

Abstract
Treatment options of glioblastoma multiforme are limited due to the blood–brain barrier (BBB). In this study, we investigated the utility of intranasal (IN) delivery as a means of transporting stem cell–based antiglioma therapeutics. We hypothesized that mesenchymal stem cells (MSCs) delivered via nasal application could impart therapeutic efficacy when expressing TNF-related apoptosis-inducing ligand (TRAIL) in a model of human glioma. 111In-oxine, histology and magnetic resonance imaging (MRI) were utilized to track MSCs within the brain and associated tumor. We demonstrate that MSCs can penetrate the brain from nasal cavity and infiltrate intracranial glioma xenografts in a mouse model. Furthermore, irradiation of tumor-bearing mice tripled the penetration of 111In-oxine–labeled MSCs in the brain with a fivefold increase in cerebellum. Significant increase in CXCL12 expression was observed in irradiated xenograft tissue, implicating a CXCL12-dependent mechanism of MSCs migration towards irradiated glioma xenografts. Finally, MSCs expressing TRAIL improved the median survival of irradiated mice bearing intracranial U87 glioma xenografts in comparison with nonirradiated and irradiated control mice. Cumulatively, our data suggest that IN delivery of stem cell–based therapeutics is a feasible and highly efficacious treatment modality, allowing for repeated application of modified stem cells to target malignant glioma.
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive form of primary brain tumor. Patient prognosis is poor even with aggressive interventions including surgical resection and radiation. Tumors typically recur after treatment and the median survival time following diagnosis is ~15 months.1,2 The blood–brain barrier (BBB) limits the ability of systemically delivered anticancer pharmaceuticals to reach the brain, hence complicating the treatment of GBM due to lack of accessibility to the tumor bed. Direct delivery of chemotherapeutic drugs to the tumor site, through methods such as convection-enhanced delivery, allows for high concentration of the drug at the appropriate location. However, this method is invasive, risks damaging surrounding normal brain tissue, and at present, remains to be fully optimized for clinical applications.3
Previous work has demonstrated that stem cells, specifically neural stem cells (NSCs) and mesenchymal stem cells (MSCs), have a tropism for brain tumors.4,5 This property has generated much interest in utilizing stem cells as vehicles for targeted drug delivery. As is the case in CNS drug delivery, stem cell delivery is also hampered by the presence of the BBB. Because of the BBB, few stem cells reach the brain following intravenous delivery and have a propensity to accumulate in the lungs or other organs.6,7 Intra-arterial delivery has been shown to deliver larger numbers of cells to the brain compared with intravenous delivery;7,8,9 however, this method has also been associated with a high incidence of mortality and impaired cerebral blood flow in rats.9,10 Attempts have been made to increase the efficiency of systemic delivery by disrupting all or portions of the BBB,11 but this could potentially leave the CNS vulnerable to toxins or infection.
Recent publications have explored the nasal system as a novel stem cell delivery route to the brain. MSCs delivered into the nasal cavity have been shown to migrate through the cribriform plate and into brain tissue via the olfactory and trigeminal pathways.12 Not only were stem cells located in varying and relatively remote regions of the brain such as the cerebellum, but the delivery of MSCs appeared to have a therapeutic effect in animal models of Parkinson's disease and ischemic brain injury.13,14 NSCs have also been shown to penetrate into mouse brain and reach the tumor bed in experimental glioma models after intranasal (IN) application.15 Thus, accumulating evidence suggests that IN delivery of stem cells might be a viable approach for treatment of CNS pathology. Moreover, complications associated with intravascular delivery, such as obstruction by the BBB, pulmonary embolism and infarctions could also be avoided using this approach. In addition, IN delivery offers a practical advantage over direct intracranial application of stem cells into resection cavity during surgery or convection-enhanced delivery, since it might allow for multiple treatment regimens and can also be utilized in patients with inoperable tumors.
In this study, we examined if MSCs delivered via the nasal cavity can reach intracranial human glioma xenografts in mice and be therapeutically relevant when expressing TNF-related apoptosis-inducing ligand (TRAIL). TRAIL has been shown to promote apoptosis in a variety of cancers including glioma,16 with minimal or no effect on normal cells.17 The therapeutic efficacy of stem cells modified to express TRAIL has been previously demonstrated in glioma.18,19 However, in these studies the delivery method of the stem cells to the brain was limited either to injection via tail vain or to direct intracranial inoculation. IN delivery of therapeutic stem cells is a potentially advantageous treatment modality because it represents a noninvasive, nontoxic delivery method; once optimized, it can allow for repeated application of therapeutic stem cells alone or in combination with traditional treatment methods such as radiation and chemotherapy.
Results
MCSs migrate in the brain of animals bearing intracranial glioma xenograft
It has been previously shown that MSCs display tropism to solid tumors.20 In this study, we first confirmed that MSCs are capable of migrating towards intracranial glioma xenografts. Figure 1a shows that MSCs-fluc injected in the left hemisphere are capable of migrating quickly towards the right, tumor-bearing side of the brain. On average, 11% of implanted MSCs were detected in the right hemisphere as soon as 2 hours following MSC inoculation. Number of MSCs remained steady at the 24-hour time point but significantly decreased in both hemispheres through day 11 (P < 0.05). Migration of MSCs towards the tumor-bearing side of the brain was further confirmed using live animal imaging. Figure 1b shows distinct pattern of MSCs migration from the injection (left) site toward the right side of the brain in tumor bearing but not control animals. Thus, collectively this data demonstrate that MSCs are capable of migrating towards intracranial tumor in a mouse model.
Figure 1.

MCSs migrate in the brain of animals towards intracranial glioma xenograft. (a) Quantitative luciferase assay of migration of hMSCs-fluc in the brain of nude mice. MSCs-fluc were inoculated into the left hemisphere of brain of animals with U87 glioma xenografts established in the right hemisphere of brain. Luciferase activity in the left and right hemispheres of brain was measured at various time points to determine dynamics of MSCs migration toward the tumor-bearing side of the brain. (b) Imaging of MSCs-fluc migration within the brain of control and tumor-bearing mice. Arrow shows site of injection of MSCs in the left hemisphere of the brain. T-tumor injection site. Data are presented as mean + SEM. Paired t-tests were applied to examine differences in relative luciferase activity between the left and right side of the brain at each time point (n = 4/group). Unpaired t-tests were utilized to examine changes in luciferase activity from day 0 to day 11 in each side of the brain. * P < 0.05, ** P < 0.01. MSCs, mesenchymal stem cells.
MSCs penetrate into mouse brain after IN inoculation
Next, we investigated if MSCs could penetrate and be tracked in the brain after IN administration. Live animal imaging was performed after application of MSCs-fluc into the nasal cavity of the animals. Figure 2a shows the presence of a fluc signal in the nasal cavity of both control and tumor-bearing animals. A significant decrease in signal intensity in the nasal cavity was observed from day 0 to day 1, and therefore, imaging required longer exposure times for detection of MSCs on following days. No significant advancement of MSCs into the brain beyond the orbital arches was detected at various time points either in control or tumor-bearing animals using this technique. Taking into consideration the limitations of this method and in order to demonstrate that MSCs are able to advance in the brain of the animals after IN application, the brains of animals were removed 3 hours after IN inoculation of MSCs-fluc and imaged for the presence of fluc and green fluorescent protein (GFP) signal (tumors expressed GFP). Figure 2b shows images of MSCs in the nasal cavity of the (panel A) live animal and luciferase signal over the olfactory bulbs and frontal lobes of the (panel B) brain of tumor-bearing animal. No signal was detected in control, phosphate-buffered saline (PBS)–treated animals. Figure 2b (panel C) shows the localization of xenograft tumors in the right hemisphere of both animals. Finally, panels D and E in Figure 2b show that MSCs are also capable of rapid penetration into the brain of mice after IN inoculation in control, nontumor-bearing animals in agreement with a previous report showing penetration MSCs in the brain after IN application in rats.12,13
Figure 2.

MSCs penetrate into mouse brain after IN inoculation. (a) Live animal imaging after intranasal inoculation of MSCs-fluc. Control (no tumor) and U87 glioma-bearing animals were imaged live at various time points after IN application of MSCs-fluc. (b) Imaging of MSCs-fluc in the brain of the control and tumor-bearing animals after intranasal inoculation. In order to demonstrate the advancement of MSCs into the brain after IN application, the brains of animals were removed 3 hours after IN inoculation of MSCs-fluc and imaged for the presence of fluc and GFP signal corresponding to U87 cells. (A) live animal imaging without (left) and with inoculated MSCs (right); (B) imaging of brain without (left) and with inoculated MSCs (right); (C) imaging of U87-GFP tumor in the right hemisphere of the brain; (D) imaging of MSCs in control, nontumor-bearing animals; (E) imaging of brain of nontumor animals for GFP signal. MSCs, mesenchymal stem cells.
Distribution of 111In-oxine–labeled MSCs in the brain after IN delivery
This part of the study was designed to quantitatively evaluate the ability of MSCs to penetrate into the brain of tumor-bearing animals after IN delivery. Figure 3a shows that MSCs treated with 111In-oxine for 10, 20, and 30 minutes were labeled at a similar level, therefore a 20 minutes labeling time point was chosen for following experiments. Taking into consideration the short half-life of 111In-oxine (2.8 days), we determined the sensitivity at which MSCs could be potentially detected within the brain 24 hours after labeling. Figure 3b shows that radioactivity corresponding to as little as one labeled cell could be detected using this method. It has previously been shown that MSC tropism to tumor is increased following irradiation of the tumor.20,21 With this in mind, we next investigated the distribution of 111In-oxine–labeled MSCs in the brain of tumor-bearing control and irradiated animals. Figure 3c shows the distribution of 111In-oxine–labeled MSCs in the olfactory bulbs; the left, nontumor-bearing hemisphere of the brain; the right, tumor-bearing hemisphere of the brain, and in the cerebellum of control and irradiated animals. No difference in the presence of MSCs was detected in the olfactory bulbs of control and irradiated animals, whereas the presence of MSCs was two- (P = 0.03, n = 4) and threefold (P = 0.057, n = 4) higher in the left and right hemispheres of irradiated animals respectively. Furthermore, we observed approximately a fivefold (P = 0.03, n = 4) increase in the presence of MSCs in the cerebellum of irradiated animals. On average, the total number of MSCs penetrating in the brain of irradiated animals was 2.8 times higher (P = 0.028, n = 4) than in control nonirradiated tumor-bearing animals (Figure 3d). 111In-oxine was also detected in other organs (Figure 3e); specifically, the highest quantities were in the lung and stomach. Thus, these data quantitatively validate and further indicate that MSCs are capable of penetrating into the brain of mice after IN inoculation and their penetration is significantly enhanced by irradiation.
Figure 3.
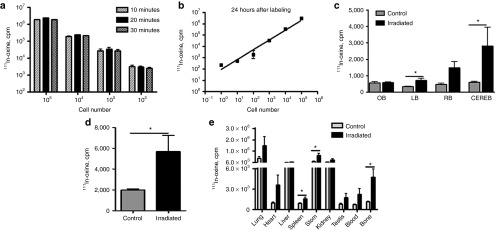
Radiolabeling of MSCs with 111In-oxine and their distribution in the brain and other organs after IN delivery. (a) Labeling of MSCs with 111In-oxine. MSCs were incubated with 111In-oxine at 10, 20, and 30 minutes in CO2 incubator at 37 °C in order to determine an optimal labeling time. (b) Sensitivity of assay. In order to determine if the level of MSCs labeling with 111In-oxine is sufficient to detect low number of cells, MSCs were titrated from 1 to 106 cells per tube and the radioactivity in corresponding samples was measured using a gamma counter. (c) Distribution of MSCs through varying parts of the brain of control and irradiated tumor-bearing animals. 111In-oxine–labeled MSCs were inoculated in the brain of nonirradiated and irradiated tumor-bearing animals. Animals were killed 24 hours after IN inoculation and the brain and other organs were collected for measurement of 111In-oxine activity. OB-olfactory bulbs, LB-left hemisphere of the brain, RB-right hemisphere of the brain, CEREB-cerebellum. (d) Comparison of MSC presence in the whole brain of control and irradiated animals. (e) Distribution of MSCs through the organs of control and irradiated tumor-bearing animals. Data are presented as mean + SEM. Two-way analysis of variance followed by Bonferroni's post hoc test has been applied to examine the effect of incubation time on labeling efficiency of MSCs. The presence of 111In-oxine–labeled MSCs in each organ has been compared between control versus irradiated animals using Mann–Whitney tests. *P < 0.05. MSCs, mesenchymal stem cells.
Detection of MSCs in the tumor bed after IN inoculation
In order to further localize MSCs within the brain, MSCs were labeled with micron-size paramagnetic iron oxides (MPIOs) before IN delivery to tumor-bearing mice. Supplementary Figure S1a demonstrates the efficiency of MSCs labeling with MPIOs at various cell-to-particle ratios. The ratio of 1:15 generated about 60% of positive cells in allophycocyanin channel in the absence of toxicity (data not shown) and, therefore, was chosen as optimal for stem cell labeling. Supplementary Figure S1b shows the presence of flash red fluorescent beads in labeled MSCs counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (right panel); the left panel shows control, nonlabeled cells. Next, we IN inoculated MPIOs labeled MSCs in control and tumor-bearing mice. Prussian blue staining was performed to detect iron particles in multiple brain tissue sections from animals collected at day one and three following IN stem cell inoculation. Prussian blue staining was detected in all animals that had received MPIOs-labeled MSCs (Figure 4a; panels B–F). The vast majority of the positive signal was detected in immediate proximity to or within the tumor bed of control and irradiated animals. Single clusters of positive staining were detected in other brain regions (data not shown). Collectively, these data indicate that stem cells are capable of migrating to intracranial tumor when delivered IN in control and irradiated animals.
Figure 4.

Tracking MPIO-labeled MSCs in the brain of the mice after IN injection using MRI. (a) Prussian blue staining of brain tissue sections from (a–c) control nonirradiated or (d–f) treated with irradiation mice bearing U87 xenografts at day 1 and 3 after IN administration of MPIO-labeled MSCs. a represents a negative control staining of tissue sections from the brain of the animals treated with PBS. f represents higher magnification of e (scale bar is 50 µm). The tumor mass is outlined. Arrows indicate positive Prussian blue staining. Scale bar for a–e is 200 µm. (b–c) T1- and T2-weighted MRI images of control animals (no MSCs), nonirradiated or irradiated tumor-bearing animals were taken at day 2 after IN inoculation of MPIO-labeled MSCs. b shows coronal images. (A,B) No MPIO-labeled MSC-induced signal was detected in the brain of control mouse (no MSCs, no tumor). Sequential T1-weighted images demonstrate the path of migration of MSCs towards the tumor in (C,D) nonirradiated animal and (E–G) irradiated animal. (H,I,J) T2-weighted sequential images of irradiated animal were taken to visualize the tumor and MPIO-induced signal. Red arrows show signal corresponding to the migrating MSCs. Yellow arrow shows tumor on T2-weighted image (J). (c) Axial T1-weighted brain images in (A–E) irradiated tumor-bearing animal. MSCs, mesenchymal stem cells.
Tracking of MSCs in the brain after IN injection using magnetic resonance imaging
MPIOs were previously utilized to track the migration of various cell types in vivo, including stem cells.22,23,24,25,26 In order to achieve sufficient resolution to visualize labeled MSCs within the mouse brain, multi-slice high resolution T2 weighted, Rapid Acquisition with Relaxation Enhancement spin echo images and multi-slice T1 weighted a Fast Low Angle Shot (FLASH) gradient echo sequences were acquired in both coronal (Figure 4b; panel B) and axial planes of the mouse brain (panel C). Figure 4b demonstrates the path of migration of MPIO-labeled MSCs towards the tumor (red arrows) on sequential T1-weighted images of nonirradiated (panels C,D) and irradiated animals (panels E–G). In order to identify the tumor mass, sequential T2-weighted images of an irradiated animal were taken and presented in Figure 4b (panels H–J). The signal induced by MPIOs is shown by red arrows. The yellow arrow indicates the tumor mass. MPIO-induced signal was more apparent in irradiated animals. This signal appeared in the basal region and sequentially extended toward the tumor. This result was confirmed on T2-weighted images. On axial images of the brain of an irradiated animal (Figure 4c; panels A–E and Supplementary Video S1), the MPIO-induced signal appears as a dark spot in the basal region of the brain, visualized in all sequential images and finally appears in the area corresponding to the location of the tumor (Figure 4c; panel E). Animals undergoing magnetic resonance imaging (MRI) were killed and their brains collected for Prussian blue staining. Positive Prussian blue staining in the tumor region confirmed the results of the MRI (data not shown). These findings further confirmed the direct migration of IN delivered MSCs towards the tumor through brain parenchyma in control and irradiated animals.
MSCs-expressing TRAIL kill glioma cells in vitro and improve survival of irradiated animals after IN delivery
To investigate if IN delivered MSCs could be therapeutically beneficial, MSCs modified to express TRAIL and irradiation were utilized as therapeutic modalities. Figure 5a shows that MSCs-TRAIL, but not control MSCs (vector control), express TRAIL protein at ~100 ng in a cell-bound fraction per 1 × 106 cells, and about 80 ng of protein is released into the culture media during a 24 hours incubation. Next, we investigated the sensitivity of various glioma cell lines to MSCs-TRAIL. For this, U87, GBM43, GBM12, and GBM39 glioma cells expressing fluc were cocultured either with control or MSCs-TRAIL at the range of 1:0.05–1:1 of glioma cells to MSCs ratios for 24 hours. Figure 5b shows that glioma cell lines possess different sensitivity to MSCs-TRAIL and are not affected by control MSCs. The IC50 is expressed as the ratio of glioma cells to MSCs-TRAIL and was calculated to be 0.049, 0.38, 0.66, and 1.19 for GBM43, GBM39, U87, and GBM12, respectively. Next, U87 and GBM43 glioma cell lines were chosen to further confirm that MSCs-TRAIL can cause apoptosis in glioma cells. Figure 5c demonstrates a 2.5- to 3-fold increase in cleaved caspase-3 expression in U87 and GBM43 glioma cell lines cocultured with MSCs-TRAIL compared with control MSCs, whereas no change in cleaved caspase-3 expression was detected between untreated (PBS) and control MSCs treated glioma cells. Lastly, we investigated the effect of IN delivered MSCs-TRAIL on the survival of animals bearing intracranial U87 glioma xenografts. The MSCs-TRAIL group (n = 10) demonstrated 11% improvement in survival in comparison with PBS (n = 8, P < 0.05), but not with control MSCs group of animals (Supplementary Figure S2). Irradiation treatment of control animals resulted on average in 28% improvement in median survival. The MSCs-TRAIL plus irradiation group of animals demonstrated 45% and on average 13% improvement in median survival compared with control nonirradiated and control irradiated animals respectively (n = 6, P < 0.001) (Figure 5d). These data indicate that MSCs delivered IN could be therapeutically beneficial in intracranial glioma animal models. However, various treatment modalities and glioma models need to be further investigated to validate this approach.
Figure 5.

Effect of MSCs expressing TRAIL on various glioma cells in vitro and survival of animals bearing intracranial glioma xenografts after IN delivery. (a) The level of TRAIL expression in MSCs was measured using ELISA as described in Materials and Methods section. (b) Killing effect of MSCs-TRAIL on various glioma cell lines. Control or MSCs-TRAIL were cocultured with U87, GBM39, GBM43, and GBM12 expressing fluc at 1:0.05 to 1:1 of glioma to MSC ratios. At 24 hours, luciferase activity in glioma cells was measured in triplicates. Two-way analysis of variance followed by Bonferroni post hoc test was utilized to evaluate the difference between control MSCs and MSCs-TRAIL on the toxicity in glioma cell lines. (c) Changes in caspase-3 expression in U87 and GBM43 glioma cell lines followed by 8-hour coculture with control or MSCs-TRAIL were evaluated by flow cytometry using rabbit anti–caspase-3 antibody. Samples were run in triplicates. Statistical difference among groups was evaluated using two-way analysis of variance followed by Bonferroni post hoc test. (d) Effect of intranasally administered MSCs-TRAIL on survival of mice bearing intracranial U87 glioma xenografts and treated for 5 days with 2 Gy gamma irradiation (total dose 10Gy), n = 6/group. Survival curves were generated by the Kaplan–Meier method, Logrank test was applied to determine statistical difference on survival time distribution between groups. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. MSCs, mesenchymal stem cells; RLU, relative luciferase units.
Changes in CXCL12 expression in U87 glioma xenograft tissue as a potential mechanism for increased penetration of MSCs in the mouse brain after gamma irradiation
In this set of experiments, we attempted to uncover potential mechanisms governing the therapeutic effect of the combined treatment of irradiation and MSCs-TRAIL following IN delivery. The expression of TRAIL receptors, DR4 and DR5, was analyzed in control and irradiated U87 glioma xenograft tissues. GFP expressing U87 glioma tissues were removed from the brain of animals 24 hours after irradiation under the guidance of a fluorescent microscope and samples were processed and analyzed for DR4 and DR5 expression by quantitative PCR and flow cytometry. Figure 6a shows that the expression of DR4 and DR5 mRNA remained unchanged in xenograft tissue after irradiation in our experimental setting. Similarly, no change in the expression of DR4 and DR5 was found on the surface of U87 glioma cells obtained from the glioma xenografts (Figure 6b).
Figure 6.

Effect of irradiation on expression of TRAIL receptors and the CXCL12 in U87 glioma xenografts. (a) Expression of mRNA for DR4 and DR5 in U87 glioma xenograft tissue of control and irradiated mice. (b) Expression of DR4 and DR5 receptors in U87 glioma xenograft tissue of control and irradiated mice was analyzed using flow cytometry, and expressed as mean fluorescence intensity (MFI). Samples were analyzed in triplicates. (c) Expression of mRNA for CXCL12 in U87 glioma xenograft tissue of control and irradiated mice. (d) Expression of CXCL12 protein in U87 glioma xenograft tissue of control and irradiated mice was analyzed by ELISA. Samples were analyzed in duplicates from each control and irradiated mice (n = 3). Student's t-tests have been utilized to evaluate the difference between groups. Data are presented as mean ± SEM. *P < 0.05.
In has been previously shown that certain chemokines and cytokines secreted from the tumor bed can act as chemo-attractant for stem cells.5,27 Based on this, we next used an array-based RT-PCR approach to evaluate the effects of the radiation therapy on the chemokines and cytokines milieu at the tumor sites that can enhance the migratory capacity of the implanted MSCs. For this, glioma tissue from three control or irradiated animals was pooled together and processed for array analysis per manufacturer's recommendations. Supplementary Table S1 summarizes changes in gene expression above threefold in U87 xenograft tissue after irradiation. The CXCL12 or stromal cell–derived factor-1 α (SDF-1 α) has been shown to contribute to tropism of MSC towards glioma.28,29 In our study, CXCL12 was identified as a chemokine potentially involved in enhanced migration of MSCs towards glioma tissue after irradiation. The changes in expression of mRNA in U87 glioma xenografts after irradiation were confirmed in a separate qPCR reaction (Figure 6c). ELISA was utilized to confirm the changes of CXCL12 expression in U87 glioma xenograft tissue obtained from three control and three irradiated animals (Figure 6d). In summary, our data identify CXCL12 as a chemokine potentially responsible for enhanced migration of MSCs in the brain of irradiated animals. However, further studies validating the role of CXCL12 in driving the migration of IN delivered MSCs towards intracranial glioma xenografts still need to be performed.
Discussion
The BBB hampers many therapeutic agents from reaching the tumor bed, thus making treatment of GBMs a challenging task. Additionally, the invasive nature of GBMs allows them to escape localized therapies such as surgery. Recently, stem cells have gained significant interest as therapeutic carriers for the targeted treatment of brain tumors due to their inherent tumor tropism. However, current clinical method for delivery of stem cells relies on the placement of cells in the resection cavity during surgery. In that respect, IN delivery of therapeutic stem cells to the CNS is an attractive, noninvasive, alternative method, allowing stem cells to bypass the BBB. Therefore, in the present study, we investigated if IN delivery of MSCs is a viable approach for the treatment of brain tumors in an experimental mouse model. We showed for the first time that IN delivery of TRAIL expressing MSCs can prolong median survival in an animal model of orthotropic human glioma xenograft.
In this study, we employed several methods to demonstrate the feasibility of IN delivery of MSCs to the brain in tumor-bearing animals. First, we showed that MSCs are capable of migrating within the brain towards tumor in agreement with previously published reports.5,30,31 Next, using bioluminescent imaging we demonstrated penetration of MSCs from the nasal cavity into the brain of mice. It is worth noting that luciferase signal was easily detectable in the nasal cavity and in the brain within several hours after injection, but detection of MSCs in nasal cavity at later time points required longer exposure, suggesting rapid elimination of MSCs. Similarly, the fast disappearance of signal corresponding to labeled MSCs from the nasal cavity was observed in a rodent model of Parkinson's disease.32 It is likely that fast penetration of MSCs from the nasal cavity into brain tissue is due to direct passage of MSCs through the cribriform plate via the olfactory and trigeminal pathways.12 111In-oxine–labeled MSCs were also detected in the brain of mice. Moreover, radiation treatment of tumor-bearing animals enhanced MSCs penetration into the brain. As proliferation and clearance of MSCs could differ in control and irradiated animals, it is unlikely that it contributed to the observed effect at 24 hours after MSCs delivery, and rather can be explained by higher penetration of MSCs in the brain of irradiated animals. Interestingly, we also found a fivefold increase in accumulation of 111In-labeled MSCs in the cerebellum of irradiated animals. This finding is of importance given the high frequency of cerebellar brain tumors in children33 and warrants the evaluation of IN delivery of therapeutic stem cells in experimental model of medulloblastoma and other cerebellar tumors. Histological evaluation of the brain tissues further confirmed the presence of MSCs in the glioma xenograft and adjacent tissue of control and irradiated animals. The accumulating evidence of the ability of stem cells to penetrate the brain via the nasal cavity12,13,14,15,32 makes it prudent that we are able to understand the mechanism underlying this phenomenon. Elucidating this mechanism may allow us to increase the number of stem cells that reach the brain after IN delivery.
Recent reports indicate that stem cells modified with iron particles can be tracked in the brain using MRI.24,34 In our study, MSCs were labeled with MPIOs in order to visualize their migration towards tumor following IN delivery. Using MRI, we were able to detect MPIO-induced signals extending from the basal frontal lobe towards the tumor in control and irradiated animals after IN inoculation of MSCs. To our knowledge, this is the first report utilizing MRI to confirm migration of IN delivered MSCs towards glioma xenografts. Previously, methods of fluorescent microscopy, [H3]-thymidine and near-infrared live imaging were utilized to track MSCs in the brain after IN delivery.12,13,14,32 In addition to the fact that each of these methods has relatively limited sensitivity, more notably there is a lack of clinical relevance and applicability. MRI studies monitoring the distribution of stem cells in the CNS will be necessary to evaluate the feasibility of IN delivery of therapeutic stem cells for treatment of brain tumors and other CNS disorders on a clinical level.
In our study, we demonstrated that IN delivered MSCs expressing TRAIL improved survival of the animals bearing intracranial glioma xenografts in combination with irradiation. It has been shown that radiation treatment can sensitize cancer cells to TRAIL treatment and upregulation of TRAIL receptors, DR4 and DR5, has been implicated in this process.35,36 However, no change in the expression of DR4 and DR5 in U87 glioma tissue upon radiation treatment was detected suggesting that a different mechanism is responsible for the therapeutic effect of MSCs expressing TRAIL. Enhanced migration of MSCs towards irradiated tumors has been demonstrated by several groups,20,21,37 but the exact mechanism of this tropism remains unknown. We found that along with other factors, CXCL12 (e.g., SDF-1) is upregulated after the radiation treatment suggesting that the SDF-1/CXCR4 axis and other mechanisms may contribute to migration of MSCs into the brain and towards the glioma tissue in irradiated animals. Recently, Kim and coauthors demonstrated that IL-8 is involved in migration of umbilical cord blood-derived MSCs towards irradiated U87 glioma cells in vitro.21 In contrast, no upregulation in IL-8 gene expression in irradiated glioma xenograft tissue was found in our study, which might relate to differences in experimental setting. Future studies should be carried out to elucidate the mechanisms regulating the migration of stem cell into the brain of tumor-bearing animals via the nasal cavity.
IN delivery of stem cells appears to be promising approach for therapy of CNS disease. This approach shows a clinical and practical advantage over the intracranial application of stem cells into resection cavity during surgery and the convection-enhanced delivery, since it is not associated with the time of the surgery, is noninvasive in nature, and allows for multiple treatment regiments. It also can avoid complications associated with intravascular delivery as well as increase number of stem cells delivered to the tumor site through the repeated application of stem cells in nasal cavity. Moreover, this method could be also potentially utilized to treat patients with inoperable brain tumors. Recently, two groups reported a beneficial effect of unmodified MSCs delivered via nasal cavity in rat models of ischemia and Parkinson disease.13,14 NSCs have been shown to accumulate in intracranial glioma xenografts after IN delivery,15 but the authors did not evaluate preclinical efficacy of IN delivered NSCs. Here, we demonstrated the therapeutic effect of MSCs-TRAIL in combination with radiation treatment in intracranial orthotopic glioma model. We found that only a low number of MSCs can penetrate the brain via nasal cavity in mice, which could be the limiting factor for treatment of brain tumors via IN delivery of stem cells. To overcome this limitation, stem cells could be manipulated to increase their migration from nasal cavity into the brain by various means. The overexpression of CXCR4 in MSCs has been shown to enhance in vivo mobilization and engraftment of MSCs into ischemic area of myocardium.38,39 Hypoxic preconditioning of MSCs has been shown to improve their homing in ischemic area of the brain after IN delivery.40 Treatment of rat MSCs with insulin-like growth factor-1 (IGF-1) has been shown to induce an increase in MSCs migration in response to SDF-1 via CXCR4 receptor signaling.41 Thus, understanding the mechanisms which drive the migration of stem cells will allow for the development of new strategies to enhance penetration of stem cells into the brain via the nasal cavity and ultimately efficient migration towards the tumor where they can execute their therapeutic effect. In our study, we did not investigate the dynamics of the presence of therapeutic MSCs in the tumor burden. This knowledge would be necessary for development of approaches enhancing the homing of therapeutic MSCs in the tumor tissue after their IN application. Additionally, nasal delivery of therapeutic stem cells for treatment of brain tumors has to be evaluated in solid and infiltrative glioma models closely resembling patient tumors as well as in primates, given a known difference between the rodent's and the human olfactory systems,42 to make this approach clinically sound.
Conclusions
Our study has demonstrated for the first time that noninvasive IN delivery of MSCs is a promising approach for the therapeutic treatment of experimental glioma. Using complementing methods, we were able to demonstrate (i) migration of MSCs into the brain and towards glioma xenografts after IN application, (ii) MSC presence in the tumor by histological evaluation and by MRI, and (iii) improvement in survival of animals using TRAIL modified MSCs in combination with radiation treatment. Ultimately, further studies utilizing various therapeutic modalities in different animal models are necessary to validate IN delivery of stem cells as a recognized approach for the treatment of experimental brain tumors.
Materials and Methods
Cell lines. U87MG glioma cell line expressing EGFRvIII (mutant Epidermal growth factor receptor) was kindly provided by Dr Frank Furnari (the University of California at San Diego, San Diego, CA) and is designated elsewhere in the text as U87 glioma cell line. GBM43, GBM12, and GBM39 patient derived glioma cell lines expressing firefly luciferase (fluc) were kindly provided by Dr David James (the University of California at San Francisco, San Francisco, CA). Human bone marrow derived MSCs were provided by Dr Michael Mathis (the Louisiana Health Sciences Center, New Orleans, LA). Modifications of MSCs to express fluc (MSCs-fluc)43 and membrane-bound TRAIL (MSCs-TRAIL) were performed as previously described for NSCs.44
Luciferase assay. Killing effect of MSCs-TRAIL on glioma cells was measured using a luciferase assay. Briefly, 5 × 103 of fluc-expressing glioma cells were cocultured with MSCs-TRAIL, and in 24 hours, cells were lysed. To quantify migration of MSCs towards tumor xenografts, U87 cells were implanted in the right hemisphere of nude mice. One week later, MSCs-fluc were inoculated in the left hemisphere of the brain and mice were killed at various time points. The left and right hemispheres of the brain were homogenized in equal volume of lysis buffer and cleared by centrifugation. Luciferase activity in the cell lysates and homogenates was measured per manufacturer's recommendation (Promega, Madison, WI).
MSC labeling with 111In- oxine. The procedure for labeling MSCs was performed as previously described45 with slight modifications. MSCs at 106/ml were mixed with 50 µCi of 111In-oxine (GE Healthcare, Pittsburgh, PA) and incubated for 10, 20 and 30 minutes at 37 °C in 5% CO2. After two PBS washes, cells were counted in a gamma counter. The efficiency of the 111In-oxine labeling was calculated as a percentage of the label retained by the cells relative to the total added radioactivity.
MSCs labeling with MPIOs. For labeling of MSCs, a MPIO particles (Bangs Laboratories, Fishers, IN) were added to about 80% confluent MSC's culture in the presence of Fugene reagent (Roche Applied Science, Indianapolis, IN) for overnight incubation. For fluorescent microscopy, cells grown in Lab-Tek chamber (Thermo Scientific, Rochester, NY) slides were fixed with 4% paraformaldehyde and counterstained with DAPI nucleic acid stain (Invitrogen, Grand Island, NY). Slides were analyzed using Olympus IX70 inverted microscope and images were obtained using MetaMorph software (Molecular Devices, LLC, Sunnyvale, CA). The efficiency of MSCs labeling with flash red labeled MPIOs was determined by flow cytometry as percent of positive cells.
Detection of MPIO-labeled MSCs with Prussian Blue staining. For Prussian blue staining, frozen 16 µm brain sections were fixed in 4% paraformaldehyde and after three washes with PBS were stained with potassium ferrocyanide trihydrate (Sigma-Aldrich, St Louis, MO) and counterstained with nuclear Fast Red stain (Vector Laboratories, Burlingame, CA) as described.34
Flow cytometry. Glioma cells cocultured with control or MSCs-TRAIL for 8 hours were stained for cleaved caspase-3 as previously described.44 Samples were analyzed using BD FACSCanto-A flow cytometer (BD Biosciences, San Jose, CA) and FACSDiVa software version 6.3 (BD Biosciences, San Jose, CA).
PCR-based array. Glioma xenograft tissue was processed for total RNA purification using the RNeasy Plus kit (Qiagen, Valencia, CA). For comparison of control and irradiated samples, 2 µg of total RNA was treated to remove genomic DNA and reverse transcribed using SABiosciences protocol developed for PCR-based arrays. Human Cytokines/Chemokines array was used in this study (SABiosciences, Valencia, CA). PCR array reaction was performed using Bio-Rad Opticon 2 system and data were analyzed using SABioscience software (SABiosciences). In order to verify upregulation of gene of interest, quantitative PCR was repeated using primers purchased from RealTimePrimers (Real Time Primers, LLC, Elkins Park, PA).
ELISA. CXCL12 expression in glioma xenograft tissue was determined using human CXCL12/SDF-1 α Quantikine ELISA Kit (R&D Systems, Minneapolis, MN). Shortly, glioma xenograft tissue was lysed in M-Per protein extraction reagent (Thermo Scientific, Hanover Park, IL) in the presence of halt protease inhibitors cocktail (Pierce, Rockford, IL). All samples were equilibrated to apply equal amount of protein to ELISA plates and bound CXCL12 was revealed per manufacturer's recommendation. The amount of CXCL12 in the samples was measured using a calibration curve and the concentration of CXCL12 per mg of protein was determined. TRAIL expression in MSCs was measured as previously described.44
Bioluminescent imaging of MSCs in vivo. In order to visualize MSCs-fluc in animals, mice were injected intra peritoneally (i.p.) with 200 µl (4.5 mg per animal) D-luciferin sodium salt (Gold Biotechnology, St Louis, MO). In addition, 3 µl of substrate was placed in each nostril to help with detection of MSCs in the animal's nasal cavity. Signal was recorded as photon counts using Xenogen IVIS 200 system. Localization of glioma cells expressing GFP was also detected using Xenogen IVIS 200 system.
Tracking MPIO-labeled stem cells in the brain of tumor-bearing mice using MRI. MPIO-labeled MSCs (1 × 106) were IN inoculated in control and tumor-bearing mice as described in the “animal studies” section. Two days later, MR images were acquired on a 33 cm horizontal bore Bruker 9.4 T small animal scanner with a Bruker console (Bruker-Biospin, Billerica, MA) equipped with a 12 cm shielded gradient set with a maximum strength of 600 mT/m available through the University of Chicago Core Facility. Mice were anesthetized with 2% isoflurane in oxygen and fixed in a prone position during scanning. In order to achieve sufficient resolution to visualize-labeled MSCs within the mouse brain, multi-slice high resolution T2 weighted, Rapid Acquisition with Relaxation Enhancement (RARE) spin echo images (TR/TEeffective = 4000/26.6 ms, field of view (FOV) = 2.56 cm, matrix size = 256 × 256, slice thickness = 0.5 mm, number of excitation (NEX) = 2, RARE factor = 4) were acquired in axial and coronal planes. Then multi-slice T1-weighted A Fast Low Angle Shot (FLASH) gradient echo sequence (TR/TE = 340/3.7 ms, flip angle = 20°, FOV = 2.56 cm, matrix size = 128 × 128, slice thickness = 0.5 mm, NEX = 4) were also acquired in axial and coronal planes.
Animal studies. All animals were maintained and cared for in accordance the NIH Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee protocol. The U87-EGFRvIII glioma xenografts are known to be more aggressive than parental U87MG glioma cells in an in vivo model46,47 and, therefore, were used as glioma model. Intracranial xenografts were established as previously described.44 One week later, anesthetized animals were placed in a supine position and nasal cavity of each animal was treated with total of 100 U of hyaluronidase12 as four repeated inoculations with 5-minute intervals (3 µl in each nostril) after which either sterile PBS, or 5 × 105 of MSCs (3 µl in each nostril with 5-minute intervals) were applied for four times. This treatment was repeated weekly for a total four times through the course of experiment. For irradiation, the entire body of animals excluding the head was covered with lead shields and exposed to 2 Gy of daily dose of radiation for 5 consecutive days. Twenty four hours later, animals were treated either with sterile PBS, or 5 × 105 of MSCs as described above. All mice were followed to assess their survival. In order to understand distribution of MSCs in control and irradiated animals, animals were killed 24 hours later after IN application 111In-oxine–labeled MSCs (106 per animal); brain and other organs were extracted and radioactivity was measured using a gamma counter.
Statistical analysis. Differences between groups were evaluated using parametric or nonparametric Student's t-test or two-way analysis of variance followed by Bonferroni's post hoc correction as appropriate. For the in vivo data, survival curves were generated by the Kaplan–Meier method and the log-rank test was applied to compare the distributions of survival times. All reported P values were two-sided and were considered to be statistically significant at <0.05.
SUPPLEMENTARY MATERIAL Figure S1. Labeling of MSCs with MPIOs. Figure S2. Effect of MSCs-TRAIL on survival of mice bearing intracranial U87 glioma xenografts. Table S1. Changes in mRNA expression for selected cytokines and chemokines in U87 glioma xenograft tissue of nude mice after irradiation. Video S1. Migration of MPIO-labeled MSCs towards the tumor through brain parenchyma.
Acknowledgments
This work was supported by the Elsa U. Pardee Foundation (I.V.B.), ACS IL. Div. (I.V.B.) and in part by the NIH grants R01 CA138587 (M.S.L.), NIH R01 CA122930 (M.S.L.), NIH R01 NS077388 (M.S.L.), NIH U01 NS069997 (M.S.L.), by pilot research funding provided by the Virginia and D. K. Ludwig Fund for Cancer Research via the Imaging Research Institute in the Biological Sciences Division of the University of Chicago. K.S.A. is chief scientific officer at TheraBiologics. The other authors declare no conflict of interest.
Supplementary Material
Labeling of MSCs with MPIOs.
Effect of MSCs-TRAIL on survival of mice bearing intracranial U87 glioma xenografts.
Changes in mRNA expression for selected cytokines and chemokines in U87 glioma xenograft tissue of nude mice after irradiation.
Migration of MPIO-labeled MSCs towards the tumor through brain parenchyma.
References
- Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, et al. Cancer surveillance series [corrected]: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91:1382–1390. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Serwer LP, James CD. Challenges in drug delivery to tumors of the central nervous system: an overview of pharmacological and surgical considerations. Adv Drug Deliv Rev. 2012;64:590–597. doi: 10.1016/j.addr.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Lee DH, Ahn Y, Kim SU, Wang KC, Cho BK, Phi JH, et al. Targeting rat brainstem glioma using human neural stem cells and human mesenchymal stem cells. Clin Cancer Res. 2009;15:4925–4934. doi: 10.1158/1078-0432.CCR-08-3076. [DOI] [PubMed] [Google Scholar]
- Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen RS, Narkilahti S, Huhtala T, Liimatainen T, Suuronen T, Närvänen A, et al. The SPECT imaging shows the accumulation of neural progenitor cells into internal organs after systemic administration in middle cerebral artery occlusion rats. Neurosci Lett. 2008;440:246–250. doi: 10.1016/j.neulet.2008.05.090. [DOI] [PubMed] [Google Scholar]
- Li L, Jiang Q, Ding G, Zhang L, Zhang ZG, Li Q, et al. Effects of administration route on migration and distribution of neural progenitor cells transplanted into rats with focal cerebral ischemia, an MRI study. J Cereb Blood Flow Metab. 2010;30:653–662. doi: 10.1038/jcbfm.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendharkar AV, Chua JY, Andres RH, Wang N, Gaeta X, Wang H, et al. Biodistribution of neural stem cells after intravascular therapy for hypoxic-ischemia. Stroke. 2010;41:2064–2070. doi: 10.1161/STROKEAHA.109.575993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak P, Zhang J, Gilad AA, Kedziorek DA, Ruiz-Cabello J, Young RG, et al. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke. 2008;39:1569–1574. doi: 10.1161/STROKEAHA.107.502047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Ayala-Grosso CA, Ganguly M, Jordão JF, Aubert I, Hynynen K. Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PLoS ONE. 2011;6:e27877. doi: 10.1371/journal.pone.0027877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, Klopfer T, et al. Intranasal delivery of cells to the brain. Eur J Cell Biol. 2009;88:315–324. doi: 10.1016/j.ejcb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, Buadze M, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011;14:3–16. doi: 10.1089/rej.2010.1130. [DOI] [PubMed] [Google Scholar]
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Nasal administration of stem cells: a promising novel route to treat neonatal ischemic brain damage. Pediatr Res. 2010;68:419–422. doi: 10.1203/PDR.0b013e3181f1c289. [DOI] [PubMed] [Google Scholar]
- Reitz M, Demestre M, Sedlacik J, Meissner H, Fiehler J, Kim SU, et al. Intranasal delivery of neural stem/progenitor cells: a noninvasive passage to target intracerebral glioma. Stem Cells Transl Med. 2012;1:866–873. doi: 10.5966/sctm.2012-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, Yong VW, et al. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–1170. [PubMed] [Google Scholar]
- Kuijlen JM, Bremer E, Mooij JJ, den Dunnen WF, Helfrich W. Review: on TRAIL for malignant glioma therapy. Neuropathol Appl Neurobiol. 2010;36:168–182. doi: 10.1111/j.1365-2990.2010.01069.x. [DOI] [PubMed] [Google Scholar]
- Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL, et al. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2002;62:7170–7174. [PubMed] [Google Scholar]
- Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–9623. doi: 10.1158/0008-5472.CAN-08-0451. [DOI] [PubMed] [Google Scholar]
- Klopp AH, Spaeth EL, Dembinski JL, Woodward WA, Munshi A, Meyn RE, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67:11687–11695. doi: 10.1158/0008-5472.CAN-07-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Oh JH, Park SA, Ryu CH, Lim JY, Kim DS, et al. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells. 2010;28:2217–2228. doi: 10.1002/stem.543. [DOI] [PubMed] [Google Scholar]
- Heyn C, Ronald JA, Mackenzie LT, MacDonald IC, Chambers AF, Rutt BK, et al. In vivo magnetic resonance imaging of single cells in mouse brain with optical validation. Magn Reson Med. 2006;55:23–29. doi: 10.1002/mrm.20747. [DOI] [PubMed] [Google Scholar]
- Adamina M, Rosenthal R, Weber WP, Frey DM, Viehl CT, Bolli M, et al. Intranodal immunization with a vaccinia virus encoding multiple antigenic epitopes and costimulatory molecules in metastatic melanoma. Mol Ther. 2010;18:651–659. doi: 10.1038/mt.2009.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro EM, Gonzalez-Perez O, Manuel García-Verdugo J, Alvarez-Buylla A, Koretsky AP. Magnetic resonance imaging of the migration of neuronal precursors generated in the adult rodent brain. Neuroimage. 2006;32:1150–1157. doi: 10.1016/j.neuroimage.2006.04.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valable S, Barbier EL, Bernaudin M, Roussel S, Segebarth C, Petit E, et al. In vivo MRI tracking of exogenous monocytes/macrophages targeting brain tumors in a rat model of glioma. Neuroimage. 2007;37 suppl. 1:S47–S58. doi: 10.1016/j.neuroimage.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Wu YL, Ye Q, Foley LM, Hitchens TK, Sato K, Williams JB, et al. In situ labeling of immune cells with iron oxide particles: an approach to detect organ rejection by cellular MRI. Proc Natl Acad Sci USA. 2006;103:1852–1857. doi: 10.1073/pnas.0507198103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA, et al. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol Cancer Res. 2008;6:1819–1829. doi: 10.1158/1541-7786.MCR-08-0146. [DOI] [PubMed] [Google Scholar]
- Park SA, Ryu CH, Kim SM, Lim JY, Park SI, Jeong CH, et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int J Oncol. 2011;38:97–103. [PubMed] [Google Scholar]
- Xu F, Shi J, Yu B, Ni W, Wu X, Gu Z. Chemokines mediate mesenchymal stem cell migration toward gliomas in vitro. Oncol Rep. 2010;23:1561–1567. doi: 10.3892/or_00000796. [DOI] [PubMed] [Google Scholar]
- Bexell D, Gunnarsson S, Tormin A, Darabi A, Gisselsson D, Roybon L, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. 2009;17:183–190. doi: 10.1038/mt.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Hu J, Zhou L, Mao Y, Yang B, Gao L, et al. In vivo tracking of superparamagnetic iron oxide nanoparticle-labeled mesenchymal stem cell tropism to malignant gliomas using magnetic resonance imaging. Laboratory investigation. J Neurosurg. 2008;108:320–329. doi: 10.3171/JNS/2008/108/2/0320. [DOI] [PubMed] [Google Scholar]
- Bossolasco P, Cova L, Levandis G, Diana V, Cerri S, Lambertenghi Deliliers G, et al. Noninvasive near-infrared live imaging of human adult mesenchymal stem cells transplanted in a rodent model of Parkinson's disease. Int J Nanomedicine. 2012;7:435–447. doi: 10.2147/IJN.S27537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. Childhood brain tumors. Pediatr Rev. 2013;34:63–78. doi: 10.1542/pir.34-2-63. [DOI] [PubMed] [Google Scholar]
- Thu MS, Najbauer J, Kendall SE, Harutyunyan I, Sangalang N, Gutova M, et al. Iron labeling and pre-clinical MRI visualization of therapeutic human neural stem cells in a murine glioma model. PLoS ONE. 2009;4:e7218. doi: 10.1371/journal.pone.0007218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini P, Schmid A, Jendrossek V, Faltin H, Daniel PT, Budach W, et al. Irradiation specifically sensitises solid tumour cell lines to TRAIL mediated apoptosis. BMC Cancer. 2005;5:5. doi: 10.1186/1471-2407-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S, Singh TR, Srivastava RK. Ionizing radiation enhances the therapeutic potential of TRAIL in prostate cancer in vitro and in vivo: Intracellular mechanisms. Prostate. 2004;61:35–49. doi: 10.1002/pros.20069. [DOI] [PubMed] [Google Scholar]
- Zielske SP, Livant DL, Lawrence TS. Radiation increases invasion of gene-modified mesenchymal stem cells into tumors. Int J Radiat Oncol Biol Phys. 2009;75:843–853. doi: 10.1016/j.ijrobp.2008.06.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Fan GC, Zhou X, Zhao T, Pasha Z, Xu M, et al. Over-expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2008;44:281–292. doi: 10.1016/j.yjmcc.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider HKh, Jiang S, Idris NM, Ashraf M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ Res. 2008;103:1300–1308. doi: 10.1161/CIRCRESAHA.108.186742. [DOI] [PubMed] [Google Scholar]
- Wei N, Yu SP, Gu X, Taylor TM, Song D, Liu XF, et al. Delayed intranasal delivery of hypoxic-preconditioned bone marrow mesenchymal stem cells enhanced cell homing and therapeutic benefits after ischemic stroke in mice. Cell Transplant. 2013;22:977–991. doi: 10.3727/096368912X657251. [DOI] [PubMed] [Google Scholar]
- Li Y, Yu X, Lin S, Li X, Zhang S, Song YH. Insulin-like growth factor 1 enhances the migratory capacity of mesenchymal stem cells. Biochem Biophys Res Commun. 2007;356:780–784. doi: 10.1016/j.bbrc.2007.03.049. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Gheusi G, Vincent JD. Information processing in the mammalian olfactory system. Physiol Rev. 2005;85:281–317. doi: 10.1152/physrev.00008.2004. [DOI] [PubMed] [Google Scholar]
- Balyasnikova IV, Franco-Gou R, Mathis JM, Lesniak MS. Genetic modification of mesenchymal stem cells to express a single-chain antibody against EGFRvIII on the cell surface. J Tissue Eng Regen Med. 2010;4:247–258. doi: 10.1002/term.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balyasnikova IV, Ferguson SD, Han Y, Liu F, Lesniak MS. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011;310:148–159. doi: 10.1016/j.canlet.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildehaus FJ, Haasters F, Drosse I, Wagner E, Zach C, Mutschler W, et al. Impact of indium-111 oxine labelling on viability of human mesenchymal stem cells in vitro, and 3D cell-tracking using SPECT/CT in vivo. Mol Imaging Biol. 2011;13:1204–1214. doi: 10.1007/s11307-010-0439-1. [DOI] [PubMed] [Google Scholar]
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Labeling of MSCs with MPIOs.
Effect of MSCs-TRAIL on survival of mice bearing intracranial U87 glioma xenografts.
Changes in mRNA expression for selected cytokines and chemokines in U87 glioma xenograft tissue of nude mice after irradiation.
Migration of MPIO-labeled MSCs towards the tumor through brain parenchyma.