

Abstract
Traf2 and NcK interacting kinase (TNiK) contains serine-threonine kinase and scaffold domains and has been implicated in cell proliferation and glutamate receptor regulation in vitro. Here we report its role in vivo using mice carrying a knock-out mutation. TNiK binds protein complexes in the synapse linking it to the NMDA receptor (NMDAR) via AKAP9. NMDAR and metabotropic receptors bidirectionally regulate TNiK phosphorylation and TNiK is required for AMPA expression and synaptic function. TNiK also organizes nuclear complexes and in the absence of TNiK, there was a marked elevation in GSK3β and phosphorylation levels of its cognate phosphorylation sites on NeuroD1 with alterations in Wnt pathway signaling. We observed impairments in dentate gyrus neurogenesis in TNiK knock-out mice and cognitive testing using the touchscreen apparatus revealed impairments in pattern separation on a test of spatial discrimination. Object-location paired associate learning, which is dependent on glutamatergic signaling, was also impaired. Additionally, TNiK knock-out mice displayed hyperlocomotor behavior that could be rapidly reversed by GSK3β inhibitors, indicating the potential for pharmacological rescue of a behavioral phenotype. These data establish TNiK as a critical regulator of cognitive functions and suggest it may play a regulatory role in diseases impacting on its interacting proteins and complexes.
Introduction
Central to understanding the molecular basis of cognitive functions are the signaling mechanisms connecting neurotransmitter receptors to intracellular pathways regulating transcription, translation, and changes in electrical properties of neurons. It has become apparent that many of the proteins that participate in these pathways are physically organized within the cytoplasm into multiprotein complexes that act as molecular machines exploiting their different protein components to perform regulatory functions (Husi et al., 2000; Scott and Pawson, 2009). Within many signaling complexes are protein kinases that phosphorylate the nearby proteins and thereby orchestrate a variety of cellular functions (Scott and Pawson, 2009). How neuronal signaling complexes function is poorly understood and there are very few examples of studies where the dysfunction of signaling complexes has been studied following a mutation in the intact animal.
Toward these issues, we were intrigued by Traf2 and NcK interacting kinase (TNiK), a protein with both scaffolding and kinase domains that had been implicated in postsynaptic signaling as well as in regulation of cell proliferation (Mahmoudi et al., 2009; Shitashige et al., 2010). TNiK is expressed in the nervous system but its role in vivo is currently unknown. A recent study showed that activation of NMDA receptors (NMDARs) regulates phosphorylation of TNiK (Coba et al., 2009). Moreover, knockdown of TNiK in primary cultured neurons decreases surface GluA1 levels (Hussain et al., 2010) and alters the synchrony of network activity (MacLaren et al., 2011), suggestive of a postsynaptic signaling function at excitatory synapses. TNiK has also been implicated in controlling dendritic outgrowth mediated by a ternary complex involving the E3 ubiquitin ligase Nedd4-1, Rap2A, and TNiK (Kawabe et al., 2010). In non-neuronal cells TNiK modulates cell proliferation by regulating activation of Wnt signaling cascade through its ability to interact with β-catenin and phosphorylate the transcription factor Tcf7l2 (Mahmoudi et al., 2009; Shitashige et al., 2010). It is unknown if TNiK plays any role in neurogenesis or brain development. Finally, a link between TNiK and schizophrenia has also been suggested based on the observation that TNiK binds Disrupted in Schizophrenia 1 (DISC1) in vitro resulting in decreased TNiK levels and kinase activity (Wang et al., 2010). Human genetic studies have not identified mutations in TNiK, although several association studies have suggested TNiK to be involved in schizophrenia, attention deficit hyperactivity disorder, and general cognitive function (Potkin et al., 2009; Shi et al., 2009; Ayalew et al., 2012; Elia et al., 2012).
Here we address the in vivo role of TNiK by examining mice carrying a knock-out mutation in TNiK and show the mutation leads to dysregulation of key synaptic and nuclear signaling mechanisms. We identify complexes of proteins associated with TNiK in the postsynaptic density and the nucleus and show that the TNiK mutation has a dramatic impact on the regulation of GSK3β and phosphorylation of proteins within the complexes. We assessed the requirement of TNiK for synaptic plasticity, neuronal development and specific aspects of higher order cognitive processing using a computerized touchscreen apparatus (Bussey et al., 2012) and find evidence that TNiK plays a role in multiple cognitive functions through both synaptic and nuclear signaling pathways.
Materials and Methods
Generation of TNiK mutant mice.
The targeting vector was constructed using the AB2.2 genomic DNA BAC clone. The vector containing 6.9 and 2.9kb of 5′ and 3′ homology arms, respectively, replaced 2.6kb of TNiK genomic DNA (X28438374 to X28440972; Ensembl Build 55) containing part of exon 6 and 7 that encoded the kinase domain with IRES-lacZ-neo reporter cassette. The targeting construct was electroporated into E14TG2a embryonic stem (ES) cells. G418 (neo)-resistant clones were screened for homologous recombination by long-range PCR using the Expand Long Template PCR system (Roche Cat 11681842001) with PCR primer × (5′-GAGCTATTCCAGAAGTAGTGAG-3′) and primer Y (5′-CAGAGGTCTTGTCTATTCTTC-3′) that correspond to sequence in the IRES-lac-Z-neo cassette and sequence outside the 2.9 kb flanking region, respectively. The correctly targeted ES cells were injected into C57BL/6 blastocysts to create chimeric mice, which were bred with 129S5 mice to generate heterozygous (+/−) TNiK mutant mice. Those F1 heterozygous mice had been backcrossed with 129S5 mice 1–2 times before being used for intercrossing. Genotyping PCR consisted of a 540 bp product amplified from the wild-type (wt) allele using a forward primer A (CAACTGTCTTCTCATTAGTGG) in the wt sequence deleted by targeted mutation and a reverse primer B (GACCTGAGAGTTGTGAGCTG) downstream of the cassette. A 700bp product was amplified from the targeted allele using primer B with forward primer X, within the selection cassette. After enzymatic amplification for 35 cycles (45 s at 94°C, 45 s at 55°C, and 1 min at 72°C), the PCR products were size fractionated on a 2% agarose gel in 1 × Tris borate-EDTA buffer. Animals were treated in accordance with the UK Animal Scientific Procedures Act 1986.
Biochemical assays.
Postsynaptic density (PSD) and nuclear fractions from mouse hippocampus were prepared using a three-step protocol as previously described (Coba et al., 2009). In brief, tissue was homogenized in ice-cold buffer (10 mm Tris-HCl, pH 7.4/320 mm sucrose, 20 mm β-glycerol phosphate, 1 mm sodium orthovanadate, and 50 mm sodium fluoride) and protease inhibitors (Roche-Complete protease inhibitor mixture). The homogenates were centrifuged at 800 × g for 10 min at 4°C and pellet resuspended in 50 mm HEPES, pH 7.4, 40 mm β-glycerol phosphate, 1 mm sodium orthovanadate, and 50 mm sodium fluoride and protease inhibitors (Roche-Complete protease inhibitor mixture). The homogenates were centrifuged at 16,000 × g for 15 min at 4°C and supernatants used for nuclear extract assays. Data presented as mean ± SEM and statistical significance determined using unpaired Student's t tests.
Hippocampal slice electrophysiology.
Synaptic physiology was assessed using a multi-electrode array system and whole-cell voltage-clamp techniques as described previously (Kopanitsa et al., 2006; Carlisle et al., 2008). Eight MEA1060-BC set-ups were run in parallel and monopolar stimulation of Schäffer collateral/commissural fibers through array electrodes was performed by STG2008 stimulus generator (Multi Channel Systems). For long-term potentiation (LTP) experiments, a single principal recording electrode was picked in the proximal part of CA1 stratum radiatum. To stimulate control and test pathways, two stimulation electrodes were assigned on the subicular side and on the CA3 side of stratum radiatum, respectively. The distance from the recording electrode to the test stimulation electrode was 400–510 μm and to the control stimulation electrode 280–510 μm. To evoke orthodromic field EPSPs (fEPSP), test and control pathways were activated in succession at a frequency of 0.02 Hz. Baseline stimulation strength was adjusted to evoke a response that corresponded to 40% of the maximal attainable fEPSP at the recording electrode located in proximal stratum radiatum. Amplitude of the negative part of fEPSPs was used as a measure of the synaptic strength. To induce LTP, 10 bursts of baseline strength stimuli were administered at 5 Hz to test pathway with four pulses given at 100 Hz per burst (total 40 stimuli). LTP plots were scaled to the average of the first five baseline points. Normalization of LTP values was performed by dividing the fEPSP amplitude in the tetanized pathway by the amplitude of the control fEPSP at corresponding time points. Normalized LTP values averaged across the period of 61–65 min after theta-burst stimulation was used for statistical comparison.
Paired stimulation with an interpulse interval of 50 ms was used to observe paired-pulse facilitation (PPF) in baseline conditions in the test pathway before LTP induction. PPF was calculated by dividing the negative amplitude of fEPSP obtained in response to the second pulse by the amplitude of fEPSP amplitude evoked by the preceding pulse. To assess changes in basal synaptic transmission, input–output relationships were initially compared by mixed model repeated-measures ANOVA and Bonferroni post hoc test implemented in Prism 5 (GraphPad Software) using individual slice data as independent observations. Since several slices were routinely recorded from every mouse, fEPSPmax, PPF, and LTP values between wt and mutant mice were then compared using two-way nested ANOVA design with genotype (group) and mice (subgroup) as fixed effects. Fisher's F statistic was calculated as mean of squaresGenotype/mean of squaresResidual and genotype effect was considered significant if corresponding probability for the group F statistic was <0.05.
Conventional extracellular recording techniques were used to examine the induction of long-term depression (LTD) by 900 pulse trains of 1 Hz presynaptic fiber stimulation (Carlisle et al., 2008). For whole-cell voltage-clamp techniques, slices were bathed in an oxygenated (95% O2-5% CO2) artificial CSF containing the following (in mm): 124 NaCl, 2.4 KCl, 25 NaHCO3, 1 NaH2PO4, 4.0 CaCl2, 4 MgSO4, 0.1 picrotoxin, and 10 glucose. EPSCs were recorded using electrodes and were filled with a solution containing 102 mm cesium gluconate, 17.5 mm CsCl, 10 mm TEA-Cl, 5 mm QX314, 4.0 mm Mg-ATP, 0.3 mm Na-GTP, and 20 mm HEPES, pH 7.2. Tetrodotoxin (TTX) (1 μm) was added to the external solution and cells were voltage-clamped at −80 mV to record miniature EPSCs (mEPSCs). Spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs) were recorded using electrodes containing 140 mm CsCl, 4 mm NaCl, 1 mm MgCl2, 0.1 mm EGTA, 4 mm Mg-ATP, 0.3 mm Na-GTP, 5 mm QX314, and 10 mm HEPES, pH, 7.2. AMPA and NMDAR-mediated synaptic currents were blocked with 3.0 mm kynurenate and 1.0 μm TTX was added to the extracellular solution in experiments recording mIPSCs. Unpaired Student's t tests were used to determine statistical significance of differences between whole-cell voltage-clamp measurements in mutant and wt neurons.
Microarray analysis.
Hippocampi (TNiK−/− n = 11; wt n = 33) from male and female mice aged 98–174 d were dissected and snap frozen in liquid nitrogen. RNA was extracted using a Qiagen miRNeasy kit. RNA integrity was verified using the Agilent 2100 Bioanalyzer and all samples had RIN values >7. Unpooled samples were prepared for array analysis using the Illumina TotalPrep-96 RNA Amplification Kit and hybridized to Illumina Mouse WG-6 v2.0 Beadchips. The data were read into the Bioconductor using Lumi (Du et al., 2008) and re-annotated using a previously described dataset (Barbosa-Morais et al., 2010). A Variance Stabilizing Transformation was applied followed by Quantile Normalization. Differential expression between wt and TNiK−/− mice was determined using the empirical Bayes linear model approach implemented in the Limma package (Smyth et al., 2005). Multiple hypothesis testing was corrected for using the Bonferroni–Hochberg procedure. Gene set enrichment analysis was undertaken using DAVID (Huang da et al., 2009) on those genes with adjusted p < 0.05. The dataset has been uploaded to ArrayExpress with accession E-MTAB-1202.
Histology.
For 5-bromo-4-chloro-3-indolyl-β-galactoside (X-gal) staining, free-floating sections were stained overnight in X-gal (Malford), mounted on pretreated slides, dehydrated, and mounted with DPX mountant. For immunohistochemistry, free-floating sections were incubated in either anti-doublecortin (1:250; Cell Signaling Technology 4604), anti-NeuroD1 (1:500; Abcam ab60704) or anti-Ki67 (1:300; Abcam ab15580). Sections were incubated with either biotinylated anti-mouse or anti-rabbit secondary antibody (1:200; DAKO) for peroxidase staining or with anti-mouse Alexa Fluor 633 or anti-rabbit Alexa Fluor 568 (1:1000) for immunofluorescence. For peroxidase staining, signal was amplified using an avidin–biotin system (Vector Laboratories) before staining in 3,3-diaminobenzidine (Sigma). For quantitation of cell numbers, a minimum of two sagittal brain sections per animal was analyzed from four pairs of TNiK−/− mice and wt littermates. Total numbers of immunopositive cells in the hippocampus were counted by an experimenter blinded to genotype and statistical significance determined using unpaired Student's t tests.
Cognitive testing in the rodent touchscreen operant system.
Testing was conducted in a touchscreen-based automated operant system that consisted of an operant chamber (21.6 × 17.8 × 12.7 cm) made of clear Perspex walls and a stainless steel grid floor, housed within a sound- and light-attenuating box (40 × 34 × 42 cm) (Med Associates). A pellet dispenser delivering 14 mg dustless pellets (BioServ) into a magazine, a house light, and a tone generator was located at one end of the chamber. At the opposite end of the chamber was a flat-screen monitor equipped with an infrared touchscreen (16 × 21.2 cm) (Craft Data Limited). The touchscreen was covered by a black Perspex “mask” that either had six windows (for two-choice spatial discrimination) or three windows (for object-location paired associates learning) positioned in front of the touchscreen allowing the presentation of stimuli to be spatially localized and prevented the mouse from accidentally triggering the touchscreen. Stimuli presented on the screen were controlled by custom software (“MouseCat,” L.M. Saksida), and responses made via nose pokes at the stimuli were detected by the touchscreen and recorded by the software. Before testing, the body weight of mice was slowly reduced and then maintained at or >85% of free-feeding body weight. Animals were pretrained through several stages to learn to touch stimuli on the screen to obtain a reward as described previously (Brigman et al., 2008).
Two-choice spatial discrimination pattern separation.
Adult mice (wt n = 8, TNiK−/− n = 12) were initially trained to touch one of two illuminated stimuli (squares). Responses at one location (correct) resulted in a reward; responses at the other location (incorrect) resulted in a 5 s timeout (signaled by extinction of the house light). During training, the location of the two illuminated stimuli was separated by an intermediate degree of separation (two empty/dark locations; Creer et al., 2010). Animals were required to complete seven of eight consecutive correct touches (acquisition) following which the reward contingences were reversed so that the previous incorrect location now became correct, and animals had to complete seven of eight consecutive correct touches in the other location (reversal). The correct starting location was counterbalanced between animals in each genotype and in addition, the reversal component of the task decreased the likelihood of animals developing a side bias or the use of nonspatial strategies. Mice were given a maximum of 60 trials/session/d until they reached a training criterion of completing a minimum of one acquisition and one reversal on 3 of 4 consecutive days.
Following training, pattern separation was assessed by varying the distance between the choice locations, either situated by a high degree of separation (i.e., four empty/dark locations between the two illuminated locations) or a low degree of separation (i.e., no empty/dark location between the two illuminated locations). Mice were given unlimited trials/session/d in a 60 min period and were allowed a maximum of one reversal. Spatial separations were held constant during each testing session per day but were varied across testing days. Mice were tested for four sessions on each separation, and the order of separation tested was counterbalanced between animals in each genotype across days.
Average number of trials taken to reach the criterion of seven of eight consecutive correct touches for acquisition and reversal for each separation was calculated. Group differences were analyzed using either an independent samples t test or a mixed between- and within-subjects ANOVA with genotype as the between-subjects factor and separation as the within-subjects factor. A paired samples t test was used for post hoc analysis to assess significant between × within-subjects interaction effect. All values reported represent mean ± SEM.
Object-location paired associates learning.
Mice (wt n = 8, TNiK−/− n = 12) were tested for the ability to associate between objects (shapes) and locations on the touchscreen (Talpos et al., 2009). There were three objects (flower, plane, and spider) and three correct spatial locations (left, middle, and right, respectively). For each trial, only two objects were presented; one object in its correct location (S+) and the other object in one of two incorrect locations (S−). There were six possible trial types (Talpos et al., 2009), so that the flower was rewarded only when presented in the left location, the plane was rewarded only when presented in the middle location, and the spider was rewarded only when presented in the right location. A nose poke to the correct S+ resulted in delivery of a reward and incorrect responses resulted in a 5 s timeout period, followed by a correction procedure whereby the trial was repeated until the mouse made a correct choice. Nose pokes to response windows in which no stimulus was presented were ignored. Mice were given 36 trials/session/d for 50 sessions. Group differences were analyzed using a mixed between-within subjects ANOVA with genotype as the between-subjects factor and session block as the within-subjects factor. All values reported represent mean ± SEM.
Results
TNiK in glutamate receptor function in vivo
The TNiK gene was replaced with a LacZ reporter using homologous recombination in mouse ES cells (Fig. 1A,B), producing viable homozygous (TNiK−/−) mice that showed no detectable TNiK protein on immunoblots of brain extracts (Fig. 1C). We observed normal Mendelian transmission in TNiK+/− intercrosses (χ2 test, p = 0.23) and no detectable abnormalities in body size or gross brain morphology (Fig. 1E). In wt mice, TNiK is widely expressed in brain, including the cortex and hippocampus, where strongest staining was observed in dentate gyrus granule cells (Fig. 1D). As previously reported, TNiK protein was expressed in the PSD (Kawabe et al., 2010; Wang et al., 2010) and the nucleus (Fig. 2A)(Mahmoudi et al., 2009).
Figure 1.
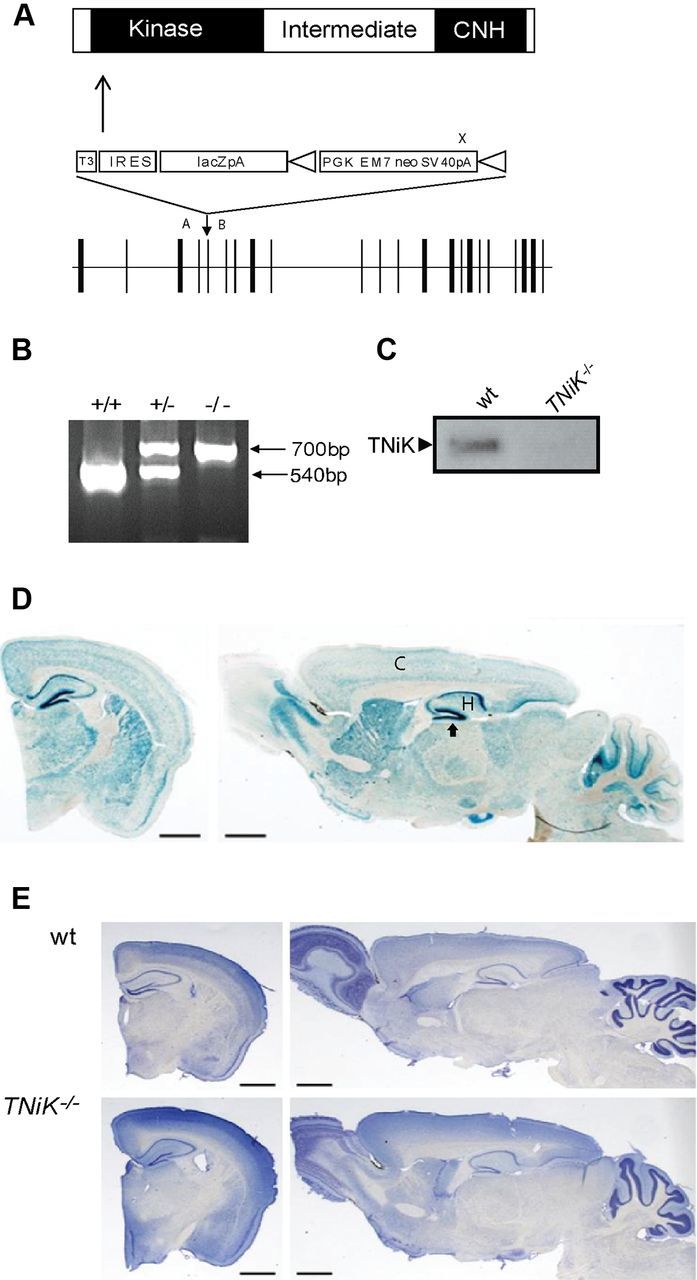
TNiK knock-out mice. A, Targeting vector for the generation of TNiK knock-out mice. Linear structure of TNiK protein showing kinase, intermediate, and citron homology (CNH) domain. Arrow indicates N-terminal point of targeted deletion. Middle, Shows gene targeting vector containing the IRES-lacZ-neo reporter cassette. Bottom, Shows the genomic locus (exons; vertical bars). Vector sequences include: T3, triple termination sequence; IRES; LacZpA, β-galactosidase reporter with polyA sequence; and triangles flanking PGKEM7neoSV40pA (selection cassette) are loxP recombination sites. PCR genotyping primer sites, A, B, and X. B, Genotyping heterozygous and homozygous mice. A 540 bp product was amplified from the wt allele using primers A and B and a 700 bp product amplified from the targeted allele using primers B and X. C, Immunoblot with TNiK antibodies of total hippocampal lysates shows absence of TNiK protein in TNiK−/− mice. D, Brain expression of TNiK. Coronal and sagittal brain section stained with X-gal in TNiK−/− mice. C, cortex; H, hippocampus. Arrow to dentate gyrus. Scale bar, 1.4 mm. E, Coronal and sagittal brain sections stained with Nissl from adult wt and TNiK−/− mice. Scale bar, 1.4 mm.
Figure 2.

Postsynaptic TNiK complexes and increased GSK3β signaling in TNiK−/− mice. A, Immunoblot showing TNiK, DISC1, β-catenin, and GSK3β in total hippocampal lysates, PSD, and nucleus fraction. The transcription factor NeuroD1 was found only in the nuclear fraction and absent from the PSD. B, TNiK, NMDAR, and DISC1 complexes. PSD lysates (input) were immunoprecipitated with antibodies to GluN1, GLuN2B, TNiK, DISC1, and control IgG (labels above the blots) and immunoblotted with antibodies to TNiK and GluN1 (labels to left of blots). C, TNiK, CRMP2, and GSK3β complexes. PSD lysates were immunoprecipitated with antibodies to TNiK, GSK3β, and control IgG and immunoblotted with antibodies to CRMP2 and GSK3β. Labeled as in B. D, IP with GluN1 antibodies and control IgG, and Western blot against TNiK in wt and AKAP9−/− mice (left) showed a 52% reduction (p < 0.05) (right). Reciprocal IP with TNiK and GluN1 antibodies and Western blot against GluN1 in wt and AKAP9 mutant mice (left) observed a 63% reduction (p < 0.05; right). The total levels of TNiK and GluN1 were not altered in AKAP9−/− mice (lanes 1 and 6). E, Unaltered GluN1, GluN2B, and PSD95 complexes in AKAP9−/− mice. Wt and AKAP9 mutant input extracts are indicated and immunoprecipitating antibody is shown above the blot. F–I, Glutamate receptors modulate bidirectional phosphorylation of TNiK. F, TNiK S735 phosphorylation measured by immunoblot of PSD lysates from hippocampal slices stimulated with NMDA. G, TNiK S735 phosphorylation measured by immunoblot of PSD lysates from hippocampal slices stimulated with the metabotropic agonist dihydroxyphenylglycine (DHPG). H, TNiK S735 phosphorylation measured by immunoblot of PSD lysates from hippocampal slices stimulated with AMPA. I, NMDAR activation in hippocampal slices decreases TNiK S735 phosphorylation; activation of metabotropic glutamate receptors type I (mGluRI) with DHPG produced an increase in S735 phosphorylation, while AMPA activation had no effect. J, PSD lysates were immunoblotted with antibody to GluA1. Histogram shows 35% decrease (p < 0.05).
As a starting point for understanding the biochemical role of TNiK and the impact of a knock-out mutation, we characterized the protein interactions and complexes formed by TNiK. Since TNiK binds DISC1 (Camargo et al., 2007; Wang et al., 2010), which binds GSK3β (Mao et al., 2009), expression of all three proteins was identified in the nucleus and in the PSD (Fig. 2A). Using immunoprecipitations (IP) from PSD extracts, these three proteins were co-isolated in NMDAR complexes (Fig. 2B) together with CRMP2 (Fig. 2C), a GSK3β substrate (Cole et al., 2006). These findings are consistent with previous reports showing that CRMP2 binds DISC1 (Camargo et al., 2007) and NMDAR GluN2B subunits (Al-Hallaq et al., 2007). How TNiK is recruited into complexes with the NMDAR is unknown: a direct interaction between TNiK and NMDAR subunits has not been reported in numerous yeast two-hybrid screens using NMDAR GluN1 and GluN2 subunits as bait, thus it seems likely that TNiK interacts with NMDARs via an intermediate protein. A-kinase anchoring protein 9 (AKAP9)(Lin et al., 1998) is a candidate since it binds TNiK (Camargo et al., 2007) and the cytoplasmic domain of GluN1. To test this, AKAP9 mutant mice were generated and characterized, and as shown in Figure 2D, TNiK's association with GluN1 was reduced to 52% (p < 0.05) and the reverse IP confirmed this result with a 63% decrease (p < 0.05; Fig. 2D). Since the total levels of GluN1 and TNiK remained unchanged in extracts from AKAP9 mutants (Fig. 2D) and the interaction between TNiK and PSD-95 or GluN2B was not altered in AKAP9 mutants (Fig. 2E), these results support the view that AKAP9 links TNiK to GluN1 in vivo.
To understand the functional coupling of NMDAR and other glutamate receptors to TNiK, we examined the phosphorylation of S735, a proposed autophosphorylation site indicative of TNiK activity (Shitashige et al., 2010; Wang et al., 2010). In PSD extracts from hippocampal slices treated with NMDA, S735 was robustly dephosphorylated (42%, p < 0.01). In contrast, mGluR1 agonist increased S735 phosphorylation (241%, p < 0.01) and AMPAR activation had no effect (p > 0.05) (Fig. 2F–I). In PSD extracts of TNiK−/− mice, a decrease in AMPAR GluA1 levels (35%, p < 0.05; Fig. 2J) was observed, consistent with a recent report in primary cultures with TNiK knockdown (Wang et al., 2010). Since these data show biochemical interactions between TNiK and glutamate receptors, we examined excitatory and inhibitory synaptic transmission and synaptic plasticity in the hippocampus of TNiK−/− mice. NMDAR- and AMPAR-mediated synaptic currents, long-term plasticity (LTP and LTD), and sIPSCs and mIPSCs were normal in the hippocampal CA1 region of TNiK−/−] mice (Fig. 3A–F). We noted, however, that evoked fEPSPs were slightly but significantly enhanced at stronger stimulation intensities compared with wt littermates (Fig. 3B; F(9,522) = 6.48, p < 0.0001). We also observed a significant increase in PPF (188 ± 3% in wt compared with 196 ± 3% in TNiK−/− mice, F(1,40) = 5.36, p = 0.026) and a significant decrease in the frequency of AMPAR-mediated mEPSCs in TNiK−/− cells (TNiK−/− mice: 4.1 ± 0.5 Hz; wt: 5.3 ± 3 Hz; p < 0.05) indicating the probability of neurotransmitter release from presynaptic terminals of excitatory synapses may be reduced in TNiK−/− mice (Fig. 3G).
Figure 3.
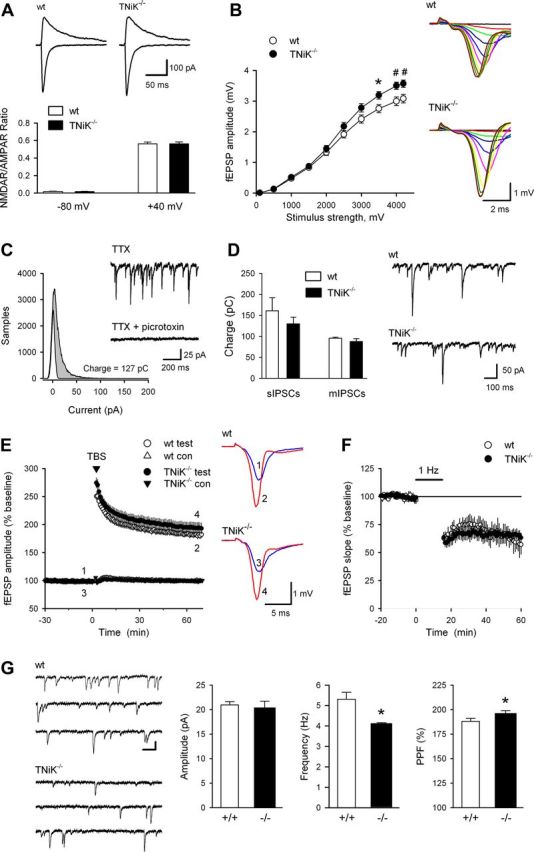
Synaptic physiology in TNiK−/− mice. A, NMDAR-mediated EPSCs are normal in TNiK−/− mice. Traces (top) show EPSCs recorded at −80 and +40 mV in pyramidal cells from a wt (left) and TNiK−/− mouse (right). Calibration bars are 100 pA and 25 ms. The ratios of NMDAR-mediated EPSCs to AMPAR-mediated EPSCs measured at −80 and +40 mV (bottom) are not significantly different in TNiK−/− mice (n = 17; N = 3) compared with wt littermates (n = 17; N = 3). B, Baseline synaptic transmission was slightly but significantly enhanced in TNiK−/− mice. Input–output relationships illustrate averaged fEPSP amplitudes in slices from TNiK−/− (n = 28; N = 10) and wt mice (n = 32; N = 10) in response to stimulation of Schäffer collaterals by biphasic voltage pulses of 0.1–4.2 V (*p < 0.05, #p < 0.01). Amplitudes of fEPSPs evoked by maximum 4.2 V stimulation were significantly increased in TNiK mice (F(1,40) = 9.18; p = 0.004, two-way nested ANOVA, main genotype effect). Representative families of fEPSP traces are shown at right. Calibration bars are 1 mV and 2 ms. C, Spontaneously occurring IPSCs were recorded in cells voltage-clamped at −70 mV and all-points histograms were generated from 10 s long segments of data. Points corresponding to baseline noise were estimated from the Gaussian curve centered on 0 pA. The difference between this area and the remaining area under the histogram corresponds to the currents generated by the IPSCs (shaded area) and was used to calculate total charge transfer. Traces show mIPSCs recorded before and after bath application of picrotoxin (100 μm). D, Comparison of sIPSC and mIPSC charge transfer in wt (open bars) and TNiK−/− cells (filled bars). Spontaneous IPSCs (recorded in the absence of TTX) in cells from TNiK−/− mice (n = 10 cells from 4 mice) are not significantly different from that seen in wt cells (n = 10 cells from 4 mice, p = 0.41). Total charge transfer due to mIPSCs is also similar in TNiK−/− (n = 13 cells from 4 mice) and wt mice (n = 15 cells from 3 mice, p = 0.39). Traces show sIPSCs recorded from CA1 pyramidal cells in slices from wt and TNiK−/− mice. E, Theta-burst stimulation elicited pathway-specific LTP of synaptic transmission in hippocampal CA1 area. Normalized magnitude of this potentiation 60–65 min after LTP induction was identical in mutant mice (189 ± 5%; n = 28; N = 10; p = 0.94) and their wt littermates (189 ± 4%; n = 32; N = 10). Traces show examples of test pathway fEPSPs evoked immediately before and 1 h after theta-burst stimulation. F, Hippocampal LTD induced by 1 Hz stimulation (900 pulses) is normal in TNiK−/− mice. At 45 min post 1 Hz stimulation fEPSPs were reduced to 65 ± 4% of baseline in slices from TNiK−/− mice (n = 11; N = 5) compared with 60 ± 9% of baseline in slices from wt mice (n = 5; N = 3). G, Frequency of spontaneous mEPSCs is significantly reduced in TNiK−/− mice. Traces (left) show consecutive sweeps of mEPSCs recorded in a wt cell (top, n = 14; N = 3) and a TNiK−/− cell (bottom, n = 14; N = 3). Calibration bars are 100 ms and 15 pA. Histograms (right) show the amplitude, and frequency of mEPSPs as well as PPF of fEPSPs (50 ms interpulse interval) in TNiK−/− mice compared with wt mice (*p < 0.05).
Postsynaptic signaling complexes not only regulate glutamate receptor function and synaptic plasticity, but also transcriptional and other signaling pathways. Thus in the absence of a major requirement for TNiK in synaptic transmission and plasticity, and because of the previous data suggesting TNiK can regulate Wnt pathway in non-neuronal cells, we examined the requirement for TNiK in gene expression. mRNA was isolated from the hippocampus of wt and TNiK−/− mice and the transcriptome profile analyzed. A total of 372 annotated genes were found to be differentially expressed with adjusted p values <0.05 (see ArrayExpress database, http://www.ebi.ac.uk/arrayexpress, under accession number E-MTAB-1202) with many mRNAs involved in neurogenesis identified. Gene set enrichment analysis of TNiK-dependent genes revealed the three most significantly overrepresented gene ontology terms related to neurogenesis (“regulation of neuron differentiation,” p = 0.00153; “regulation of neurogenesis,” p = 0.002; “regulation of nervous system development,” p = 0.0038).
Since GSK3β plays a key role in regulating Wnt pathway and is physically associated with TNiK (Fig. 2C), we asked whether GSK3β was altered in TNiK−/− mice. There was a dramatic increase in GSK3β levels (143%, p < 0.05; Fig. 4A) in the PSD and nucleus, which was associated with an increase in phosphorylation of postsynaptic (CRMP2 threonine 514 increased 183%, p < 0.01; PSD-95 serine 418 and threonine 19 increased 157%, p < 0.01 and 141%, p < 0.01, respectively; Tau serine 396 increased 211%, p < 0.01) (Fig. 4B) and nuclear substrates (NeuroD1 S274 increased 390%, p < 0.05) (Fig. 4C). No changes were observed in the levels of GluN1, PSD-95, CRMP2, Tau, or NeuroD1 in TNiK−/− mice (p > 0.05; Fig. 4A) and the hyperphosphorylation of the GSK3β sites on these proteins was reversed within minutes of applying a pharmacological inhibitor of GSK3β to hippocampal slices (data not shown).
Figure 4.
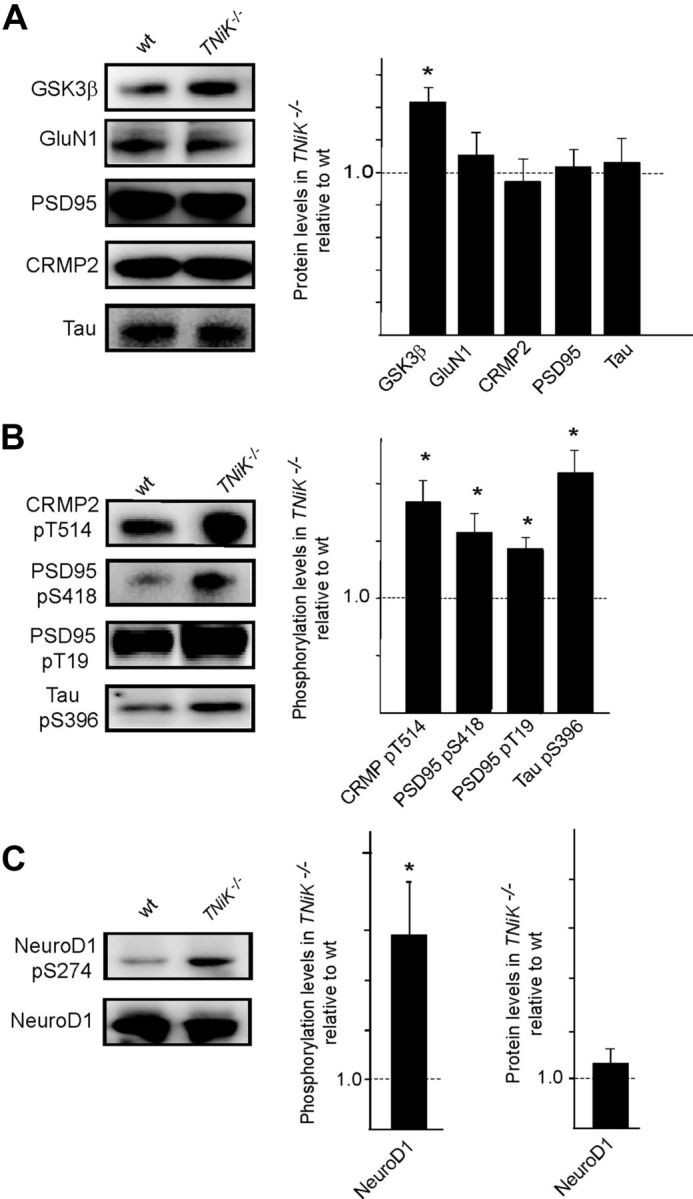
Elevated GSK3β levels in TNiK−/− mice. A, Immunoblots of GSK3β, GluN1, PSD-95, CRMP2, and Tau in PSD lysates from wt and TNiK−/− mice. Histograms show protein levels in TNiK−/− mice PSD normalized to wt levels. B, PSD lysates were immunoblotted with phospho-specific antibodies showing increased phosphorylation of CRMP2 threonine 514, PSD-95 serine 418 and threonine 19, and tau serine 396 in TNiK−/− mice. C, Hippocampal lysates were immunoblotted with phospho-specific antibodies to serine 294 on NeuroD1 (top) and total NeuroD1 (bottom) and levels in TNiK−/− mice quantified relative to wt in histograms. Images show representative Western blot assays from at least triplicate experiments. Asterisk indicates significant difference to wt (p < 0.05).
GSK3β antagonist reversal of locomotor hyperactivity
Spontaneous locomotor hyperactivity has been reported in mice overexpressing GSK3β (Mohn et al., 1999; Prickaerts et al., 2006; Shen et al., 2008); therefore we tested locomotor activity in four test environments (open field, novel object exploration, elevated plus maze, and locomotor activity chamber). A robust increase in locomotor activity was observed in TNiK−/− mice in all tests (Fig. 5A). Next, the ability of this effect to be rapidly reversed with a GSK3β inhibitor was tested. TNiK−/−and wt mice were placed in an activity chamber for an initial period of 30 min to assess baseline spontaneous locomotor activity where as expected, TNiK−/− mice showed significantly elevated locomotor behavior compared with wt (Fig. 5B; p < 0.001). Within 30 min following administration of the GSK3β inhibitor SB216763, the locomotor activity of TNiK−/− mice was significantly reduced (p < 0.05) to the same levels as wt mice. Furthermore, there was no significant difference between wt mice treated with vehicle or SB216763, indicating a unique sensitivity to GSK3β inhibition (p < 0.05; Fig. 5C) was conferred by the TNiK mutation. These results suggest the mechanism underlying the locomotor phenotype in TNiK−/− mice is the result of aberrant GSK3β signaling.
Figure 5.

Inhibition of GSK3β reverses hyperactivity in TNiK−/− mice. A–C, Locomotor activity was assessed in an open field chamber and total distance moved measured (A). Representative tracks of wt (A, left) and TNiK−/− (A, right) mice in the open field. GSK3β inhibition on locomotor activity (B) was assessed by placing TNiK−/−and wt mice in an activity chamber for an initial period of 30 min, then mice were injected (indicated by arrow) with either vehicle or a GSK3β inhibitor (SB 216763; 10 mg/kg body weight, i.p.) and monitored for a further period of 30 min starting 10 min after injection. Sensitivity to GSK3β (C) was calculated as a ratio of change in locomotor activity between administration of vehicle and GSK3β inhibitor. *p < 0.05; **p < 0.01; ***p < 0.005; a = TNiK−/− vehicle versus wt vehicle; b = TNiK−/− vehicle versus TNiK−/− GSK3β inhibitor.
TNiK is required for dentate gyrus neurogenesis
Because the S274 phosphorylation on NeuroD1 is known to inhibit its transcriptional activity (Chae et al., 2004) and NeuroD1 is required for dentate gyrus neurogenesis through Wnt signaling (Miyata et al., 1999; Kuwabara et al., 2009), and both Wnt/β-catenin defects result in loss of the dentate gyrus (Miyata et al., 1999; Liu et al., 2000), this raises the possibility that TNiK−/− mice could have dentate gyrus abnormalities. Moreover, inhibition of Wnt (Wexler et al., 2008; Clelland et al., 2009), NeuroD1 (Miyata et al., 1999), or DISC1 (Mao et al., 2009) and upregulation of GSK3β (Mao et al., 2009; Sirerol-Piquer et al., 2011) all result in impairments in dentate gyrus neurogenesis. We therefore examined neuronal cell numbers and markers of neurogenesis in the hippocampus of developing and mature mice. In young adult (6- to 8-week-old) TNiK−/− mice there was a reduction in the numbers of dentate gyrus granule cells (29%, p < 0.001; Fig. 6A), but no difference was observed in CA3 and CA1 pyramidal neurons (cell counts CA3 wt = 101.0 ± 4.2; TNiK−/− = 99.6 ± 3.9, p = 0.82; CA1 wt = 141.5 ± 4.4, TNiK−/− = 144.3 ± 4.5, p = 0.66). Examining cells expressing doublecortin (DCX), a widely used marker of adult neurogenesis and newly born neurons, and NeuroD1 (a neuronal precursor marker expressed in differentiating neurons) (von Bohlen Und Halbach, 2007), we observed significant reductions of both (DCX, 55%, p < 0.001; NeuroD1, 41%, p < 0.001; Fig. 6A,B). The cell proliferation marker Ki67 (Kee et al., 2002) was also reduced (70%, p < 0.01; Fig. 6B). The decrease in dentate neurons in TNiK−/− mice was also observed by postnatal day 7 (43%, p < 0.001) (NeuN staining; Fig. 6C) as was the reduction in cells expressing NeuroD1 (37%, p < 0.01; Fig. 6D) and DCX (77%, p < 0.001; Fig. 6E). Commensurate with a requirement for TNiK during these developmental stages, we examined the postnatal expression profile of TNiK and found strong expression in principal excitatory neurons in all hippocampal subfields from P7 (Fig. 7).
Figure 6.
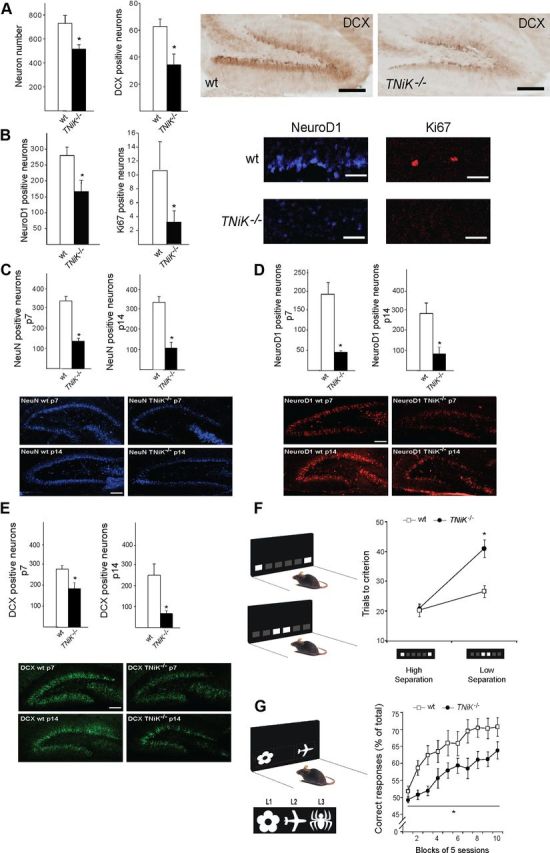
Impaired dentate gyrus neurogenesis and cognition in TNiK−/− mice. A, B, Histograms comparing dentate gyrus granule cell number and cells stained with markers relevant to neurogenesis (see text) in wt and TNiK−/− mice brain sections. Representative images of dentate gyrus cells in wt and TNiK−/− brain sections stained with markers for DCX (A), NeuroD1, and Ki67 (B). Scale bars: (in A) 200 and (in B) 60 μm. C–E, Hippocampus development in TNiK−/− mice. Number of NeuN-positive neurons at P7 (upper, left histogram) and P14 (lower, right histogram) in wt and TNiK−/− mice (C). Number of NeuroD1-positive neurons at P7 (upper, left histogram) and P14 (lower, right histogram) in wt and TNiK−/− mice (D). Number of DCX-positive neurons at P7 (upper, left histogram) and P14 (lower, right histogram) in wt and TNiK−/− mice (E). Scale bars: 100 μm. F, Pattern separation was assessed using a two-choice spatial discrimination task in the touchscreen-based operant system. Mice were trained to discriminate the correct spatial location between two illuminated squares, located in two of six possible locations. Pattern separation was tested by varying the distance between the choice locations, either situated far apart (high degree of separation; top) or close together (low degree of separation; bottom). TNiK−/− mice performed as well as wt mice when stimuli were separated by a high degree of separation; however, they showed a significant impairment (p < 0.05) in discrimination when the locations were in close proximity. As a control measure, analysis of response reaction times showed no significant differences between groups at either separation (high separation, wt = 5.0 ± 1.1 s, TNiK−/− = 5.2 ± 0.8s; low separation, wt = 4.9 ± 0.7 s, TNiK−/− = 5.0 ± 0.9 s). G, TNiK−/− mice show a deficit in object-location associative learning (significant effect of genotype, p < 0.05) where mice were tested for the ability to associate between three objects (flower, plane, and spider) with their correct spatial locations on the screen [left (L1), middle (L2), and right (L3), respectively].
Figure 7.

Postnatal expression profile of TNiK. A, Sagittal sections showing expression pattern of TNiK using X-gal staining in TNiK−/− mice at P7, 14, and 20. Scale bar, 100 μm. B, Detailed expression pattern of TNiK using X-gal staining of hippocampal area. Scale bar, 200 μm. DG, dentate gyrus; GCL, granule cell layer; ML, molecular layer; PCL, pyramidal cell layer.
TNiK mutants show cognitive deficits in touchscreen tests
The role of TNiK in behavior is unknown and the abnormalities in the dentate gyrus pointed to the possibility that there could be a role for TNiK in pattern separation. Pattern separation is a specific cognitive function where the ability to form distinct representations from similar inputs closely presented in space and/or time is necessary for decreasing the interference between memories. Impairments in dentate gyrus neurogenesis induced either by X-irradiation or Wnt inhibition lead to impairments in pattern separation (Clelland et al., 2009) and conversely, enhanced pattern separation performance correlates with increased neurogenesis (Creer et al., 2010; Sahay et al., 2011). We therefore asked whether TNiK was required for pattern separation using the same two-choice spatial discrimination task in the touchscreen apparatus that was used to show the role of Wnt (Clelland et al., 2009). Mice were initially trained to discriminate the correct spatial location between two illuminated squares (located in two of six possible locations) and then pattern separation was tested by varying the distance between the choice locations, either situated far apart (high degree of separation) or close together (low degree of separation; Fig. 6F). TNiK−/− mice showed no significant difference to wt mice during initial training (trials to training criterion: wt = 458.1 ± 45.8, TNiK−/− = 576.3 ± 74.2, p = 0.248) and when tested with stimuli separated by a high degree of separation (p > 0.05; Fig. 6F). However, TNiK−/− mice showed a significant impairment in discrimination when the locations were in close proximity (genotype × separation interaction, p < 0.01; low separation, p < 0.05).
Previous studies have shown a reduction in dentate gyrus neurogenesis impairs pattern separation without impacting object-location paired associates learning, which can also be measured in the touchscreen apparatus (Clelland et al., 2009). Moreover, intrahippocampal infusions with NMDA or AMPA antagonists impair performance in this task (Talpos et al., 2009). Since TNiK is involved in NMDAR and AMPA function, it was of interest to assess object-location paired associates learning in TNiK mutants. The task measures the ability to associate three objects (flower, plane, and spider) with their correct spatial locations on the screen [left (L1), middle (L2), and right (L3)], respectively (Fig. 6G). We found that TNiK−/− mice were significantly impaired in learning the association of an object and its location (effect of genotype, p = 0.016; Fig. 6G).
Discussion
Our data provide the first direct evidence showing TNiK in the nervous system plays key roles at the synapse and nucleus, and is essential for cognition. We found that TNiK was required for two different forms of learning—pattern separation and object-location paired associates learning—that both rely on processing visual and spatial information. Pattern separation is important for keeping similar memories separate and is known to require the function of the dentate gyrus (Gilbert et al., 2001; Leutgeb et al., 2007; Bakker et al., 2008; Hunsaker and Kesner, 2008). Deletion of the essential NR1 subunit of the NMDAR specifically in dentate granule cells (McHugh et al., 2007) or disruption of neurogenesis through interference with Wnt signaling or irradiation (Clelland et al., 2009) all result in impaired pattern separation. Conversely, increased neurogenesis has been reported to correlate with enhanced pattern separation ability (Creer et al., 2010; Sahay et al., 2011). Although we observe that TNiK−/− mice show a marked impairment in neurogenesis of dentate gyrus granule cells and exhibit deficits in pattern separation, we cannot exclude the possibility that some other signaling deficit or neuroanatomical change within the granule cells might also contribute to this specific cognitive impairment. In addition to pattern separation, TNiK was required for learning object-location paired associates, which is not dependent on dentate gyrus neurogenesis (Clelland et al., 2009), but performance of this task is dependent on NMDAR and AMPAR function (Talpos et al., 2009). These findings suggest that loss of TNiK leads to additional dysfunction outside of the dentate gyrus and thus has wider cognitive impacts.
The molecular functions of TNiK are summarized in Figure 8, highlighting a role for TNiK in both nuclear and postsynaptic complexes. Several lines of evidence indicate that TNiK mediates its role in neurogenesis through nuclear signaling complexes involving Wnt and GSK3β substrates. In the dentate gyrus subgranular zone, Wnt proteins (Grove et al., 1998; Lee et al., 2000) and β-catenin (Gao et al., 2007) are essential for neurogenesis. One of the major effectors of Wnt/β-catenin pathways is the activation of the basic helix-loop helix transcription factor NeuroD1 (Kuwabara et al., 2009), and similar to Wnt/β-catenin defects, the absence of NeuroD1 during hippocampus development results in loss of cells in the dentate gyrus (Miyata et al., 1999; Liu et al., 2000). Furthermore, in adult stages, the conditional deletion of NeuroD1 shows it is indispensable for survival and maturation of adult-born neurons (Gao et al., 2009) and defines NeuroD1 as a critical regulator of adult neurogenesis. The transcription factor activity of NeuroD1 has also been shown to be modulated by heterodimerization with the schizophrenia susceptibility gene TCF4 (Brzózka et al., 2010) and by phosphorylation of S274 by GSK3β (Chae et al., 2004), which inhibits NeuroD1 transcription activity. We found that deletion of TNiK resulted in enhanced phosphorylation of NeuroD1 S274, which would be expected to contribute to the observed reduction in neurogenesis and number of dentate gyrus neurons. Moreover, we found that NeuroD1-positive cells were decreased in the dentate gyrus of TNiK−/− mice and this decrease in precursor/progenitor cells was paralleled with a decrease in the number of newly born neurons and total number of dentate gyrus granule cells.
Figure 8.

Molecular functions of TNiK. The molecular functions of TNiK in the PSD and nucleus are shown. Proteins are shown as colored shapes and grouped into their synaptic and nuclear locations. Black lines connecting proteins show known protein–protein interactions. Arrows show regulatory interactions: red, phosphorylation (phosphorylated amino acid residue indicated with number and letter); blue, dephosphorylation; green, regulates protein level.
In non-neuronal cells, TNiK has been described as a critical component of transcriptional regulatory mechanisms, mediated by Wnt signaling, through binding and phosphorylation of the HMG box transcription factor Tcf7l2. TNiK forms a ternary complex with Tcf7l2 and β-catenin, modulating the transcriptional activation of Wnt target genes (Mahmoudi et al., 2009). In agreement with this, our results show that the deletion of TNiK results in a dysregulation of mRNAs involved in Wnt pathways. For example, CCND1 is a characteristic Wnt/β-catenin-modulated gene (Mahmoudi et al., 2009) and was downregulated in the TNiK−/− mice. CCND1 was also reported to decrease as a result of DISC1 knockdown in vivo (Mao et al., 2009) and DISC1 and β-catenin, both TNiK interacting proteins, regulate Wnt signaling and neuronal progenitor proliferation (Kuwabara et al., 2009; Mao et al., 2009). Moreover, DISC 1 induces a premature cell cycle exit and neuronal differentiation through GSK3β activity (Mao et al., 2009) and deletion of TNiK leads to an impairment in cell proliferation as demonstrated by the decrease in the total numbers of Ki67+ cells. These data indicate that TNiK is a key regulator of neurogenesis and cell proliferation through a combination of mechanisms, including modulation of transcription factor phosphorylation, activation of Wnt pathway and nuclear protein complexes.
The finding that TNiK was critical for constraining GSK3β was unexpected. In the absence of this constraint, there was increased phosphorylation of both nuclear substrates controlling cellular proliferation and synaptic substrates in the signal transduction complexes formed with scaffold proteins and glutamate receptors. GSK3β controls both neurogenesis and rapid synaptic signaling events (Jope and Roh, 2006; Beaulieu et al., 2009; Mao et al., 2009; Hur and Zhou, 2010; Sirerol-Piquer et al., 2011). Aberrant GSK3β signaling has also been implicated in and been a prominent therapeutic target (Jope and Roh, 2006; Beaulieu et al., 2009) for a variety of cognitive disorders including neuropsychiatric, neurodegenerative, and more recently, neurodevelopmental disorders such as Fragile X. Interestingly, Fmr1 knock-out mice show elevated GSK3β levels, aberrant neurogenesis, and impairments in spatial discrimination similar to that observed in TNiK−/− mice (Min et al., 2009; Luo et al., 2010; Guo et al., 2012). This is noteworthy as Fmr1 is expressed widely in hippocampal subfields and moreover, loss of Fmr1 results in a phenotype where the dentate gyrus is particularly impacted, similar to that of TNiK−/− mice. Our findings support the view that TNiK acts in concert with GSK3β and DISC1 to regulate formation of neural circuits. DISC1 has been linked to Wnt signaling and neuronal progenitor proliferation (Kuwabara et al., 2009; Mao et al., 2009; Ishizuka et al., 2011) and in agreement with our biochemical data, TNiK directly binds DISC1 and β-catenin (Mahmoudi et al., 2009; Wang et al., 2010) forming complexes with GSK3β. DISC1 knockdown induces a premature cell cycle exit and neuronal differentiation through GSK3β activity (Mao et al., 2009) and TNiK knock-out leads to an impairment in cell proliferation as demonstrated by the decrease in the total numbers of Ki67+ cells. It will be important in future work, using conditional and domain specific mutations in both TNiK and GSK3β, to dissect the multifunctional roles of these interacting kinases. These genetic approaches combined with pharmacological tools will allow the developmental and cell type-specific functions to be elucidated.
We observed several relevant electrophysiological deficits in CA3–CA1 synapses of TNiK−/− mice showing a requirement for TNiK in normal excitatory synaptic function. The frequency of mEPSCs recorded in CA1 pyramidal cells of TNiK−/− mice was significantly lower than that seen in wt cells. Decreases in mEPSC frequency can arise from a reduction in the number of excitatory synapses and/or a decrease in the probability of transmitter release from the presynaptic terminal. We found that PPF in the hippocampal CA1 region was significantly enhanced in slices from TNiK−/− mice suggesting that TNiK may influence presynaptic processes involved in glutamatergic synaptic transmission. At the biochemical level, we found that TNiK was required for GluA1 expression, which is consistent with recent studies using siRNA-mediated TNiK knockdown in primary neuronal cultures (Hussain et al., 2010; Wang et al., 2010). The change in GluA1 did not appear to have a major impact on synaptic transmission as we did not observe reductions in mEPSC amplitudes or evoked AMPAR-mediated synaptic responses in hippocampal slices from TNiK−/− mice. It is not surprising that the 35% decrease in GluA1 levels in TNiK−/− mice is not associated with changes in basal synaptic transmission in the hippocampal CA1 region. For example, previous studies have found that both fEPSPs and mEPSCs are normal in GluA1 knock-out mice (Zaminillo et al., 1999), although this was not seen in all studies (Andrásfalvy et al., 2003). This is most likely due to the fact that functional AMPARs can be made from hetero and homomeric combinations of other subunits (GluA2–4). In addition, ∼95% of the extrasynaptic pool of AMPARs normally contain GluA1 subunits (Lu et al., 2009). Thus there is likely a considerable reserve pool of other AMPAR subunits that could compensate for the modest decrease in total GluA1 levels in TNiK mutants.
TNiK was physically linked to the NMDAR complex and its autophosphorylation site was regulated by both NMDAR and mGluR (in opposing directions). Moreover, in the absence of TNiK there was an elevation of GSK3β phosphorylation on postsynaptic substrates. Plasticities induced by transcription and translation-independent LTP and LTD protocols were normal in TNiK−/− mice. We cannot rule out a potentially important role for TNiK in other forms of LTP such as transcription- or translation-dependent forms of late-phase LTP, which may be relevant to the potential link with fragile X mental retardation protein noted above.
A number of circumstantial lines of evidence suggest TNiK is involved in neuropsychiatric diseases. In addition to its interaction with DISC1, there are links between TNiK and other genes implicated in schizophrenia. NeuroD1 heterodimerizes with the schizophrenia susceptibility protein TCF4 (Brzózka et al., 2010) and TNiK forms a ternary complex with Tcf7l2 (another schizophrenia susceptibility protein; Hansen et al., 2011) and β-catenin, modulating the transcriptional activation of Wnt target genes. We find that TNiK binds and regulates glutamate receptor complexes, which are involved in a range of different cognitive disorders including schizophrenia, autism, and forms of mental retardation (Fernández et al., 2009; Bayés et al., 2011). A recent large-scale study of de novo copy number variation in schizophrenia found a highly significant enrichment in mutations affecting the MAGUK-associated signaling complexes that include PSD-95, TNiK, and proteins dependent on TNiK function (Kirov et al., 2012). We observed that loss of TNiK impairs object-location paired associates learning, which has been evaluated in humans using computer-automated tests such as the object-place paired associates learning (PAL) test from the Cambridge Neuropsychological Test Automated Battery that is widely used in clinical settings (Barnett et al., 2010). Moreover, schizophrenic patients show robust impairments on PAL and performance on this test correlates with symptom severity (Barnett et al., 2005, 2010). Aberrant neurogenesis is also emerging as a neuropathological change in schizophrenia (Reif et al., 2006, 2007; Toro and Deakin, 2007; DeCarolis and Eisch, 2010) and while no study has yet explicitly examined pattern separation in schizophrenic patients, its importance as a cognitive function of potential relevance to schizophrenia has been argued (Tamminga et al., 2010). It is now emerging that human cognitive disorders arise from mutation or disruption in many postsynaptic signaling proteins, including those interacting with TNiK, and future studies may determine whether TNiK plays a role in modulating these disease phenotypes.
Notes
Supplemental Figure 1, Tables 1 and 2, and details of antibodies used in WB, IP, IHC, and IF assays are available at http://www.genes2cognition.org/publications/tnik. This material has not been peer reviewed.
Footnotes
M.P.C., N.H.K., J.N., M.V.K., N.G.S., E.J.T., D.G.F., K.A.E., L.E.S., N.A., and S.G.N.G. were supported by the Wellcome Trust, Medical Research Council (UK), European Union Seventh Framework Programme under grant agreement numbers HEALTH-F2-2009-241498 (“EUROSPIN” project), HEALTH-F2–2009-242167 (“SynSys project”), and HEALTH-F2-2009-241995 (“GENCODYS project”). M.P.C. was also supported by Human Frontiers Science Program. T.J.O. and T.I. were supported by National Institutes of Health–National Institute of Mental Health (Grant #MH609197).We thank T. Ryan, C. Pettit, J. Bussell, and the Research Support Facility staff for technical assistance and Mike Croning for the website and G2Cdb support and members of the Genes to Cognition Programme for critical comments on this manuscript. We are grateful to M. Goedert for Tau antibodies; N. Brandon for DISC1 antibody; and P. Somogyi, J. Gogos, T. W. Robbins, R. Barker, and M. Caron for advice.
References
- Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci. 2007;27:8334–8343. doi: 10.1523/JNEUROSCI.2155-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrásfalvy BK, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR1-deficient mice. J Physiol. 2003;552:35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, Geyer M, Tsuang MT, Salomon D, Schork NJ, Fanous AH, O'Donovan MC, Niculescu AB. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17:887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Kirwan CB, Miller M, Stark CE. Pattern separation in the human hippocampal CA3 and dentate gyrus. Science. 2008;319:1640–1642. doi: 10.1126/science.1152882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Dunning MJ, Samarajiwa SA, Darot JF, Ritchie ME, Lynch AG, Tavaré S. A re-annotation pipeline for Illumina BeadArrays: improving the interpretation of gene expression data. Nucleic Acids Res. 2010;38:e17. doi: 10.1093/nar/gkp942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett JH, Sahakian BJ, Werners U, Hill KE, Brazil R, Gallagher O, Bullmore ET, Jones PB. Visuospatial learning and executive function are independently impaired in first-episode psychosis. Psychol Med. 2005;35:1031–1041. doi: 10.1017/s0033291704004301. [DOI] [PubMed] [Google Scholar]
- Barnett JH, Robbins TW, Leeson VC, Sahakian BJ, Joyce EM, Blackwell AD. Assessing cognitive function in clinical trials of schizophrenia. Neurosci Biobehav Rev. 2010;34:1161–1177. doi: 10.1016/j.neubiorev.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Bayés A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, Grant SG. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A. Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem. 2008;15:50–54. doi: 10.1101/lm.777308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzózka MM, Radyushkin K, Wichert SP, Ehrenreich H, Rossner MJ. Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol Psychiatry. 2010;68:33–40. doi: 10.1016/j.biopsych.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Holmes A, Lyon L, Mar AC, McAllister KA, Nithianantharajah J, Oomen CA, Saksida LM. New translational assays for preclinical modelling of cognition in schizophrenia: the touchscreen testing method for mice and rats. Neuropharmacology. 2012;62:1191–1203. doi: 10.1016/j.neuropharm.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, O'Dell TJ. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J Physiol. 2008;586:5885–5900. doi: 10.1113/jphysiol.2008.163469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JH, Stein GH, Lee JE. NeuroD: the predicted and the surprising. Mol Cells. 2004;18:271–288. [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, Grant SG. Neurotransmitters drive combinatorial multistate postsynaptic density networks. Science Signaling. 2009;2:ra19. doi: 10.1126/scisignal.2000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernández F, Eickholt BJ, Nikolic M, Sutherland C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J Biol Chem. 2006;281:16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creer DJ, Romberg C, Saksida LM, van Praag H, Bussey TJ. Running enhances spatial pattern separation in mice. Proc Natl Acad Sci U S A. 2010;107:2367–2372. doi: 10.1073/pnas.0911725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarolis NA, Eisch AJ. Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology. 2010;58:884–893. doi: 10.1016/j.neuropharm.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Elia J, et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 2012;44:78–84. doi: 10.1038/ng.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández E, Collins MO, Uren RT, Kopanitsa MV, Komiyama NH, Croning MD, Zografos L, Armstrong JD, Choudhary JS, Grant SG. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol Syst Biol. 2009;5:269. doi: 10.1038/msb.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Arlotta P, Macklis JD, Chen J. Conditional knock-out of beta-catenin in postnatal-born dentate gyrus granule neurons results in dendritic malformation. J Neurosci. 2007;27:14317–14325. doi: 10.1523/JNEUROSCI.3206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ables JL, Lagace DC, Nave KA, Goebbels S, Eisch AJ, Hsieh J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci. 2009;12:1090–1092. doi: 10.1038/nn.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PE, Kesner RP, Lee I. Dissociating hippocampal subregions: double dissociation between dentate gyrus and CA1. Hippocampus. 2001;11:626–636. doi: 10.1002/hipo.1077. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S, Limon J, Yip L, Ragsdale CW. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM, Zhao X. Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet. 2012;21:681–691. doi: 10.1093/hmg/ddr501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T, et al. At-Risk Variant in TCF7L2 for Type II diabetes increases risk of schizophrenia. Biol Psychiatry. 2011;70:59–63. doi: 10.1016/j.biopsych.2011.01.031. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hunsaker MR, Kesner RP. Evaluating the differential roles of the dorsal dentate gyrus, dorsal CA3, and dorsal CA1 during a temporal ordering for spatial locations task. Hippocampus. 2008;18:955–964. doi: 10.1002/hipo.20455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Hussain NK, Hsin H, Huganir RL, Sheng M. MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function. J Neurosci. 2010;30:14786–14794. doi: 10.1523/JNEUROSCI.4124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka K, Kamiya A, Oh EC, Kanki H, Seshadri S, Robinson JF, Murdoch H, Dunlop AJ, Kubo K, Furukori K, Huang B, Zeledon M, Hayashi-Takagi A, Okano H, Nakajima K, Houslay MD, Katsanis N, Sawa A. DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature. 2011;473:92–96. doi: 10.1038/nature09859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe H, Neeb A, Dimova K, Young SM, Jr, Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang DE, Umikawa M, Kariya K, Goebbels S, Nave KA, Rosenmund C, Jahn O, Rhee J, Brose N. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron. 2010;65:358–372. doi: 10.1016/j.neuron.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115:97–105. doi: 10.1016/s0165-0270(02)00007-9. [DOI] [PubMed] [Google Scholar]
- Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanitsa MV, Afinowi NO, Grant SG. Recording long-term potentiation of synaptic transmission by three-dimensional multi-electrode arrays. BMC Neurosci. 2006;7:61. doi: 10.1186/1471-2202-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Muotri A, Yeo G, Warashina M, Lie DC, Moore L, Nakashima K, Asashima M, Gage FH. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat Neurosci. 2009;12:1097–1105. doi: 10.1038/nn.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Pleasure SJ, Collins AE, Noebels JL, Naya FJ, Tsai MJ, Lowenstein DH. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc Natl Acad Sci U S A. 2000;97:865–870. doi: 10.1073/pnas.97.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren EJ, Charlesworth P, Coba MP, Grant SG. Knockdown of mental disorder susceptibility genes disrupts neuronal network physiology in vitro. Mol Cell Neurosci. 2011;47:93–99. doi: 10.1016/j.mcn.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Li VS, Ng SS, Taouatas N, Vries RG, Mohammed S, Heck AJ, Clevers H. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009;28:3329–3340. doi: 10.1038/emboj.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, Bauchwitz RP. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. 2009;56:463–472. doi: 10.1016/j.neuropharm.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Turner JA, Guffanti G, Lakatos A, Fallon JH, Nguyen DD, Mathalon D, Ford J, Lauriello J, Macciardi F. A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr Bull. 2009;35:96–108. doi: 10.1093/schbul/sbn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514–522. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- Reif A, Schmitt A, Fritzen S, Lesch KP. Neurogenesis and schizophrenia: dividing neurons in a divided mind? Eur Arch Psychiatry Clin Neurosci. 2007;257:290–299. doi: 10.1007/s00406-007-0733-3. [DOI] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they're apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G, St Clair D. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitashige M, Satow R, Jigami T, Aoki K, Honda K, Shibata T, Ono M, Hirohashi S, Yamada T. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res. 2010;70:5024–5033. doi: 10.1158/0008-5472.CAN-10-0306. [DOI] [PubMed] [Google Scholar]
- Sirerol-Piquer M, Gomez-Ramos P, Hernández F, Perez M, Morán MA, Fuster-Matanzo A, Lucas JJ, Avila J, García-Verdugo JM. GSK3beta overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus. 2011;21:910–922. doi: 10.1002/hipo.20805. [DOI] [PubMed] [Google Scholar]
- Smyth G, Ritchie M, Thorne N, Wettenhall J, Shi W. User's guide. Melbourne, Australia: 2005. Limma: linear models for microarray data. [Google Scholar]
- Talpos JC, Winters BD, Dias R, Saksida LM, Bussey TJ. A novel touchscreen-automated paired-associate learning (PAL) task sensitive to pharmacological manipulation of the hippocampus: a translational rodent model of cognitive impairments in neurodegenerative disease. Psychopharmacology. 2009;205:157–168. doi: 10.1007/s00213-009-1526-3. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- Toro CT, Deakin JF. Adult neurogenesis and schizophrenia: a window on abnormal early brain development? Schizophr Res. 2007;90:1–14. doi: 10.1016/j.schres.2006.09.030. [DOI] [PubMed] [Google Scholar]
- von Bohlen Und Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007;329:409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, Revilla-Sanchez R, Kelly MP, Dunlop AJ, Murdoch H, Taylor N, Xie Y, Pausch M, Hayashi-Takagi A, Ishizuka K, Seshadri S, Bates B, Kariya K, Sawa A, Weinberg RJ, et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 2011;16:1006–1023. doi: 10.1038/mp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EM, Geschwind DH, Palmer TD. Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008;13:285–292. doi: 10.1038/sj.mp.4002093. [DOI] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lubke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]