallum Cancer Centre Ethics Committee (Project No: 11/61). The approval included waiver of patient consent.
Germline BRCA1/2 testing
Mutation testing for BRCA1 and BRCA2 mutations was performed as reported previously [1]. Testing of index cases in kConFab families was carried out by denaturing high performance liquid chromatography or multiplex ligation-dependent probe amplification. Once the family mutation had been identified, all pathogenic (including splice site) variants of BRCA1 and BRCA2 were genotyped by kConFab in all available family members' DNA.
High-Resolution Melting (HRM) assay
Genomic DNA was extracted from formalin-fixed, paraffin embedded (FFPE) samples. A 3 μM haematoxylin and eosin (H&E) stained slide was cut from FFPE blocks and stained to identify tumour enriched areas. From the relevant area on the FFPE block, a 2 mm punch biopsy core was taken. The cores were then dewaxed and hydrated through gradient alcohol. Genomic DNA was then extracted using the DNeasy Tissue kit (Qiagen, Valencia, CA, USA)) following proteinase K digestion at 56°C for three days.
The PIK3CA, AKT1, BRAF and KRAS primer sequences are shown in Additional file 3: Supplementary table 2. PIK3CA exon 9 and 20 primers produced amplicons with 104 base pairs (bp) and 102 bp, respectively. AKT1 exon 4, BRAF exon 15 and KRAS exon 4 primers produced 78 bp, 144 bp and 92 bp amplicons, respectively. PCR for HRM analysis was performed in 0.1 ml tubes on a Rotor-Gene Q (Qiagen) utilising the fluorescent DNA intercalating dye, SYTO 9 (Invitrogen, Carlsbad, CA, USA). A 20 μL final reaction volume contained 1 × PCR buffer, 0.5 to 2.0 mM MgCl2, 200 to 400 nM of forward and reverse primer, 200 μM of dNTPs, 5 μM of SYTO 9, 0.5 U of HotStarTaq polymerase (Qiagen), 5 ng of genomic DNA, Uracil-DNA glycosylase (UDG) (0.5 units/reaction), UDG buffer (New England BioLabs, Ipswich, MA, USA) and PCR grade water. The cycling and melting conditions are shown in Additional file 3: Supplementary table 2. All reactions had initial UDG treatment for FFPE artefacts at 37°C for 30 minutes [41], followed by an incubation step at 95°C for 15 minutes, denaturation step at 95°C, annealing steps at the temperatures listed in Additional file 3: Supplementary table 2, and an elongation step at 72°C. A single cycle of 97°C for one minute preceded a melt phase run between temperatures listed in Additional file 3: Supplementary table 2 and rising 0.2°C per step. Samples were run in duplicate. HRM analysis was performed on the Rotor-Gene Q Software (v1.7) (Qiagen, Valencia, CA, USA).
DNA sequencing
All samples with either or both duplicates showing abnormal melt were sequenced for detection of mutations. PIK3CA exon 9 and 20 HRM products were amplified using M13 tagged primers (Additional file 3: Supplementary table 2) initially and then M13 primers for a second step for PIK3CA exon 9 (amplicon 185 bp) and a single step PCR reaction for PIK3CA exon 20 (amplicon 149 bp) using primers listed in Additional file 3: Supplementary table 2. The composition of a total reaction mixture of 20 μL contained; 1 × PCR buffer, 2.5 mM MgCl2, 400 nM of each primer, 200 μM of dNTPs, 0.5 U of HotStarTaq polymerase (Qiagen), 5 ng of HRM DNA products and PCR grade water. The PCR conditions were as follows: an initial incubation at 95°C for 1 minute, followed by 35 cycles of 95°C for 10 seconds, 55°C for 10 seconds and 72°C for 4 minutes. The sequencing reaction was then performed using the Big Dye Terminator v3.1 chemistry according to the manufacturer's protocol (Applied Biosystems, Foster City, CA, USA) using 6 μL of the PCR products that were purified with 2 μL of ExoSapIT (GE Healthcare, Little Chalfont, Buckinghamshire, UK). After ethanol precipitation, the sequencing products were run on a 3700 Genetic Analyser (Applied Biosystems). The sequencing data were then analysed using Sequencher 4.6 (Gene Codes Corporation, Ann Arbor, MI, USA). Each mutation was confirmed by sequencing a second independent PCR reaction. The work flow is outlined in Figure 1.
Figure 1.
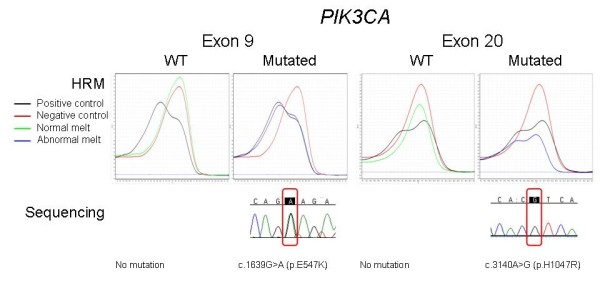
Work flow of sample analysis. Initial High-Resolution Melting (HRM) analysis was used to screen mutation by abnormal melt. Sanger sequencing was then performed to detect specific mutations.
Immunohistochemistry
Tumour-tissue microarrays (1-mm cores), with a two-fold redundancy, were prepared from archival FFPE tissue blocks. TMA sections were cut from each block at 4 μm thick intervals, dewaxed, placed through graded alcohol and then into water.
For phosphorylated 4EBP1 (p4EBP1) and phosphorylated S6 (pS6), antigen retrieval was performed using high pH antigen retrieval buffer (DAKO, Glostrup, Denmark) in pressure cooker for three minutes at 125°C. For phosphorylated AKT1 (pAKT), antigen retrieval was performed with CC1 high pH retrieval solution (Roche, Basel, Switzerland) at 100°C for 36 minutes. Staining for p4EBP1 (dilution 1:400, clone 2855, Cell Signalling Technology, Danvers, MA, USA) and pS6 (dilution 1:200, clone 2211, Cell Signalling Technology) was performed using a monoclonal and polyclonal rabbit antibodies respectively. Antigen-antibody complex was detected using the Envision FLEX system (EnVision FLEX/HRP and EnVision FLEX DAB + Chromogen, DAKO). Staining for pAKT1 (dilution 1:1,000, clone LP18, Novocastra, Newcastle upon Tyne, UK) was performed using a monoclonal mouse antibody with secondary detection using Ventana Ultraview detection reagents (Roche). Slides were then counterstained with haematoxylin, dehydrated, cleared and mounted for assessment. Phosphorylated 4EBP1 expression was assessed for both cytoplasmic and nuclear expression, nuclear expression for pAKT1 and cytoplasmic expression for pS6 (Figure 2a). A histoscore was generated by multiplying staining intensity (0, no staining; 1, weak; 2, moderate; 3, strong) by the percentage of positive tumour cells (0, 0; 1, < or = to 25%; 2, >25% to 50%; 3, >50% to 75%, 4, >75%). The histoscores ranged between 0 and 12. For subsequent analysis, histoscores were categorised into either absent (histoscore = 0) or present (1 to 12) or low (0 to 3) and high (4 to 12) to differentiate from baseline staining of adjacent normal breast epithelium.
Figure 2.
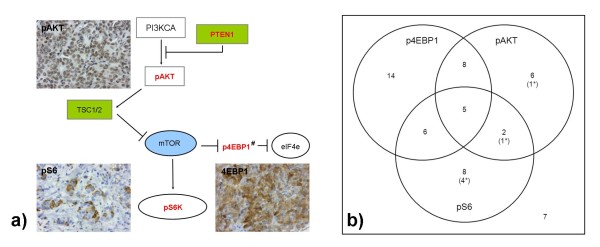
PIK3CA/mTOR pathway (# 4EBP1 is inactivated by phosphorylation) a). Immunohistochemistry was performed for phosphorylated AKT, pS6 and p4EBP1. b) Outcome of immunohistochemical staining integrating PIK3CA mutation status. Numbers indicate the amount of cases showing positive immunohistochemical staining. Mutations are signified by (n*), with n = number of cases. mTOR, mammalian target of rapamycin.
A PIK3CA mutation phenotype was defined by either a tumour harbouring a somatic PIK3CA activating mutation or showing an absence of p4EBP1 expression and moderate to strong pS6 expression (histoscore 4-12/12) on immunohistochemistry.
Statistical analysis
Comparison of groups was made using Mann-Whitney U for non-parametric continuous distributions and chi-square test for threshold data. Kaplan-Meier survival curves were plotted using breast cancer related death as the endpoint and compared using a log rank test. Analysis was performed with GraphPad Prism 5 software (GraphPad Prism version 5.04 for Windows, GraphPad Software, La Jolla, CA, USA). A two-tailed P-value test was used in all analyses and a P-value of less than 0.05 was considered statistically significant.
Results
PIK3CA is commonly mutated in familial male breast cancer
Seven PIK3CA mutations were identified and confirmed in six samples (Table 1). Four activating mutations were identified in exon 9, with two cases of E547K mutation and one sample demonstrated concurrent E542K and E547K mutations in exon 9. Three further mutations were identified in exon 20, all of which were H1047R mutations. Screening of AKT1, BRAF and KRAS showed no evidence of somatic mutations.
Table 1.
Somatic PIK3CA mutations in familial male breast cancer
| Nucleotide change | Amino acid change | BRCA status |
|---|---|---|
| c.1624G>A | p.E542K | BRCAX |
| c.1639G>A | p.E547K | BRCAX |
| c.1624G>A, c.1639G>A | p.E542K, p.E547K | BRCAX |
| c.3140A>G | p.H1047R | BRCAX |
| c.3140A>G | p.H1047R | BRCAX |
| c.3140A>G | p.H1047R | BRCA1 del exons 21_24 |
PIK3CA mutation is uncommonly seen in BRCA2 mutation carriers
One tumour arising in a BRCA1 carrier had an exon 20 PIK3CA mutation, five PIK3CA mutations occurred in BRCAX males whereas no PIK3CA mutation were identified in tumours from BRCA2 mutation carriers. There was a significant positive association between PIK3CA mutation incidence and BRCACX (17.2%) compared with BRCA2 (0%) associated tumours (P = 0.030). There was otherwise no correlation between the presence of somatic PIK3CA mutation and age of diagnosis, primary tumour size, tumour histological subtype, tumour grade, intrinsic phenotype, lymphovascular or perineural invasion (P > 0.05) (Table 2). The presence of PIK3CA mutation was not associated with a significant difference in Disease Specific Survival (DSS) (Figure 3).
Table 2.
Correlation of PIK3CA mutation status with clinicopathological parameters
| PIK3CA Mutation (n = 6) | PIK3CA Wild type (n = 51) | P-value | |
|---|---|---|---|
| Age - mean (years) | 62.2 | 63.0 | 0.899 |
| Overall DSS | 33.3% | 33.3% | 1.000 |
| Carrier mutation status | |||
| BRCA1 | 1 | 2 | 0.288 |
| BRCA2 | 0 | 25 | 0.030 |
| BRCAX | 5 | 24 | 0.194 |
| Primary tumour size (mm) | 18.8 | 19.1 | 0.948 |
| Histological type | |||
| Invasive carcinoma - no special type (IC - NST) | 6 | 37 | 0.319 |
| IC-NST with micropapillary areas | 0 | 9 | 0.575 |
| Invasive Papillary Carcinoma | 0 | 3 | 1.000 |
| Invasive Lobular Carcinoma | 0 | 2 | 1.000 |
| Tumour grade | |||
| 1 | 0 | 3 | 1.000 |
| 2 | 3 | 26 | 1.000 |
| 3 | 3 | 22 | 1.000 |
| Lymphovascular invasion | 33.3% | 39.2% | 1.000 |
| Perineural invasion | 50.0% | 41.2% | 0.689 |
| Intrinsic subtype | |||
| Luminal | 6 | 45 | 1.000 |
| HER2 | 0 | 5 | 1.000 |
| Basal | 0 | 1 | 1.000 |
| Null | 0 | 0 | 1.000 |
P-values < 0.05 in bold text.
Figure 3.

Correlation of PIK3CA mutation and disease specific survival.
Co expression and clinicopathological correlation of p4EBP1, pS6, pAKT biomarkers
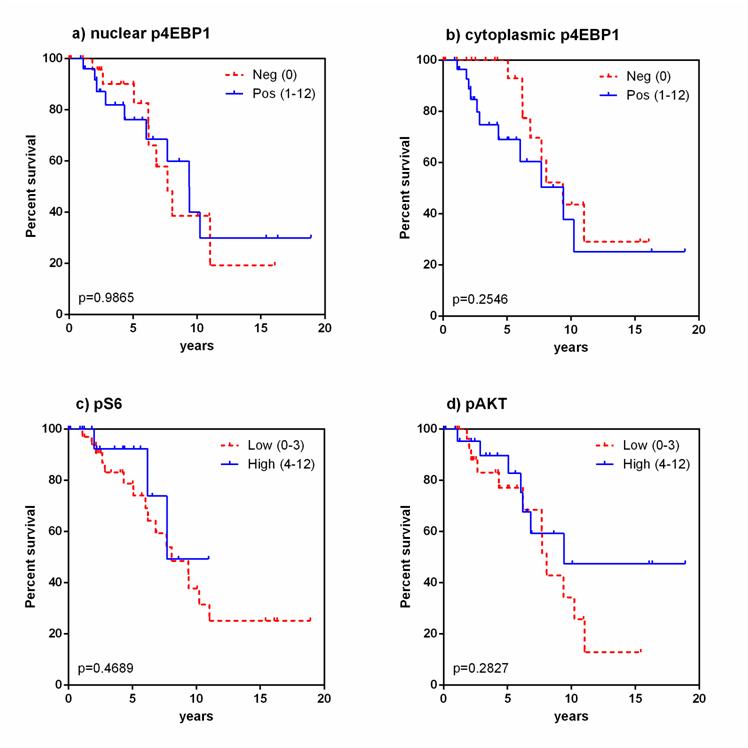
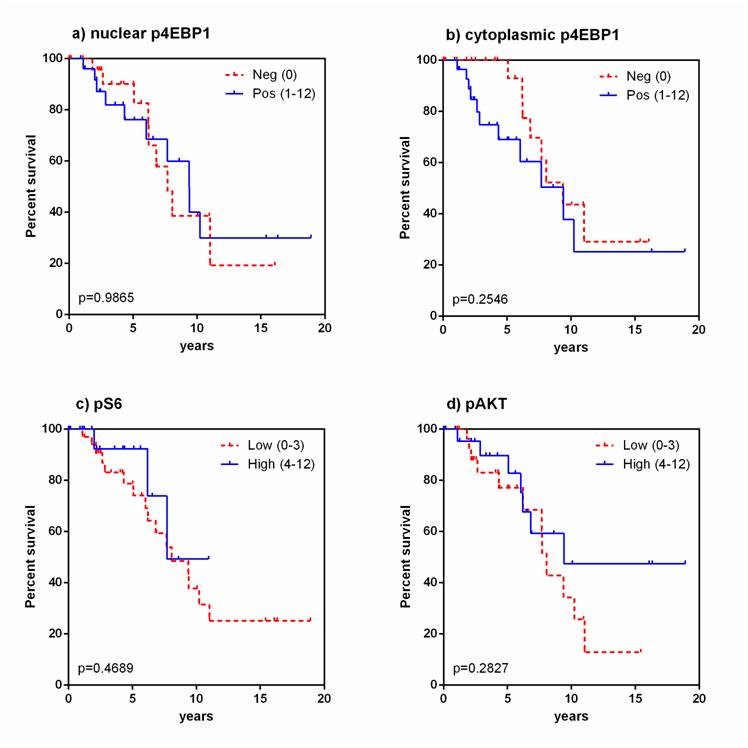
Cytoplasmic expression of p4EBP1 was present (histoscore 1 to 12) in 55.4% (31/56) of cases, nuclear p4EBP1 expression (histoscore 1 to 12) in 51.8% (29/56) of cases and either nuclear or cytoplasmic expression in 58.9% (33/56) of cases. High expression of both pS6 and pAKT1 (histoscore 4 to 12) was seen in 37.5% (21/56) of cases each. A pattern of co-expression of any of the markers was not seen (P > 0.05) (Figure 2b). Clinicopathological correlation showed that nuclear expression of p4EBP1 correlated with BRCA2 carrier status (17/25 (68.0%) P = 0.035) and inversely with BRCAX cases (11/30 (36.7%) P = 0.0184). There was no correlation between DSS and expression of any markers (Additional file 4: Supplementary figure 2).
PIK3CA mutation phenotype
All tumours with PIK3CA mutation showed differences in some downstream pathway members. Expression of p4EBP1, pS6 and pAKT was observed in 0/6 (0%), 5/6 (83.5%) and 2/6 (33.3%) of cases respectively (Figure 2b). There was significant absence of p4EBP1 nuclear (P = 0.009) or cytoplasmic (P = 0.006) staining and up-regulation of pS6 (P = 0.024) in tumours with PI3KCA somatic mutations when compared with PIK3CA wild type (Table 3).
Table 3.
Correlation of p4EBP1, pS6 and pAKT immunohistochemistry with BRCA status and clinicopathological factors
| 4EBP1 | pS6 | pAKT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytoplasmic | Nuclear | Cytoplasmic | Nuclear | |||||||||
| Present | Absent | P-value | Present | Absent | P-value | High | Low | P-value | High | Low | P-value | |
| PIK3CA Mutation | 0 | 6 | 0 | 6 | 5 | 1 | 2 | 4 | ||||
| PIK3CA Wild-Type | 31 | 19 | 0.006 | 29 | 21 | 0.009 | 16 | 34 | 0.024 | 19 | 31 | 1.000 |
| Carrier mutation status | ||||||||||||
| BRCA1 | 1 | 2 | 0.581 | 1 | 2 | 0.605 | 1 | 2 | 1.000 | 0 | 3 | 0.284 |
| BRCA2 | 15 | 10 | 0.596 | 17 | 8 | 0.035 | 9 | 16 | 0.582 | 10 | 15 | 0.786 |
| BRCAX | 15 | 13 | 0.456 | 11 | 19 | 0.018 | 11 | 17 | 1.000 | 11 | 17 | 1.000 |
| Age - mean (years) | 60.3 | 64.5 | 0.240 | 59.1 | 65.3 | 0.078 | 63.4 | 61.4 | 0.503 | 62.0 | 62.2 | 0.940 |
| Primary tumour size (mm) | 20.3 | 17.2 | 0.169 | 19.5 | 18.3 | 0.589 | 17.21 | 20.03 | 0.312 | 20.05 | 18.25 | 0.437 |
| Histological type | ||||||||||||
| Invasive carcinoma - no special type (IC - NST) | 21 | 19 | 0.563 | 22 | 19 | 0.766 | 13 | 28 | 0.212 | 15 | 26 | 1.000 |
| IC-NST with micropapillary areas | 6 | 3 | 0.716 | 3 | 6 | 0.288 | 6 | 3 | 0.066 | 3 | 6 | 1.000 |
| Invasive Papillary Carcinoma | 2 | 2 | 1.000 | 3 | 1 | 0.612 | 2 | 2 | 0.626 | 2 | 2 | 0.626 |
| Invasive Lobular Carcinoma | 2 | 1 | 1.000 | 1 | 1 | 1.000 | 0 | 2 | 0.523 | 1 | 1 | 1.000 |
| Tumour Grade | ||||||||||||
| 1 | 2 | 1 | 1.000 | 2 | 1 | 1.000 | 2 | 1 | 0.549 | 1 | 2 | 1.000 |
| 2 | 16 | 13 | 1.000 | 16 | 13 | 0.789 | 9 | 20 | 0.409 | 11 | 18 | 1.000 |
| 3 | 13 | 11 | 1.000 | 11 | 13 | 0.590 | 10 | 14 | 0.591 | 9 | 15 | 1.000 |
| LVI | 40.7% | 36.0% | 0.781 | 28.0% | 48.2% | 0.163 | 31.8% | 43.3% | 0.565 | 33.3% | 41.9% | 0.780 |
| PNI | 51.7% | 45.8% | 0.575 | 40.7% | 57.6% | 0.436 | 36.4% | 58.1% | 0.166 | 33.3% | 59.0% | 0.093 |
| Intrinsic subtype | ||||||||||||
| Luminal | 26 | 24 | 0.210 | 24 | 26 | 0.195 | 17 | 33 | 0.183 | 20 | 30 | 0.393 |
| HER2 | 4 | 1 | 0.367 | 4 | 1 | 0.353 | 3 | 2 | 0.352 | 1 | 4 | 0.640 |
| Basal | 1 | 0 | 1.000 | 1 | 0 | 1.000 | 1 | 0 | 0.375 | 0 | 1 | 1.000 |
| Null | 0 | 0 | 1.000 | 0 | 0 | 1.000 | 0 | 0 | 1.000 | 0 | 0 | 1.000 |
Discussion
This study is the first to characterise biomarkers and mutations in the PIK3CA/mTOR pathway in familial male breast cancer noting several novel observations. We identified a PIK3CA mutation rate of 10.5% in familial MBCs but an absence of common activating mutations of AKT1, KRAS and BRAF. While limited by moderate numbers in our study, the absence of KRAS mutation contrasts with the only other study performed in sporadic MBCs by Dawson et al. who reported an overall incidence of 12% [20]. Methodological reasons may be underlying these difference but in our experience, HRM is a highly sensitive and robust technique [42,43]. The absence of BRAF mutation is also somewhat expected and is supported by the stronger association between basal cell breast cancer lines and BRAF mutation [44] (since the majority of MBCs are of a luminal subtype). While a true frequency of these mutations requires further testing in a much larger cohort, these data suggest frequency is unlikely to be high and should parallel the range (0.7 to 5%) that is observed in female breast cancer.
The mutation rate of PIK3CA in this series is lower than the reported 17.9% (7/39) in the only other study performed, although this was in a population-based cohort of MBCs patients [19]. It is also less frequent than that reported in FBC (16.3% [19] to 40.0% [3]) (Table 4), which supports the notion that male breast cancer is biologically different from female breast cancer and that therapies that rely on the experience of the female disease are likely to be suboptimal. Furthermore, evidence from our data demonstrating that differences in this PIK3CA/mTOR pathway is dependent on the germline genotypes of male breast cancer, shows the basis of male breast cancer in BRCA2 mutation carriers is very different to that of BRCAX giving further credence to personalising breast cancer treatment whether male or female using individual patient and tumour characteristics. Thus, as the incidence of PIK3CA mutations in tumours from in BRCA2 carriers is likely to be negligible, these patients are unlikely to derive benefits from the PIK3CA inhibitors that are now entering clinical trials for female breast cancer [19].
Table 4.
Comparison of PIK3CA mutation studies in male and female breast cancer
| Male Breast Cancer | Female Breast Cancer | |||||
|---|---|---|---|---|---|---|
| Current Study | Benvenuti S et al. [20] | Benvenuti S et al. [20] | Buttitta F et al. [14] | Campbell IG et al. [3] | Saal H et al. [16] | |
| Study population | High risk - familial | Population based | Population based | Population based | Population based | Population based |
| Frequency | 6/57 (10.5%) | 7/39 (17.9%) | 14/86 (16.3%) | 46/180 (25.6%) | 28/70 (40.0%) | 77/292 (26.4%) |
| Mutation Locus | 3 exon 9, 3 exon 20 | 7 exon 20 | 6 exon 9, 8 exon 20 | 23 exon 9, 23 exon 20 | 15 exon 9, 9 exon 20, 3 exon 7, 1 exon 6 | 31 exon 9, 49 exon 20, 7 exon 7, 7 others |
| Clinicopathological association | Inverse correlation with BRCA2 mutation carrier status | No clinicopathological association | No clinicopathological association | Mutation seen more frequently in lobular carcinoma (46%, P < 0.001). Exon 9 more frequently seen in lobular carcinoma (30% of cases, P < 0.001). | No clinicopathological association | Association with ER positivity (P = 0.0001), PgR Positivity (0.0063) and lymph node positivity (P = 0.0375). |
P-values < 0.05 in bold text
The distribution of mutations of PIK3CA in male breast cancer reported by Benvenuti et al. (Table 4) showed exclusively exon 20 mutations in MBC, supporting the suggestion that the frequency of exon 9 and 20 mutations may be gender and tissue specific. We, however, noted an equal distribution of exon 9 and 20 mutations, which is more reflective of the distribution seen by others in FBC [3,14]. Furthermore, the E547K mutation noted in two of our BRCAX patients has only once previously been reported in a single female breast cancer suggestive of a unique hot spot preferentially within male cancers. This mutation was detected and confirmed using HRM and Sanger sequencing in duplicate for each case using methodologies optimised for FFPE material. We have extensive experience with this methodology and feel it to be well suited and robust for formalin fixed paraffin embedded material. While we also acknowledge the occurrence of artifactual changes, the E547K mutation has not been detected in over 300 FFPE tumour samples we have screened to date (unpublished data) and thus, we feel that this mutation may be particular to a subset of MBC. The E547K mutation itself is found in the highly conserved helical domain of PIK3CA and possibly confers increased catalytic activity. The mutation is not unique to breast cancer, and has also been reported previously in one colorectal adenocarcinoma [45] and in seven neuroendocrine tumours of the lung [46] lending support for a true pathogenic mutation. Targeted sequencing of further MBC, and in particular non-BRCA2 tumours, may help determine a more accurate incidence and potential relevance of this uncommon mutation. We also observed a case with two concurrent exon 9 mutations, which has not been previously reported in MBC. While there is some suggestion of a more aggressive phenotype or of tumour heterogeneity in cases with dual PIK3CA mutations [16,47,48], the clinical significance of this is also unclear due to the infrequency of this observation.
Recent data show that BRCA2 appears to be a significant driver in MBC, with a considerably higher penetrance within male BRCA2 carriers compared with males in BRCAX families and BRCA1 male mutation carriers [1]. It is also noteworthy that BRCA2 somatic mutations have also been reported in 21.8% of sporadic MBCs [22]. Furthermore, unlike in FBC, studies by Ottini et al. [49] and ourselves [1] intimate a distinct BRCA2 phenotype in MBCs, which more commonly contain areas of micropapillary histology, are of a higher grade, are PgR negative and are HER2 amplified. The genomic findings of this study emphasize that BRCA2 tumours may be a distinct subgroup in familial MBC and as such BRCA2 mutation may be a significant driver in MBC. Further support for a strong inherent BRCA2 associated drive independent of gender and estrogenic influence in male breast cancer is the association of PIK3CA mutation and ERα positive female breast cancer [14-17], a phenotype which is common to BRCA2 associated male tumours (92%) [1], but without the associated rate of PIK3CA mutation. These data suggest that gender and hormonal dimorphism may not be so significant in BRCA2 carriers and that BRCA2 male breast cancers align with the non-PIK3CA mutated ERα positive group of female breast cancer.
PIK3CA oncogenic drive, however, may be more important in non-BRCA2 MBCs where estrogenic influences may be more prominent. While our previous studies have shown that ERα and PgR positive tumours were seen at a similar frequency across all BRCA1, BRCA2 and BRCAX cohorts and more commonly than in FBC [1], based on this genotypic analysis, the mechanism and effect of PIK3CA mutation is likely to be different between the subgroups. Overall, given the association between ERα positive tumours and increased PIK3CA mutation frequency in FBC, one would assume an increased rate of PIK3CA mutation in MBCs. This is not seen and may suggest alternate receptor and PIK3CA/mTOR interaction in male breast cancer or a dose-based relationship differentiated by male cancers with low estrogen at one end of the spectrum and higher levels of estrogen in females at the opposite end. While studies have extensively examined the correlation between hormone receptor status and incidence of PIK3CA mutation, as yet there are very limited data on the effect of circulating oestradiol on PIK3CA mutation rate with some suggestion that PIK3CA/mTOR activation may contribute to tamoxifen-resistance. Further evidence of estrogen influence is also provided by Benvenuti et al. who observed a gender bias for PIK3CA mutations in colorectal cancer with a higher incidence of mutations in women (23%) compared with men (9%) [19] (Table 4), which reflect the findings of our study. Further study correlating serum oestradiol, testosterone levels and PIK3CA mutation frequency in MBCs are required to further elaborate on a possible association.
Recent in vitro studies showing increased sensitization of cancers with defects in DNA homologous recombination (as seen in BRCA1/2 deficient cancers), to PARP inhibition by targeting of PIK3CA [18,50] suggest that PIK3CA/mTOR pathway interactions result in homologous recombination steady state. Support for the model is not yet seen in vivo with only one study to date to have examined a correlation between BRCA mutation carriers' status and PIK3CA mutation incidence in FBC. Limited by numbers, Michelucci et al. describe two mutations (one codon 9 and one codon 20) in 12 BRCA2 mutation carriers and no mutations in 10 BRCA1 mutation carriers [15]. The clinical value of this dual targeting is unknown in BRCA1/2 FBCs and whether it is male or female, this study is also the first to describe a PIK3CA somatic mutation in a BRCA1 mutation carrier. The low numbers of MBCs in BRCA1 mutation carriers in our study reflects the paucity of these tumours in this particular cohort, and in BRCA1 carriers in general [51-54]. What is apparent is that BRCA1-associated tumours in males appear to be more similar to the tumours seen in post-menopausal female BRCA1 carriers, with an absence of tumours arising in young patients and an absence of an association with basal cell phenotype. Notwithstanding, carrying a BRCA1 mutation does appears to be a risk factor for MBC with a higher incidence than that of the general population but at much lower penetrance than seen in female BRCA1 carriers and it is still unclear as to the role BRCA1 plays in MBC. While the findings in this study are novel, true incidence and relevance of PIK3CA mutations in this cohort require further investigation of larger numbers of BRCA1 patients, if these can be acquired for study.
The alignment of PIK3CA mutation with elevated pS6 expression and absent p4EBP1 expression is different to the expected model. Theoretically, PIK3CA mutational activation of the pathway should only lead to an elevated pS6, as is seen, but not an elevated p4EBP1 (the phosphorylated form being inactive) and pAKT, which is not observed. This is in part likely to be due to the complexity of the PIK3CA/mTOR pathway. Indeed, a correlation between PIK3CA mutation in luminal A FBC (the phenotype most similar to MBC) and combined up-regulation of pAKT, p4EBP1 and pS6 is not seen [55]. The association seen in the series between PIK3CA mutation and elevated pS6 (P = 0.024) may suggest partial activation of the PIK3CA/mTOR pathway in MBC and reflect the variability of pS6 and p4EBP1 and pAKT levels seen in vitro with dose dependent inhibition of mTORC1 [56], or interactions of mTORC2, other pathways and feedback loops.
Nevertheless, we observed up-regulation of p4EBP1 in BRCA2 mutation carriers (68.0%) more frequently than in BRCAX carriers (36.7%), an association not reported in FBC, giving further evidence to the difference in male and female breast cancers. It may be that an alternate mechanism of PIK3CA/mTOR pathway activation may be present in BRCA2 cases linked to disordered homologous recombination, as mentioned previously, through p4EBP1 and eIF4e.
Conclusion
The results of this study indicate that somatic PIK3CA mutation are a frequent alteration in familial MBC of BRCAX families, the incidence and type of which is comparable to that seen in sporadic male and slightly lower than FBCs. Conversely, the absence of PIK3CA mutation in BRCA2 associated MBCs suggests that alternate oncogenic drivers minimally contribute to tumour drive in this group, thus supporting distinct male breast cancer types. The study has also revealed differences of MBC to FBC and between sporadic and familial MBC which are of importance in optimising treatment strategies and underlying relevance of the PIK3CA/mTOR pathway in tumour biology. Indeed, the therapeutic implications of these findings support the delineation of significant molecular pathways, such as PIK3CA/mTOR and MAPK cascades for subsequent targeted therapies within specific populations.
Abbreviations
BOADICEA: Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm; Bp: base pairs; CK: cytokeratin; DSS: disease specific survival; EGFR: epidermal growth factor receptor; ERα: estrogen receptor alpha; FBC: female breast cancer; FFPE: formalin fixed paraffin embedded; H&E: haematoxylin and eosin; HRM: high resolution melt; IC-NST: invasive carcinoma of no special type; MAPK: mitogen activated pathway kinase; MBC: male breast cancer; mTOR: mammalian target of rapamycin; mTORC: mammalian target of rapamycin complex; pAKT: phosphorylated AKT; PARP: poly ADP ribose polymerase; PI3K: phosphatidylinositol 3-kinase; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; PgR: progesterone receptor; pS6: phosphorylated S6; p4EBP1: phosphorylated 4EBP1; SISH: silver in situ hybridization; TMA: tissue microarray; UDG: uracil DNA glycosylase.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AD performed the manuscript review, contributed to study concept and design and the HRM assay design. DB performed p4EBP1 and pS6 immunohistochemistry. HD developed HRM assays, and assisted in performing and interpreting Sanger sequencing. The kConFab Investigators performed germ-line BRCA1/2 testing on all patients, and acquired clinical data. NJ performed pAKT immunohistochemistry. SBF prepared the manuscript and contributed to study concept and design. SD performed HRM and Sanger Sequencing of samples, interpretation of pAKT, p4EBP1 and pS6 immunohistochemistry, statistical analysis and manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
Supplementary figure 1: BOADICEA scores for patients included in study. Probability (Prob) score (0 to 1) is generated for BRCA1 and BRCA2 mutations for each case, stratified by known BRCA status.
Supplementary table 1. REMARK criteria leading to cases recruitment.
Supplementary table 2. HRM and Sequence specific PIK3CA, AKT1, KRAS and BRAF primers.
{kind=link}
Supplementary figure 2. Disease specific survival stratified by 1a) nuclear p4EBP1 expression, 1b) cytoplasmic p4EBP1 expression, 1c) pS6 expression and 1d) pAKT expression.
{kind=link}
Contributor Information
Siddhartha Deb, Email: Siddhartha.Deb@petermac.org.
Hongdo Do, Email: Hongdo.Do@petermac.org.
David Byrne, Email: David.Byrne@petermac.org.
Nicholas Jene, Email: Nicholas.Jene@petermac.org.
kConFab Investigators, Email: heather.thorne@petermac.org.
Alexander Dobrovic, Email: Alexander.Dobrovic@petermac.org.
Stephen B Fox, Email: Stephen.Fox@petermac.org.
Acknowledgements
We wish to thank all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (funded 2001 to 2009 by NHMRC and currently by the National Breast Cancer Foundation and Cancer Australia #628333) for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by grants from the National Breast Cancer Foundation, the National Health and Medical Research Council (NHMRC) and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Those authors belonging to the kConFab investigators team that specifically worked on this study include Heather Thorne (case accrual and database management), Eveline Niedermayr (BOADICEA score generation, data management) and Amber Willems-Jones (BRCA1/2 testing). We also wish to thank the Victorian Cancer Biobank, Victorian Cancer Council, for funding contribution towards the project. We thank Kelly Waldeck, Peter MacCallum Cancer Centre, for donation of the phosphorylated 4EBP1 and pS6 antibodies.
References
- Deb S, Jene N. Kconfab Investigators. Fox SB. Genotypic and phenotypic analysis of male breast cancer shows under representation of the HER2 and basal subtypes in BRCA-associated carcinomas. BMC Cancer. 2012;15:510. doi: 10.1186/1471-2407-12-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson C, Holmqvist M, Bergkvist L, Hedenfalk I, Lambe M, Fjällskog ML. Similarities and differences in the characteristics and primary treatment of breast cancer in men and women - a population based study (Sweden) Acta Oncol. 2011;15:1083–1088. doi: 10.3109/0284186X.2011.602114. [DOI] [PubMed] [Google Scholar]
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;15:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;15:2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;15:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;15:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Chen B, Tardell C, Higgins B, Packman K, Boylan JF, Niu H. BRAFV600E negatively regulates the AKT pathway in melanoma cell lines. PloS One. 2012;15:e42598. doi: 10.1371/journal.pone.0042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu PY, Li TK, Ding ST, Lai IR, Shen TL. EGF-induced Grb7 recruits and promotes Ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells. J Biol Chem. 2010;15:29279–29285. doi: 10.1074/jbc.C110.114124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;15:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;15:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J Biol Chem. 2006;15:19925–19938. doi: 10.1074/jbc.M600482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek M, Pacher M, Abraham D, Podar K, Eulitz M, Baccarini M. Dephosphorylation of Ser-259 regulates Raf-1 membrane association. J Biol Chem. 2002;15:7913–7919. doi: 10.1074/jbc.M108733200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;15:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Buttitta F, Felicioni L, Barassi F, Martella C, Paolizzi D, Fresu G, Salvatore S, Cuccurullo F, Mezzetti A, Campani D, Marchetti A. PIK3CA mutation and histological type in breast carcinoma: high frequency of mutations in lobular carcinoma. J Pathol. 2006;15:350–355. doi: 10.1002/path.1908. [DOI] [PubMed] [Google Scholar]
- Michelucci A, Di Cristofano C, Lami A, Collecchi P, Caligo A, Decarli N, Leopizzi M, Aretini P, Bertacca G, Porta RP, Ricci S, Della Rocca C, Stanta G, Bevilacqua G, Cavazzana A. PIK3CA in breast carcinoma: a mutational analysis of sporadic and hereditary cases. Diagn Mol Pathol. 2009;15:200–205. doi: 10.1097/PDM.0b013e31818e5fa4. [DOI] [PubMed] [Google Scholar]
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, Yu JS, Malmstrom PO, Mansukhani M, Enoksson J, Hibshoosh H, Borg A, Parsons R. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;15:2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;15:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, Rodríguez O, Calvo MT, Aura C, Díez O, Rubio IT, Pérez J, Rodón J, Cortés J, Ellisen LW, Scaltriti M, Baselga J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;15:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti S, Frattini M, Arena S, Zanon C, Cappelletti V, Coradini D, Daidone MG, Pilotti S, Pierotti MA, Bardelli A. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat. 2008;15:284–288. doi: 10.1002/humu.20648. [DOI] [PubMed] [Google Scholar]
- Dawson PJ, Schroer KR, Wolman SR. ras and p53 genes in male breast cancer. Mod Pathol. 1996;15:367–370. [PubMed] [Google Scholar]
- Hiort O, Naber SP, Lehners A, Muletta-Feurer S, Sinnecker GH, Zöllner A, Komminoth P. The role of androgen receptor gene mutations in male breast carcinoma. J Clin Endocrinol Metab. 1996;15:3404–3407. doi: 10.1210/jc.81.9.3404. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska E, Teresiak M, Breborowicz D, Mackiewicz A. Somatic mutations in the BRCA2 gene and high frequency of allelic loss of BRCA2 in sporadic male breast cancer. Int J Cancer. 2002;15:943–945. doi: 10.1002/ijc.10289. [DOI] [PubMed] [Google Scholar]
- Anelli A, Anelli TF, Youngson B, Rosen PP, Borgen PI. Mutations of the p53 gene in male breast cancer. Cancer. 1995;15:2233–2238. doi: 10.1002/1097-0142(19950501)75:9<2233::AID-CNCR2820750907>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Johansson I, Nilsson C, Berglund P, Lauss M, Ringner M, Olsson H, Luts L, Sim E, Thorstensson S, Fjallskog ML, Hedenfalk I. Gene expression profiling of primary male breast cancers reveals two unique subgroups and identifies N-acetyltransferase-1 (NAT1) as a novel prognostic biomarker. Breast Cancer Res. 2012;15:R31. doi: 10.1186/bcr3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson I, Nilsson C, Berglund P, Strand C, Jönsson G, Staaf J, Ringnér M, Nevanlinna H, Barkardottir RB, Borg A, Olsson H, Luts L, Fjällskog ML, Hedenfalk I. High-resolution genomic profiling of male breast cancer reveals differences hidden behind the similarities with female breast cancer. Breast Cancer Res Treat. 2011;15:747–760. doi: 10.1007/s10549-010-1262-8. [DOI] [PubMed] [Google Scholar]
- Callari M, Cappelletti V, De Cecco L, Musella V, Miodini P, Veneroni S, Gariboldi M, Pierotti MA, Daidone MG. Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res Treat. 2011;15:601–610. doi: 10.1007/s10549-010-1015-8. [DOI] [PubMed] [Google Scholar]
- Blanco A, de la Hoya M, Balmana J, Ramon y Cajal T, Teule A, Miramar MD, Esteban E, Infante M, Benitez J, Torres A, Tejada MI, Brunet J, Grana B, Balbin M, Perez-Segura P, Osorio A, Velasco EA, Chirivella I, Calvo MT, Feliubadalo L, Lasa A, Diez O, Carracedo A, Caldes T, Vega A. Detection of a large rearrangement in PALB2 in Spanish breast cancer families with male breast cancer. Breast Cancer Res Treat. 2012;15:307–315. doi: 10.1007/s10549-011-1842-2. [DOI] [PubMed] [Google Scholar]
- Falchetti M, Lupi R, Rizzolo P, Ceccarelli K, Zanna I, Calo V, Tommasi S, Masala G, Paradiso A, Gulino A, Giannini G, Russo A, Palli D, Ottini L. BRCA1/BRCA2 rearrangements and CHEK2 common mutations are infrequent in Italian male breast cancer cases. Breast Cancer Res Treat. 2008;15:161–167. doi: 10.1007/s10549-007-9689-2. [DOI] [PubMed] [Google Scholar]
- Orr N, Lemnrau A, Cooke R, Fletcher O, Tomczyk K, Jones M, Johnson N, Lord CJ, Mitsopoulos C, Zvelebil M, McDade SS, Buck G, Blancher C, Trainer AH, James PA, Bojesen SE, Bokmand S, Nevanlinna H, Mattson J, Friedman E, Laitman Y, Palli D, Masala G, Zanna I, Ottini L, Giannini G, Hollestelle A, Ouweland AM, Novakovic S, Krajc M. et al. Genome-wide association study identifies a common variant in RAD51B associated with male breast cancer risk. Nat Genet. 2012;15:1182–1184. doi: 10.1038/ng.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauty de Chalon A, Teo Z, Park DJ, Odefrey FA, Hopper JL, Southey MC. Are PALB2 mutations associated with increased risk of male breast cancer? Breast Cancer Res Treat. 2010;15:253–255. doi: 10.1007/s10549-009-0673-x. [DOI] [PubMed] [Google Scholar]
- Silvestri V, Rizzolo P, Falchetti M, Zanna I, Masala G, Bianchi S, Palli D, Ottini L. Mutation analysis of BRIP1 in male breast cancer cases: a population-based study in Central Italy. Breast Cancer Res Treat. 2011;15:539–543. doi: 10.1007/s10549-010-1289-x. [DOI] [PubMed] [Google Scholar]
- Wasielewski M, den Bakker MA, van den Ouweland A, Meijer-van Gelder ME, Portengen H, Klijn JG, Meijers-Heijboer H, Foekens JA, Schutte M. CHEK2 1100delC and male breast cancer in the Netherlands. Breast Cancer Res Treat. 2009;15:397–400. doi: 10.1007/s10549-008-0162-7. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;15:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;15:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- kConFab: Kathleen Cuningham Foundation Consortium for research into Familial Breast Cancer. http://www.kconfab.org [DOI] [PubMed]
- Antoniou AC, Hardy R, Walker L, Evans DG, Shenton A, Eeles R, Shanley S, Pichert G, Izatt L, Rose S, Douglas F, Eccles D, Morrison PJ, Scott J, Zimmern RL, Easton DF, Pharoah PD. Predicting the likelihood of carrying a BRCA1 or BRCA2 mutation: validation of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system using data from UK genetics clinics. J Med Genet. 2008;15:425–431. doi: 10.1136/jmg.2007.056556. [DOI] [PubMed] [Google Scholar]
- Mann GJ, Thorne H, Balleine RL, Butow PN, Clarke CL, Edkins E, Evans GM, Fereday S, Haan E, Gattas M, Giles GG, Goldblatt J, Hopper JL, Kirk J, Leary JA, Lindeman G, Niedermayr E, Phillips KA, Picken S, Pupo GM, Saunders C, Scott CL, Spurdle AB, Suthers G, Tucker K, Chenevix-Trench G. Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Res. 2006;15:R12. doi: 10.1186/bcr1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Reporting recommendations for tumor marker prognostic studies. J Clin Oncol. 2005;15:9067–9072. doi: 10.1200/JCO.2004.01.0454. [DOI] [PubMed] [Google Scholar]
- Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJE. WHO Classification of Tumours of the Breast. Lyon: World Health Organization, IARC; 2012. [Google Scholar]
- Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, Gilks CB, van de Rijn M, Perou CM. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;15:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- Do H, Dobrovic A. Dramatic reduction of sequence artefacts from DNA isolated from formalin-fixed cancer biopsies by treatment with uracil- DNA glycosylase. Oncotarget. 2012;15:546–558. doi: 10.18632/oncotarget.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Krypuy M, Mitchell PL, Fox SB, Dobrovic A. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer. 2008;15:142. doi: 10.1186/1471-2407-8-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krypuy M, Newnham GM, Thomas DM, Conron M, Dobrovic A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer. 2006;15:295. doi: 10.1186/1471-2407-6-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, Riaz M, Koopman DG, Ten Hagen TL, de Leeuw BH, Zwarthoff EC, Teunisse A, van der Spek PJ, Klijn JG, Dinjens WN, Ethier SP, Clevers H, Jochemsen AG, den Bakker MA, Foekens JA, Martens JW, Schutte M. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat. 2010;15:53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- Garcia-Solano J, Conesa-Zamora P, Carbonell P, Trujillo-Santos J, Torres-Moreno DD, Pagan-Gomez I, Rodriguez-Braun E, Perez-Guillermo M. Colorectal serrated adenocarcinoma shows a different profile of oncogene mutations, MSI status and DNA repair protein expression compared to conventional and sporadic MSI-H carcinomas. Int J Cancer. 2012;15:1790–1799. doi: 10.1002/ijc.27454. [DOI] [PubMed] [Google Scholar]
- Capodanno A, Boldrini L, Ali G, Pelliccioni S, Mussi A, Fontanini G. Phosphatidylinositol-3-kinase alpha catalytic subunit gene somatic mutations in bronchopulmonary neuroendocrine tumours. Oncol Rep. 2012;15:1559–1566. doi: 10.3892/or.2012.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt JA, Fuchs CS, Ogino S. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res. 2012;15:2257–2268. doi: 10.1158/1078-0432.CCR-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;15:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Ottini L, Silvestri V, Rizzolo P, Falchetti M, Zanna I, Saieva C, Masala G, Bianchi S, Manoukian S, Barile M, Peterlongo P, Varesco L, Tommasi S, Russo A, Giannini G, Cortesi L, Viel A, Montagna M, Radice P, Palli D. Clinical and pathologic characteristics of BRCA-positive and BRCA-negative male breast cancer patients: results from a collaborative multicenter study in Italy. Breast Cancer Res Treat. 2012;15:411–418. doi: 10.1007/s10549-012-2062-0. [DOI] [PubMed] [Google Scholar]
- Kimbung S, Biskup E, Johansson I, Aaltonen K, Ottosson-Wadlund A, Gruvberger-Saal S, Cunliffe H, Fadeel B, Loman N, Berglund P, Hedenfalk I. Co-targeting of the PI3K pathway improves the response of BRCA1 deficient breast cancer cells to PARP1 inhibition. Cancer Lett. 2012;15:232–241. doi: 10.1016/j.canlet.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Basham VM, Lipscombe JM, Ward JM, Gayther SA, Ponder BA, Easton DF, Pharoah PD. BRCA1 and BRCA2 mutations in a population-based study of male breast cancer. Breast Cancer Res. 2002;15:R2. doi: 10.1186/bcr419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Bulman M, Young K, Howard E, Bayliss S, Wallace A, Lalloo F. BRCA1/2 mutation analysis in male breast cancer families from North West England. Fam Cancer. 2008;15:113–117. doi: 10.1007/s10689-007-9153-9. [DOI] [PubMed] [Google Scholar]
- Korde LA, Zujewski JA, Kamin L, Giordano S, Domchek S, Anderson WF, Bartlett JM, Gelmon K, Nahleh Z, Bergh J, Cutuli B, Pruneri G, McCaskill-Stevens W, Gralow J, Hortobagyi G, Cardoso F. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol. 2010;15:2114–2122. doi: 10.1200/JCO.2009.25.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottini L, Rizzolo P, Zanna I, Falchetti M, Masala G, Ceccarelli K, Vezzosi V, Gulino A, Giannini G, Bianchi S, Sera F, Palli D. BRCA1/BRCA2 mutation status and clinical-pathologic features of 108 male breast cancer cases from Tuscany: a population-based study in central Italy. Breast Cancer Res Treat. 2009;15:577–586. doi: 10.1007/s10549-008-0194-z. [DOI] [PubMed] [Google Scholar]
- Comprehensive molecular portraits of human breast tumours. Nature. 2012;15:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C, Kahler J, Workman J, Bittner M, Dudkin L, Epstein DM, Gibson NW, Wild R, Arnold LD, Houghton PJ, Pachter JA. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther. 2011;15:1394–1406. doi: 10.1158/1535-7163.MCT-10-1099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1: BOADICEA scores for patients included in study. Probability (Prob) score (0 to 1) is generated for BRCA1 and BRCA2 mutations for each case, stratified by known BRCA status.
Supplementary table 1. REMARK criteria leading to cases recruitment.
Supplementary table 2. HRM and Sequence specific PIK3CA, AKT1, KRAS and BRAF primers.
Supplementary figure 2. Disease specific survival stratified by 1a) nuclear p4EBP1 expression, 1b) cytoplasmic p4EBP1 expression, 1c) pS6 expression and 1d) pAKT expression.