

Abstract
Rationale:
Oxidants generated by activated endothelial cells are known to induce apoptosis, a pathogenic feature of vascular injury and inflammation from multiple etiologies. The melastatin-family transient receptor potential 2 (TRPM2) channel is an oxidant-sensitive Ca2+ permeable channel implicated in mediating apoptosis; however, the mechanisms of gating of the supra-normal Ca2+ influx required for initiating of apoptosis are not understood.
Objective:
Here we addressed the role TRPM2 and its interaction with the short splice variant TRPM2-S in mediating the Ca2+ entry burst required for induction of endothelial cell apoptosis.
Methods and Results:
We observed that TRPM2-S was basally associated with TRPM2 in the endothelial plasmalemma and this interaction functioned to constitutively suppress TRPM2-dependent Ca2+ gating. ROS production in endothelial cells or directly applying ROS induced PKCα activation and phosphorylation of TRPM2 at Ser 39. This in turn stimulated a large entry of Ca2+ and activated the apoptosis pathway. A similar TRPM2-dependent endothelial apoptosis mechanism was seen in intact vessels. The PKCα-activated phospho-switch opened the TRPM2 channel to allow large Ca2+ influx by releasing TRPM2-S inhibition of TRPM2, which in turn activated caspase-3 and cleaved the caspase substrate poly(ADP-ribose) polymerase.
Conclusions:
Here we describs a fundamental mechanism by which activation of the trp super-family TRPM2 channel induces apoptosis of endothelial cells. The signaling mechanism involves ROS-induced PKCα activation resulting in phosphorylation of TRPM2-S that allows enhanced TRPM2-mediated gating of Ca2+ and activation of the apoptosis program. Strategies aimed at preventing the uncoupling of TRPM2-S from TRPM2 and subsequent Ca2+ gating during oxidative stress may mitigate endothelial apoptosis and its consequences in mediating vascular injury and inflammation.
Keywords: Apoptosis, oxidant-activated cation channel, endothelium, vascular permeability, inflammation, oxidant signaling
Introduction
TRPM2 is an oxidant-sensitive Ca2+-permeable channel expressed in many cells including neurons 1,2, microglia 3,4, multiple lung cell types 5,6, pancreas β cells 7-9, hematopoietic and immune cells 10,11, and vascular endothelial cells 5. However, the function of TRPM2 remains enigmatic. TRPM2 is activated by the generation of ROS such as H2O2 and production of adenosine diphosphate ribose (ADPR) following DNA damage and activation of the enzyme poly(ADPR) polymerase 6,12. TRPM2 has been implicated in mediating of oxidant-induced apoptosis secondary to Ca2+ influx that may initiate apoptosis program via the caspase pathway 1,13,14. Although apoptosis is important in normal biological processes and development, apoptosis of endothelial cells, which have low turnover in vessels 15, is a fundamental pathogenic feature of inflammatory and vascular diseases such as acute lung injury16 and sepsis 17. Our studies have demonstrated a key role of TRPM2 in mediating oxidative injury of the endothelium 5 resulting in disruption of endothelial barrier and tissue edema 18-20. A component of endothelial disruption seen in these studies may well have been due to TRPM2-induced apoptosis.
TRPM2 channel opening after exposure to H2O2 and other ROS is induced by the binding of ADPR to the Nudix box sequence motif (NUDT9-H) in the carboxyl-terminal domain of TRPM2 5,6,10,12,21-23. H2O2 produced in the cell 5 also activated the production of ADPR 6,10,23,24, which functioned by binding to the TRPM2 Nudix motif 6,10,12,24,25. In addition, other mechanisms of TRPM2 activation such as direct oxidative modification of the channel have been proposed 26.
Besides TRPM2 5,27, several splice variants of TRPM2 associated with TRPM2 in the plasma membrane have also been identified 28. Their role in regulating TRPM2 function and mediating oxidant-induced apoptosis, remains obscure. Of particular interest is the short splice variant (TRPM2-S), which functions as a dominant-negative to inhibit TRPM2 channel activity 14,28 but which itself lacks both the carboxyl terminus present in the long isoform TRPM2 as well as the Ca2+-permeable pore present in TRPM2 28. In cells in which both isoforms are expressed, TRPM2-S interact with TRPM2 to inhibit formation of functional homotetrameric channels 14. Here we investigated the interaction of TRPM2-S with TRPM2 and how the component cooperated to signal oxidant-induced apoptosis in endothelial cells. The study presents a new mechanism of endothelial apoptosis involving ROS-induced and PKCα phosphorylation-dependent disruption of the interaction of TRPM2 with TRPM2-S and opening of the channel to allow sufficient Ca2+ entry required for activation of the apoptosis program.
METHODS
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Isolation of mouse endothelial cells
Endothelial cells were isolated from lungs of WT, PKCα−/− (obtained from Dr Jeffrey D. Molkentin, University of Cincinnati, Cincinnati, OH) and TRPM2−/− mice (GlaxoSmithKline). The cells were used between passages 2–5.
Transfections
Human pulmonary artery endothelial cells (HPAEC; Clonetics, La Jolla, CA) were cultured in gelatin-coated flasks and used between passages 3–6. Human TRPM2-S splice variant, tagged with poly-His (His6-TRPM2-S), was inserted into a pcDNA3 expression vector (Invitrogen). Phosphorylation-defective TRPM2-S was generated by alanine substitution (S39A) and phosphorylation-mimetic TRPM2-S was generated by aspartic substitution (S39D). Transfection of TRPM2-S constructs using fuGENE HD was verified by Western blotting. Control cells received vector alone.
siRNA experiments
HPAECs were transiently transfected with TRPM2 or PKCα siRNAs (100 nmol/L; Santa Cruz Biotechnology, Santa Cruz, CA) using TransIT-TKO transfection reagent (Mirus, Madison, WI); nonspecific siRNA served as control (Ambion, Austin, TX). Transfection efficiency was >75%.
Immunoprecipitation and phosphorylation studies
Untransfected, His6-(S39A)TRPM2-S and His6-(S39D)TRPM2-S transfected HPAEC cultures were treated with 300 μmol/L H2O2 for indicated times (37°C). In some experiments, cells were pretreated with DPQ or PKC inhibitors 30 min prior to the assay. In other experiments, cells first received siRNA to suppress TRPM2 or PKCα expression. TRPM2 or PKCα immune complexes were precipitated with protein A-Sepharose beads (Sigma) for 2 h at 4°C as described 5.
Generation of H2O2 using glucose oxidase/glucose
H2O2 production in vitro was induced by glucose (1 mmol/L) and glucose oxidase, (1-2.5 mU/ml) and was measured spectrophotometrically from the generation of resorufin (absorbance, 565 nm; extinction coefficient, 58,000 M-1cm-1). Glucose oxidase produced H2O2 at a constant rate (320 nmol/L H2O2/min).
Analysis for apoptosis
Apoptosis was identified by double-fluorescent staining with Phycoerythrin (PE) Annexin V-FITC and 7-Aminoactinomycin D (7-AAD), which detected apoptotic and dead cells, respectively. Confluent endothelial monolayers, without or with PKCα inhibition or silencing were incubated in 300 μmol/L H2O2 for 6 or 24 h (37°C). Cells were washed twice with PBS and trypsinized; samples of 1 × 106 cells were incubated with 5 μl of PE-labeled Annexin V and 5 μl of 7-AAD (BD Bioscience, Rockville, MD) for 20 min at 24 °C in the dark and analyzed with a Beckman Coulter CyAn II cytometer (Beckman Coulter, Miami, FL). We also assessed apoptosis in intact lung vascular endothelium by immunofluorescence and TUNEL assay. Lungs of TRPM2−/− and wild-type mice were perfused (2 ml/min, 37°C) for 3 h with recirculation of RPMI 1640 (5 ml) containing H2O2 (300 μmol/L) or glucose oxidase. Lungs were removed, and frozen by the OCT method. Frozen lungs, sectioned (5 μm) and fixed in 3.7% formaldehyde, were permeabilized with 0.2% triton X-100. Tissue sections were blocked with 10% FBS and incubated with goat anti-VE-cadherin and rabbit anti-PARP (i.e., the cleaved 89 kDa fragment; 1:200 dilution) overnight (4°C). The sections were incubated with secondary antibodies conjugated to Alexa Fluor 488 and 594 (Invitrogen). As an alternative to cleaved PARP antibody, TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) staining was performed according to the manufacturer’s protocol (Roche Diagnostics Corp., Indianapolis, IN). Nuclei were visualized by 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Saint Louis, MO). Slides were analyzed under a Zeiss fluorescence microscope using AxioVision software. Apoptotic cells were identified by double staining (PARP + VE-cadherin, or TUNEL + VE-cadherin).
Statistical analysis
Statistical comparisons were made with the two-tailed Student’s t-test. The significance level was p < 0.05.
Institutional study approval
All studies were conducted after review by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago where the work was performed, and in accordance with the Policy on the Care, Welfare and Treatment of Laboratory Animals.
Methods in Supplement
Methods for [Ca2+]i measurements, TRPM2 protein purification, in vitro phosphorylation assay, bone marrow transplantation, murine model of endotoxin-mediated mortality, and Western blotting are given in the Online Data Supplement.
RESULTS
TRPM2 is required for H2O2-induced endothelial cell apoptosis
Apoptosis was determined by staining endothelial cells with annexin V-PE and 7-AAD followed by FACS assessment (Figure 1A-B). H2O2 in a concentration-dependent manner induced apoptosis within 24 h with an EC50 value of 136 ± 6 μmol/L (Figure 1A). Inhibition of TRPM2 by an anti-TRPM2 blocking antibody or TRPM2 siRNA silencing prevented the apoptosis (Figure 1B). The flow cytometry dot plot data demonstrating apoptosis are shown in Online Figure I. The sustained generation of H2O2 (320 nmol/L/min for 90 min) by glucose oxidase with glucose substrate also induced endothelial apoptosis, which was blocked by TRPM2 silencing or inhibition of channel activity (Figure 1B). To address in vivo relevance, we also examined apoptosis in endothelial cells of lung vessels in wild-type and TRPM2 knockout mice perfused with a solution containing H2O2 or glucose oxidase/glucose (this generated 320 nmol/L/min H2O2).
Figure 1. TRPM2 is required for H2O2-mediated apoptosis of endothelial cells.

(A-B) Confluent endothelial cell monolayers (HPAECs) challenged with H2O2 or glucose oxidase/glucose were labeled with PE Annexin V-FITC and 7-AAD and analyzed by flow cytometry. (A) Concentration-response curve for H2O2-induced apoptosis after 24-h period of H2O2 exposure. Mean apoptotic values (± SEM) obtained by flow cytometry (top panel) are plotted as % apoptotic cells vs. [H2O2] (EC50 = 136 μmol/L ; bottom panel) (n=3). (B) Left, representative flow cytometry histograms 6 or 24 h after exposure to 300 μmol/L H2O2 or glucose oxidase/glucose (90 min), with or without prior TRPM2 silencing or inhibition with blocking TRPM2 antibody. Right panel, mean percent apoptotic cells at 0, 6, or 24 h after H2O2 treatment (± SEM, n=3). Baseline values were not significantly altered by TRPM2 silencing or TRPM2 blocking Ab. (C-D) Apoptosis in lungs of TRPM2−/− and wild-type (WT) mice measured 3 h after perfusion with solution containing H2O2 (300 μmol/L) or glucose oxidase/glucose (75 min). (C) Right, immunofluorescent staining of frozen lung sections using VE-cadherin (red) and cleaved-PARP (green) antibodies and DAPI (blue)(n=3). Scale bar: 50 μm. Left, quantification of apoptotic endothelial cells (±SEM; n=6), * p ≤ 0.007 vs. WT lungs (t-test). (D) Western immunoblots (Right) and quantification of inactive or cleaved caspase-3 and active or cleaved PARP in lung homogenates (Left). GAPDH was used as loading control. (±SEM; n=3), * p ≤ 0.002, ** p ≤ 0.009 vs. WT perfused-lungs.
As Ca2+ signaling activates the apoptotic pathway through activation of caspase-3 followed by cleavage of caspase substrates, such as 113-kDa poly(ADP-ribose) polymerase (PARP) 29,30, we also determined expression of the 17-kDa and 20-kDa caspase-3 fragments and 89-kDa or 24-kDa caspase cleavage fragments of PARP 29,30. Data from lungs showed that cleaved-PARP characteristically co-localized with the endothelial cell marker VE-cadherin whereas deletion of TRPM2 (TRPM2−/− mice) markedly reduced endothelial apoptosis (Figure 1C). Western blotting confirmed the activation of caspase-3 and cleaved PARP in lungs of wild-type and not TRPM2−/− mice (Figure 1D). Using another apoptosis assay, TUNEL staining, we observed fewer endothelial cells undergoing apoptosis after H2O2 or glucose oxidase/glucose infusion in TRPM2−/− mice compared to WT (Online Figure II).
H2O2 induces PKCα phosphorylation of TRPM2-S
We next determined the role of TRPM2 and its binding partner TRPM2-S in the mechanism of apoptosis. Western blotting showed that both TRPM2-S and TRPM2 (90 and 171 kDa, respectively) were basally expressed in HPAECs (Figure 2A) as well as in multiple other endothelial cells examined (unpublished observations). TRPM2 and TRPM2-S expression was not modified by H2O2 exposure per se (Figure 2A). Using the motif scanning graphic software (Merck Genome Research Institute), we identified a putative high affinity binding site for PKCα near the N-terminus of TRPM2-S, at Ser 39. This site was predicted as a possible domain that could be phosphorylated by PKCα. We observed that PKCα was basally expressed in these cells and its expression was not modified by H2O2 (Figure 2A). Depleting PKCα using siRNA did not modify the expression of either TRPM2 or TRPM2-S (Figure 2A), whereas TRPM2 depletion as expected suppressed the expression of its splice variants (Figure 2A).
Figure 2. PKCα binding and phosphorylation of TRPM2-S.

HPAECs were transduced with siRNA to suppress the expression of TRPM2 or PKCα or pretreated with PKC inhibitors (100 nmol/L Gö6976, 1 μmol/L PKCαi or 1 μmol/L PKCβIIi). Cells were challenged with 300 μmol/L H2O2 for the indicated times at 37°C. (A) Western blots from HPAEC lysates identifying expression of TRPM2 isoforms and PKCα; GAPDH was used as loading control. Endothelial cell expression of TRPM2-S (90 kDa) and two TRPM2 splice variants (140 and 171 kDa) were suppressed by TRPM2 silencing and PKCα protein expression (82 kDa) was blocked by PKCα silencing. (B) H2O2 induced association of PKCα and TRPM2-S. PKCα was immunoprecipitated from cell lysates. Following SDS-PAGE and electrophoretic transfer, co-immunoprecipitated TRPM2 was detected using an antibody recognizing both TRPM2 and TRPM2-S. TRPM2-S associated with PKCα immediately after H2O2 exposure whereas inhibition of PKCα activation prevented the association. (C) H2O2 induced PKCα-dependent phosphorylation of 90kDa TRPM2-S splice variant. TRPM2 was immunoprecipitated from same cell lysates using an Ab that recognized either TRPM2 isoform. Top panel: Blots showing phosphorylation of TRPM2 using monoclonal anti-phospho-Ser antibody. Membrane was reblotted with anti-phospho-PKCα (Ser 657) Ab to check the amount of phosphorylated PKCα in the complex (middle panel). Immunoprecipitation of TRPM2 was verified using an anti-TRPM2 Ab (lower panel). (D) Mean densitometric values (± SEM; n=3-4) obtained in B-C showing that PKCα inhibition prevented H2O2-induced association of PKCα with TRPM2-S and phosphorylation of TRPM2-S
Figure 3. PKCα and TRPM2 cooperation mediates Ca2+ entry in endothelial cells required for signaling apoptosis.

(A) Ca2+ repletion transients generated by “Ca2+-add-back” in the presence of H2O2. Cultured HPAECs were loaded with Fura-2 Ca2+ dye, washed, and transferred to Ca2+-free medium. In control cells, H2O2 (100 μM) elicited a marked Ca2+ transient on Ca2 repletion (red trace). Ca2+ transients were blocked by TRPM2 silencing (grey trace) and reduced by Gö6976 (100 nmol/L; green trace), PKCαi (1 μmol/L; cyan trace), or after PKCα silencing (yellow trace); PKCβIIi (1 μmol/L, navy trace) and control siRNA (purple trace) had no effect. Ordinate gives [Ca2+]i as 340:380 nm ratio. (B) Summary of mean ratiometric data (± SEM) for the peak intracellular [Ca2+]i obtained in (E) (n = 3 to 5). *P ≤ 0.0002 vs. control (t-test).
To address whether H2O2 was involved in the phosphorylation at Ser39 on TRPM2-S, we used HPAEC monolayers treated with PKCα inhibitors (Gö6976 or PKCα blocking peptide, PKCαi) or transfected with siRNA to knockdown PKCα expression. PKCα was immunoprecipitated from lysates of cells exposed to H2O2, and co-immunoprecipitated TRPM2 and TRPM2-S were detected using an antibody recognizing each form (Figure 2B). H2O2 rapidly induced the association of PKCα with TRPM2-S, but not with TRPM2, and the response persisted up to 5 min (Figure 2B). Immunoprecipitation was reduced when PKCα activation was inhibited (Figure 2B). Treatment with PKCβII inhibitory peptide, used as control for non-specific effects of Gö6976 in blocking activation of both PKCα and PKCβII 31, did not modify the H2O2-induced TRPM2-S association with PKCα (Figure 2B). PKCα-TRPM2-S immunoprecipitation was also suppressed predictably by TRPM2 silencing (Figure 2B). Control experiments showed that transfection of endothelial cells with TRPM2 siRNA significantly reduced the expression of both TRPM2 and TRPM2-S (Figure 2A). Control experiments confirmed that siRNA effectively suppressed PKCα expression (Figure 2B). Treatment of cells with PKCα inhibitors (Gö6976 or PKCαi) and PKCβII blocking peptide did not modify PKCα expression (Figure 2B).
Since the above studies dealt with the role of H2O2 in activating the phosphorylation of TRPM2-S, we next examined whether key alterations could be could be replicated using a physiological stimulus to generated oxidants. Here we used tumor necrosis factor alpha (TNF-α), a generator of intracellular oxidants and potent inducer of endothelial cell apoptosis 32. We observed that TNF-α induced the association of PKCα with TRPM2-S whereas suppressing PKCα activity prevented the association (Online Figure IIIA).
To determine whether PKCα was responsible for phosphorylating TRPM2-S following H2O2 challenge, in other studies TRPM2 proteins in cell lysates were precipitated using antibodies recognizing TRPM2 and TRPM2-S, and phosphorylated proteins were visualized using anti-phospho-Ser antibody. Western blotting demonstrated that only TRPM2-S was phosphorylated, which occurred within 1 min of H2O2 exposure with maximum response seen at 2 min, while there was no phosphorylation of TRPM2 (Figure 2C). Western blotting also showed that TRPM2-S but not TRPM2 was phosphorylated within the same time frame following TNFα exposure (Online Figure IIIB). Phosphorylation of PKCα (82 kDa) was detected as a co-migrating band on the gel (seen in top blot of Figure 2C and Online Figure IIIB), an indication that the kinase was in the active state.
We next determined phosphorylation of PKCα using an antibody recognizing phosphorylated on Ser 657, the crucial PKCα catalytic domain 33. H2O2 rapidly induced phosphorylation of PKCα at this site and the phosphorylated PKCα co-migrated with TRPM2 (middle blot) (Figure 2C). Treatment with Gö6976 (but not with control PKCβIIi) inhibited not only PKCα phosphorylation but also H2O2-induced phosphorylation of TRPM2-S (Figure 2C). These results thus show time-dependent and reversible association between TRPM2-S and PKCα induced by PKCα activation (Figure 2D).
S39 phosphorylation of TRPM2-S activates TRPM2 and supra-normal Ca2+ influx
We used the Fura-2 dye to study the Ca2+ entry response activated by TRPM2 interaction with TRPM2-S, In addition, we employed the "Ca2+ add-back" protocol to rule out any indirect effects of H2O2 on Ca2+ entry secondary to Ca2+-store depletion 5. In the absence of extracellular Ca2+, H2O2 did not produce a Ca2+ transient (Figure 3A, B), indicating that H2O2 did not deplete intracellular Ca2+ stores. By contrast, extracellular Ca2+ repletion in the continued presence of H2O2 elicited a sharp and marked increase in intracellular Ca2+ concentration secondary to Ca2+ entry (Figure 3A, B). TRPM2 knockdown markedly suppressed the Ca2+ transient (Figure 3A, B), showing that H2O2-induced Ca2+ entry required TRPM2. Gö6976 significantly decreased the amplitude of Ca2+repletion-dependent transients by 66 ± 9 % and PKCαi reduced Ca2+ transient by 46 ± 10 % (Figure 3 A, B). PKCα silencing also reduced Ca2+ entry by 43 ± 6 % (Figure 3A, B). Treatment of cells with control PKCβII peptide inhibitor, however, did not modify H2O2 -activated Ca2+ entry via TRPM2 channels (Figure 3A, B). Along the same lines, TNFα-induced Ca2+ entry in endothelial cells (Online Figure IIID) was also decreased by inhibiting PKCα.
We next addressed the role of the S39 phospho-switch on TRPM2-S in mediating TRPM2 channel activity. Here we determined whether mutation of TRPM2-S at Ser 39 (S39A), the PKCα phosphorylation site disrupted Ca2+ signaling. The mutant was tagged on its C-terminus with a poly-His fusion protein. Transfected HPAECs showed protein expression of the (S39A)-TRPM2-S mutant (Figure 4A). Western blotting showed that S39A mutation of TRPM2-S abrogated the migration of TRPM2-S with PKCα on the gel (Figure 4B) as well as phosphorylation of TRPM2-S by PKCα after H2O2 challenge (Figure 4C). PKCα also did not migrate with (S39A)-TRPM2-S mutant (Figure 4C).
Figure 4. PKCα binds TRPM2-S at Ser 39 and activates apoptosis-inducing Ca2+ entry signal in endothelial cells.

The sole predicted PKCα phosphorylation site near the TRPM2-S N-terminus at Ser 39 was mutated by Ala substitution (resulting in phosphodefective mutant). HPAEC monolayers transduced with mutant TRPM2-S (tagged on its carboxy-terminal end with poly-His residues) were grown to confluence for Western blot analysis (A through C) or intracellular Ca2+ measurements using fura-2 (D). (A-C) Cells were exposed to 300 μM H2O2 for the indicated times. (A) Western blots for TRPM2, PKCα, and GAPDH expression in cells transduced with phosphodefective construct. Transfected protein was detected with an anti-His6 Ab confirming the expression of mutant TRPM2-S construct. (B) PKCα was immunoprecipitated from cell lysates with an antibody and co-immunoprecipitated TRPM2 protein was detected using an Ab recognizing both forms of TRPM2. Graph in B shows mean densitometric values (± SEM; n=3-4). Mutation of Ser 39 with Ala in TRPM2-S prevented TRPM2-S association with PKCα. (C) TRPM2 was immunoprecipitated from the same lysates and phosphorylated TRPM2 was detected using anti-phospho-Ser Ab. Graph in C shows mean densitometric values (± SEM; n=3-4). Ala substitution at Ser 39 abrogated H2O2-induced phosphorylation of TRPM2-S confirming the importance of the PKCα phosphorylation site on TRPM2-S at Ser 39. (D) Ca2+ mobilization assay was carried out using the “Ca2+ add-back” protocol. Transduction of phosphodefective TRPM2-S mutant suppressed H2O2-induced Ca2+ entry. * p = 0.0001 compared with control (t-test). (n = 3 per bar); error bars, ± SEM.
We next determined the functional significance of the failure of PKCα to bind to and phosphorylate TRPM2-S on the TRPM2-mediated Ca2+ entry. Intracellular Ca2+ transient elicited by H2O2 was markedly reduced in cells transduced with the TRPM2-S phospho-defective mutant (Figure 4D). To validate the finding that PKCα was indeed responsible for phosphorylation of TRPM2-S at S39, we treated an extract of native protein with recombinant active PKCα. We observed that active PKCα induced phosphorylation of WT TRPM2-S but not of S39A mutant (Online Figure IV).
PKCα phosphorylation of TRPM2-S induces TRPM2-S dissociation from TRPM2
We next determined whether PKCα phosphorylation of TRPM2-S in some manner interfered with TRPM2-S association with TRPM2, thus permitting TRPM2 to gate Ca2+ at sufficient level to activate the apoptosis program. As TRPM2 variants generated by alternative splicing differed only in their C terminal 28, we immunoprecipitated TRPM2 from cell lysates using an anti-TRPM2 antibody recognizing the region present solely in TRPM2 form. TRPM2-S, which in the plasma membrane basally associated with TRPM2, dissociated within minutes from TRPM2 following H2O2 addition (Figure 5A). Inhibition of PKCα activation suppressed this TRPM2-S dissociation from TRPM2 (Figure 5A). S39A mutation of TRPM2-S also suppressed the dissociation of TRPM2 from TRPM2-S (Figure 5A). PKCα-dependent phosphorylation of TRPM2-S at Ser 39 blocked the interaction of TRPM2-S with TRPM2 (summarized in Figure 5B). These results show that phosphorylation of TRPM2-S at Ser 39 was responsible for releasing the TRPM2-S inhibition of TRPM2 and thus mediated the increased Ca2+ entry needed for apoptosis.
Figure 5. PKCα phosphorylation of TRPM2-S mediates TRPM2-S dissociation from TRPM2 resulting apoptosis-inducing Ca2+ entry signal.

HPAEC monolayers were pretreated with PKCα and PKCβII (control) inhibitors 45 min prior to experiments. (A) TRPM2 was immunoprecipitated from cell lysates with an anti-TRPM2 antibody recognizing the region present only on the long isoform following H2O2 (300 μM) exposure for indicated times. Co-immunoprecipitated short isoform was then detected using an Ab that recognizes both TRPM2 and TRPM2-S. At 0 time, TRPM2 in plasma membrane was associated with its short isoform, and H2O2 application induced rapid dissociation of TRPM2-S from TRPM2. H2O2-mediated dissociation was suppressed by PKCα inhibition and did not occur when Ser 39 of TRPM2-S was substituted by Ala. (B) Western blots were quantified by densitomitry. Co-immunoprecipitated TRPM2-S was quantified as ratio to TRPM2 and plotted relative to zero time value (mean ± SEM; n = 3)..
To reinforce the crucial role of TRPM2-S S39 phosphorylation in mediating TRPM2 channel activity, we mutated TRPM2-S S39 to aspartate (S39D) to mimic the effects of phosphorylation. This poly-His tagged phosphomimetic mutant was expressed in HPAECs (Online Fig VA), and we examined its ability to associate with TRPM2, and influence Ca2+ entry. Western blotting showed that phosphomimetic of TRPM2-S promoted TRPM2-S interaction with PKCα (Online Figure VB), but impaired its association with TRPM2 (Online Figure VC) under basal condition in the absence of H2O2 as well as after H2O2 challenge. Moreover, S39D mutation of TRPM2-S enhanced TRPM2-mediated Ca2+ entry after H2O2 challenge (Online Figure VD) consistent with the key role of PKCα phosphorylation of TRPM2-S in activating TRPM2 channel activity.
To elucidate further the mechanism of PKCα regulation of TRPM2 channel activity, we next co-expressed the TRPM2-S phosphorylation mutants with either PKCα siRNA or control siRNA in endothelial cells (Online Figure VI). Expression of (S39A)TRPM2-S phospho-defective mutant in control siRNA-transduced cells as expected inhibited H2O2-elicited Ca2+ entry compared to control cells whereas expression of phospho-mimetic mutant enhanced this response. The decreased H2O2-activated Ca2+ entry caused by depletion of PKCα was restored by expression of the (S39D)TRPM2-S but not the (S39A)TRPM2-S mutant, consistent with the essential role of PKCα phosphorylation of TRPM2-S in activating TRPM2 channel activity.
PKCα mediates H2O2-induced apoptosis through activation of TRPM2
FACS analysis showed that inhibition of PKCα activation or its silencing protected the cells from H2O2-induced apoptosis (Figure 6A and B). The role of PKCα in regulating TRPM2-mediated apoptosis was also seen in endothelial cells transduced with the (S39A)-TRPM2-S mutant (Figure 6A). Using lung endothelial cells cultured from TRPM2 or PKCα knockout mice to validate the above studies in human endothelial cells, we observed normal expression of TRPM2 in endothelial cells from PKCα knockout mice as well as normal expression of PKCα in endothelial cells from TRPM2 knockout mice (Figure 6A and B). The H2O2-induced Ca2+ entry was virtually abolished in TRPM2-null cells and was reduced by 40% in PKCα-null cells (Figure 6C). As in the human cells, H2O2-mediated apoptosis in WT mouse endothelial cells was concentration-dependent (Figure 6D). Deletion of TRPM2 caused 2.4-fold rightward shift in the concentration-response curve for H2O2-induced apoptosis (EC50 shift= 213-553 μmol/L) indicating the crucial role of TRPM2 in mediating H2O2-induced apoptosis (Figure 6D). Deletion of the PKCα gene similarly inhibited apoptosis (EC50 shift = 213-512 μmol/L) (Figure 6D).
Figure 6. H2O2-induced endothelial cell apoptosis resulting from TRPM2-mediated Ca2+ influx.

HPAECs (A) and mouse lung endothelial cells (B-D) challenged with H2O2 were labeled with PE-Annexin/7-AAD. (A) Flow-cytometry histograms (Left) and summary plots of apoptosis (Right) 0, 6, and 24 h after challenge with H2O2 (300 μmol/L) or glucose oxidase/glucose to generate 320 nmol/L H2O2/min for 90 min, as a function of PKCα inhibition or silencing (± SEM, n=5). (B-D) Mouse endothelial cells were isolated from lungs of TRPM2−/−, PKCα−/−, and WT mice. (B) Western blot verifying absence of PKCα expression in PKCα−/− cells and of TRPM2 expression in TRPM2−/− cells. (C) Left, Ca2+ mobilization assay using “Ca2+ add-back” protocol with the Fluor-3 Ca2+ indicator. Right, Mean ratiometric values (± SEM) for steady-state [Ca2+]i (n=6). *p ≤ 0.0001 vs. WT cells (t-test). (D) Dose-response curve for H2O2-induced apoptosis in mouse endothelial cells detected by flow cytometry. Deletion of PKCα or TRPM2 caused ~2.5-fold rightward shift in the dose-response curve.
Deletion of PKCα gene in mice reduces TRPM2-induced endothelial cell apoptosis improves survival in endotoxemia
To address the pathophysiological significance of PKCα phosphorylation of TRPM2 channel activity in mediating apoptosis in vivo, we examined the apoptosis response in mouse lung endothelial cells and survival of mice following intraperitoneal (ip) challenge with lipopolysaccharide (LPS, 30 mg/kg), the Gram-negative bacterial endotoxin, which produces ROS in endothelial cells34,35. Because TRPM2 and PKCα expressed in myeloid cells may also play a role in ROS production and apoptosis36, we generated chimeric mice in which the PKCα- and TRPM2-deficient mice were transplanted with bone marrow cells from wild-type mice. These mice showed comparable TRPM2 and PKCα protein expression as wild-type (Fig 7A). We observed that either TRPM2 or PKCα deletion markedly reduced endothelial cell apoptosis in lungs 4 h after LPS treatment compared to wild-type mice (Figure 7B). In a positive control experiment, administration of the oxidant scavenger Tempol in mice 30 min prior to LPS also reduced oxidant-mediated LPS-induced apoptosis (Figure 7B). In addition, deletion of either TRPM2 or PKCα significantly improved survival rate of LPS-challenged mice (Figure 7C).
Figure 7. PKCα interaction with TRPM2 in mice is required lung endothelial cell apoptosis in response to LPS and contributes to mortality.

WT, TRPM2−/− and PKCα−/− mice were transplanted with bone marrow cells isolated from WT mice 8 weeks prior to Western blotting (A), apoptosis (B-C) and survival (D) studies. (A) Representative Western blots verifying expression of TRPM2, PKCα, and β-actin in bone marrow of transplanted mice (molecular masses of 171, 82, and 45 kDa, respectively). Left, protein was quantified by densitometry. TRPM2 and PKCα densities were normalized to β-actin and plotted as percentage of untreated control (mean ± SEM for n=3). (B-C) Endothelial apoptosis (B) and oxidant production (C) were determined in lungs of mice 4h after intraperitoneal injection of lipopolysaccharide (LPS, 40 mg/kg) and in lungs of mice treated with the oxidant scavenger Tempol (100 mg/kg, IP) 30 min before injection of LPS. (B) Right, immunofluorescent staining of frozen lung sections using VE-cadherin (red) antibody, TUNEL (green), and DAPI (blue)(n=3). Scale bar: 50 μm. Left, quantification of apoptotic endothelial cells (±SEM; n=3), * p ≤ 0.001 and # p ≤ 0.005 vs. LPS-treated WT lungs. TRPM2 and PKCα deletion significantly reduced LPS-induced endothelial apoptosis. (C) Lungs were homogenized and assayed for H2O2 accumulation using the horseradish peroxide-linked Amplex Red assay. H2O2 was determined spectrophotometrically from its absorbance at 570 nM and corrected for total protein (± SEM, n=3). * p ≤ 0.005 vs. LPS-treated control lungs (D) Deletion of either TRPM2 or PKCα reduced LPS-induced lethality in mice. LPS (30 mg/kg) was injected intraperitoneally and survival was assessed every 12 h during the experiment (WT, n=16; TRPM2−/−, n= 16; and PKCα−/−, n=12). Statistical analysis was performed using the log-rank test. *p ≤ 0.03 and #p ≤ 0.05 vs. WT cells.
DISCUSSION
In the present study, we addressed the role of the ROS-activated TRPM2 channel in mediating endothelial cell apoptosis. We identified that the interaction of the 171 kDa TRPM2 10,12,27,37 with its 90 kDa splice variant TRPM2-S 28 in the endothelial cell plasmalemma. This interaction functioned constitutively to restrain TRPM2 Ca2+ entry. However, ROS-induced activation of PKCα and resulting phosphorylation of TRPM2-S at Ser39 released the TRPM2-S inhibition of TRPM2 to induce the large Ca2+ influx required for activation of the caspase apoptosis program.
PKCα phosphorylation of TRPM2-S and the dissociation of TRPM2-S from TRPM2 increased the Ca2+ concentration in endothelial cells to 4-fold the baseline levels within the range of the Ca2+ burst required to signal apoptosis, which has the intracellular Ca2+ concentration threshold of 200-500 nmol/L 38. Inhibition of PKCα by preventing the phosphorylation of TRPM2-S reduced Ca2+ entry through TRPM2 by half this level, well below the Ca2+ threshold required for activation of caspase-mediated apoptosis.
A stop codon (TAG) on the TRPM2 gene is located at the splice junction between exons 16 and 17; hence, alternative splicing resulted in deletion of the 4 C-terminal transmembrane domains in TRPM2-S, the putative Ca2+ -permeable pore region28. The observation that TRPM2-S served as a dominant negative for TRPM2 and its uncoupling from TRPM2 was required for the full gating of Ca2+ identifies TRPM2-S as an important intrinsic negative regulator of endothelial cell apoptosis. H2O2 exposure or oxidants generated by mediators such as TNF-α induced the interaction between PKCα and TRPM2-S permitting the channel to open for Ca2+ entry. Thus, TRPM2-S only functioned to induce apoptosis when PKCα was activated and induced TRPM2-S phosphorylation. That TRPM2-S mediated Ca2+ gating through heterodimerization with TRPM2 is reminiscent of the finding in melanocytes that another splice variant member of the trp gene family TRPM1 interacted with full-length long form of TRPM1 to suppressed its activity 39.
PKCα activation was shown to be crucial for the mechanism of H2O2-induced apoptosis through its binding to and phosphorylation of TRPM2-S. Mutation of the sole PKCα phosphorylation site on Ser39 of TRPM2-S N-terminus to Ala resulted in failure of PKCα to phosphorylate TRPM2-S. This mutation in turn prevented TRPM2-S dissociation from TRPM2 and hence the apoptosis-inducing Ca2+ entry signal. Both K562 myeloid leukemia cell line that do not express the TRPM2-S 14 and Jurkat t-lymphocyte cell line that expresses the short isoform at very low levels 14,40 also did not undergo apoptosis secondary to TRPM2 activation 14,40, consistent with the critical role of TRPM2-S as the apoptosis-suppressing partner of TRPM2. Although PKCα activation contributed to TRPM2-induced endothelial apoptosis in the present through phosphorylation of TRPM2-S, it is important to note that PKCα signaling in other signaling pathways that can induce endothelial injury. We have shown that PKCα phosphorylation of p120-catenin mediates disassociation of p120-catenin from VE-cadherin that resulted in disassembly of adherens junctions (AJs) and disruption of vascular endothelial barrier function 41. These studies showed the role of PKCα in mediating phosphorylation of p120-catenin in response to endotoxin and resultant increased lung vascular permeability 41. Thus, PKCα can function in a complex matter at multiple levels to induce endothelial dysfunction either via injury or through activating the apoptosis program.
The signaling pathway downstream of Ca2+ entry leading to cell death involves the activation of intrinsic “executioner” caspases (caspase-9) and extrinsic caspases (caspase-8) that activate the effector caspases −3 and −7 14,42,43. These cleave cellular substrates, disrupting survival pathways and inducing membrane blebbing, cell shrinkage, and apoptotic body formation. PARP is part of a protective mechanism involved in repair of DNA damage 44 and DNA stability 29. Inactivation of PARP by cleavage of the enzymatic domain following oxidant activation of TRPM2 also caused apoptosis14 similar to that seen with PKCα-induced uncoupling of TRPM2-S from TRPM2 in the present study.
We have uncovered in these studies a novel mechanism of TRPM2 activation resulting in Ca2+ entry secondary to PKCα-induced phosphorylation of TRPM2-S. A question arises about the relationship of this mechanism with TRPM2 activation induced by the generation of ADPR following the activation of poly(ADPR) polymerase 6,12. It is possible that both mechanisms function to activate TRPM2 secondary ROS stimulation (see Model Figure 8). ADPR generation following activation of poly(ADPR) polymerase may help to amplify the Ca2+ entry response. However, in the event that both TRPM2-S and TRPM2 are co-expressed as they are in endothelial cells, it is likely as the present results show that TRPM2-S functions by restraining the activity of TRPM2 (and hence suppresses apoptosis). However, when TRPM2-S is not expressed or poorly expressed, ADPR binding to Nudix box sequence would by default be the primary mechanism of TRPM2 activation, but it is not clear whether Ca2+ entry by this mechanism is sufficient to activate the pro-apoptotic caspases.
Figure 8. PKCα-induced phosphorylation of TRPM2-S mediates oxidant-induced TRPM2 channel opening and supra-normal Ca2+ entry required for activation of the apoptosis program.
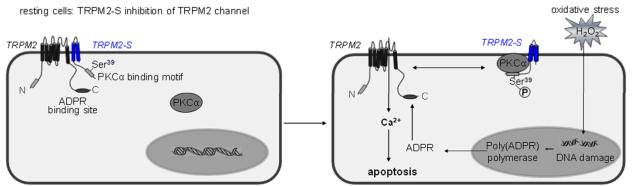
In resting cells, TRPM2-S associates with TRPM2 to restrict Ca2+ entry (the channel is in a closed conformation state). Oxidative stress induces PKCα activation resulting in binding and phosphorylation of TRPM2-S on Ser 39. TRPM2-S uncoupling from TRPM2 in turn opens the channel and Ca2+ entry resulting in activation of executioner caspases and endothelial apoptosis. In this model the generation of the endogenous TRPM2 agonist, ADPR may also contribute to opening TRPM2 channel.
In summary, we identified a fundamental relationship between oxidant-activated TRPM2 channel and its associated short splice variant TRPM2-S in the gating of large Ca2+ influx and the critical role of loss of this interaction in mediating oxidant-induced apoptosis of endothelial cells. We demonstrated that apoptosis induced by this mechanism contributed to the mortality seen in endotoxin challenged mice. PKCα functioned to induce phosphorylation of TRPM2-S, which prevented its association with TRPM2, and thereby activated Ca2+ gating and caspases. Thus, disabling TRPM2-S and TRPM2 interaction such as by inhibiting PKCα activation represents a novel strategy for abrogating apoptosis and resultant vascular injury and inflammation associated with apoptosis in diseases such as acute lung injury and vascular inflammation.
Novelty and Significance
What Is Known?
Oxidants induce injury to the vascular endothelium resulting in endothelial denudation, permeability alterations, edema formation and inflammation.
Endothelial cell loss via apoptosis is a crucial feature of vascular injury and is implicated in the mechanism of lung injury induced by sepsis and other vascular diseases.
Activation of the cation (primarily Ca2+) permeable melastatin transient receptor potential 2 (TRPM2) channel during oxidative stress is linked to cell death.
What New Information Does This Article Contribute?
Oxidant-activation of TRPM2 mediates lung endothelial cell apoptosis and is critically regulated by PKCα.
TRPM2 in endothelial cells is normally impermeable to Ca2+ because of its binding to the short splice variant TRPM2-S.
Oxidants induce PKCα phosphorylation of TPPM2-S at Ser 39, which functions by releasing TRPM2-S inhibition of TRPM2 channel and thereby induces Ca2+ entry and sequestration to activate the apoptotic program.
Endothelial cell apoptosis is a crucial feature of vascular injury that leads to disruption of the endothelial barrier and to inflammation. Understanding the molecular mechanisms regulating apoptosis is vital for identifying novel targets for treating vascular injury and inflammatory disorders. We have uncovered a role of PKCα phosphorylation of the short splice variant of TRPM2, TRPM2-S, which acts as a phospho-switch to regulate channel activity and results in calcium overload. Phosphorylation of TRPM2-S at Ser 39 caused release of TRPM2-S inhibition of TRPM2 and thereby activated Ca2+ gating. This unique mechanism of TRPM2 channel activation was crucial for induction of oxidant mediated apoptosis of endothelial cells. Thus, oxidant-induced gating of Ca2+ via TRPM2 and apoptosis are critically dependent on PKCα phosphorylation of TRPM2-S at a specific site identifying a novel potential anti-inflammatory target.
Supplementary Material
Acknowledgments
We thank M. Ushio–Fukai for her insights. We also thank B.A. Miller (Pennsylvania State University School of Medicine) for kindly supplying the GFP-TRPM2-S construct, and GlaxoSmithKline for providing the Trpm2−/− C57BL/6 mice used in these experiments. We are also grateful to Dr Jeffrey Molkentin for PKCα−/− mice.
Sources of Funding
This work was supported by National Institutes of Health grant P01 HL077806-07 to A.B.M., and by grant 10SDG2610057 from American Heart Association Midwest affiliate to C.M.H
Nonstandard Abbreviations and Acronyms
- ADPR
adenosine diphosphate ribose
- DPQ
3,4-Dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline
- FACS
Fluorescence-activated cell sorting
- GO
glucose oxidase
- HPAEC
human pulmonary aortic endothelial cells
- NUDT9-H
nucleoside diphosphate type motif 9 protein
- PE-Annexin
phycoerythrin Annexin V
- PARP
poly(ADPR) polymerase
- PKCα
protein kinase C-α
- PKCαi
protein kinase C-α inhibitor peptide
- PKCβII
protein kinase βII
- PKCβIIi
protein kinase βII inhibitor peptide
- ROS
reactive oxygen species
- S39A
serine 39 mutated to alanine
- TRPM2
Melastatin-like transient receptor potential 2
- TRPM2-S
Melastatin-like transient receptor potential 2 short variant
- VE-cadherin
vascular endothelial cadherin
- WT
wild-type
- 7-AAD
7-Aminoactinomycin D
Footnotes
Disclosures
None.
References
- 1.Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch; 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- 2.Nazıroğlu M. TRPM2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochem Res. 2011;36:355–366. doi: 10.1007/s11064-010-0347-4. [DOI] [PubMed] [Google Scholar]
- 3.Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, Shimizu N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–131. doi: 10.1006/geno.1998.5551. [DOI] [PubMed] [Google Scholar]
- 4.Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–C137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- 5.Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 6.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 7.Uchida K, Dezaki K, Damdindorj B, Inada H, Shiuchi T, Mori Y, Yada T, Minokoshi Y, Tominaga M. Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes. 2011;60:119–126. doi: 10.2337/db10-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–1815. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii M, Shimizu S, Hara Y, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat [beta]-cell line RIN-5F. Cell Calcium. 2006;39:487–494. doi: 10.1016/j.ceca.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Kühn F, Heiner I, Lückhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflügers Arch. 2005;451:212–219. doi: 10.1007/s00424-005-1446-y. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol. 2006;209:31–41. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 13.McNulty S, Fonfria E. The role of TRPM channels in cell death. Pflugers Arch. 2005;451:235–242. doi: 10.1007/s00424-005-1440-4. [DOI] [PubMed] [Google Scholar]
- 14.Zhang WY, Hirschler-Laszkiewicz I, Tong Q, Conrad K, Sun SC, Penn L, Barber DL, Stahl R, Carey DJ, Cheung JY, Miller BA. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am J Physiol Cell Physiol. 2006;290:C1146–1159. doi: 10.1152/ajpcell.00205.2005. [DOI] [PubMed] [Google Scholar]
- 15.Walsh K, Isner JM. Apoptosis in inflammatory–fibroproliferative disorders of the vessel wall. Cardiovascular Res. 2000;45:756–765. doi: 10.1016/s0008-6363(99)00270-9. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:237–243. doi: 10.1164/ajrccm.161.1.9810007. [DOI] [PubMed] [Google Scholar]
- 17.Hotchkiss RS, Tinsley KW, Swanson PE, Karl IE. Endothelial cell apoptosis in sepsis. Crit Care Med. 2002;30(5 Suppl):S225–228. doi: 10.1097/00003246-200205001-00009. [DOI] [PubMed] [Google Scholar]
- 18.Kwan H-Y, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochim Biophys Acta. 2007;1772:907–914. doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Dietrich A, Gudermann T. Another TRP to endothelial dysfunction. Circ Res. 2008;102(3):275–277. doi: 10.1161/CIRCRESAHA.107.170548. [DOI] [PubMed] [Google Scholar]
- 20.Hecquet CM, Ahmmed GU, Malik AB. TRPM2 channel regulates endothelial barrier function. doi: 10.1007/978-1-60761-500-2_10. [DOI] [PubMed] [Google Scholar]
- 21.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 22.Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 23.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 24.Fonfria E, Marshall ICB, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller BA. Inhibition of TRPM2 function by PARP inhibitors protects cells from oxidative stress-induced death. Br J Pharmacol. 2004;143(5):515–516. doi: 10.1038/sj.bjp.0705923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J biol chem. 2002;277(26):23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 27.Hecquet CM, Malik AB. Role of H2O2-activated TRPM2 calcium channel in oxidant-induced endothelial injury. J Thromb Haemost. 2009;101:619–625. [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, Miller BA. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 29.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 30.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 31.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 32.Robaye B, Mosselmans R, Fiers W, Dumont JE, Galand P. Tumor necrosis factor induces apoptosis (programmed cell death) in normal endothelial cells in vitro. Am J Pathol. 1991;138:447–453. [PMC free article] [PubMed] [Google Scholar]
- 33.Bornancin F, Parker PJ. Phosphorylation of protein kinase C-α on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J Biol Chem. 1997;272:3544–3549. doi: 10.1074/jbc.272.6.3544. [DOI] [PubMed] [Google Scholar]
- 34.Barnard ML, Matalon S. Mechanisms of extracellular reactive oxygen species injury to the pulmonary microvasculature. J Appl Physiol. 1992;72:1724–1729. doi: 10.1152/jappl.1992.72.5.1724. [DOI] [PubMed] [Google Scholar]
- 35.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 36.Di A, Gao X-P, Qian F, et al. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol. 2012;13(1):29–34. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inamura K, Sano Y, Mochizuki S, Yokoi H, Miyake A, Nozawa K, Kitada C, Matsushime H, Furuichi K. Response to ADP-Ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol. 2003;191:201–207. doi: 10.1007/s00232-002-1057-x. [DOI] [PubMed] [Google Scholar]
- 38.Sergeev IN. Calcium signaling in cancer and vitamin D. J Steroid Biochem Mol Biol. 2005;97:145–151. doi: 10.1016/j.jsbmb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Xu X-ZS, Moebius F, Gill DL, Montell C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc Natl Acad Sci. 2001;98:10692–10697. doi: 10.1073/pnas.191360198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR, Guse AH. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem. 2006;281:2489–2496. doi: 10.1074/jbc.M506525200. [DOI] [PubMed] [Google Scholar]
- 41.Vandenbroucke St Amant E, Tauseef M, Vogel SM, Gao X-P, Mehta D, Komarova YA, Malik AB. PKCα activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ Res. 2012;111:739–749. doi: 10.1161/CIRCRESAHA.112.269654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denning TL, Takaishi H, Crowe SE, Boldogh I, Jevnikar A, Ernst PB. Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells. Free Radic Biol Med. 2002;33:1641–1650. doi: 10.1016/s0891-5849(02)01141-3. [DOI] [PubMed] [Google Scholar]
- 43.Ma S, Ochi H, Cui L, Zhang J, He W. Hydrogen peroxide induced down-regulation of CD28 expression of Jurkat cells is associated with a change of site α-specific nuclear factor binding activity and the activation of caspase-3. Exp Gerontol. 2003;38:1109–1118. doi: 10.1016/s0531-5565(03)00166-9. [DOI] [PubMed] [Google Scholar]
- 44.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.