

Abstract
Here, we developed a clinically translatable platelet gene therapy approach for hemophilia B. Platelet-targeted FIX (2bF9) expression was introduced by transplantation of hematopoietic stem cells (HSCs) transduced with 2bF9 lentivirus (LV). Sustained therapeutic levels of platelet-FIX expression were obtained in FIXnull mice that received 2bF9 LV-transduced HSCs. Approximately 6–39% of the platelets expressed FIX in the transduced recipients, which was sufficient to rescue the bleeding diathesis in FIXnull mice in tail clipping models. Sequential bone marrow transplantation demonstrated that platelet-FIX expression in the secondary recipients was sustained, leading to phenotypic correction. Notably, none of the transduced recipients developed anti-FIX antibodies after platelet gene therapy. Only one of the nine recipients developed a low titer of inhibitory antibodies (1.6 BU/ml) after challenge with rhFIX. These data suggest that platelet gene therapy can not only restore hemostasis but also induce immune tolerance in hemophilia B mice, indicating that this approach may be a promising strategy for gene therapy of hemophilia B in humans.
Introduction
Hemophilia B is an X-linked, recessive bleeding disorder resulting from a factor IX (FIX) deficiency. The clinical severity of hemophilia B correlates with the circulating levels of FIX. Patients with severe hemophilia B have <1% of normal values of functional FIX and have frequent bleeding episodes. FIX protein replacement therapy is effective for the disease, but it is constrained by the short half life of the clotting factors, requiring frequently repeated infusions of relatively large doses. Furthermore, up to 5% of the patients remain at risk for developing neutralizing antibodies against FIX, rendering FIX protein replacement therapy useless. Currently, there is no effective approach available to induce immune tolerance in hemophilia B patients.1 Anaphylactic reaction to the infused FIX protein in hemophilia B inhibitor patients is a problem which limits the use of protein replacement therapy and increases the risk of morbidity and mortality.2,3,4,5,6,7
Gene therapy is an attractive alternative for hemophilia B treatment because it may provide a cure, if successful. Substantial progress has been achieved in the last two decades. Preclinical trials in animal models have shown promising benefits from FIX gene therapy.8,9,10,11 Studies on liver-directed adeno-associated virus (AAV)2-mediated FIX gene therapy demonstrate the long-term correction of the hemophilia B phenotype in the dog model.8 Phase 1 and Phase 2 clinical trials in severe hemophilia B patients show that a single infusion of AAV8 vector encoding the codon-optimized FIX under control of a liver-specific promoter leads to long-term expression of therapeutic levels of FIX.12,13,14 Current clinical trials using AAV8-mediated liver-restricted FIX expression are very encouraging. However, for individuals with severe liver disease or neutralizing antibodies to AAV that are present in 30–50% of the population,15,16 an alternative gene therapy approach might be desired.
We have previously demonstrated that lentivirus (LV)-mediated platelet-specific gene therapy can correct the murine hemophilia A phenotype with neither inhibitory nor noninhibitory antibody development.17,18 This approach has not been explored in the hemophilia B model. The current work investigates whether this strategy could potentially be adapted to FIX gene therapy of autologous patient hematopoietic stem cells (HSCs) that could then be transplanted back into the patient for a potentially life-long cure of the disease. Our results demonstrate that platelet-specific expression of FIX induced by lentiviral gene delivery to HSCs restores hemostasis and induces immune tolerance in hemophilia B (FIXnull) mice.
Results
PCR and quantitative real-time PCR (qPCR) analysis of 2bF9 proviral DNA in 2bF9 LV-transduced recipients
We constructed a 2bF9 LV vector that expresses human FIX (hFIX) under control of the platelet-specific integrin αIIb promoter. Platelet-FIX expression was introduced by 2bF9 LV transduction of HSCs followed by syngeneic transplantation. 2bF9 proviral DNA was detected in all recipients, including all primary (1°) and all secondary (2°) recipients, that received 2bF9 LV-transduced HSCs (Figure 1a). The average copy number of 2bF9 expression cassette per cell in recipients was quantitated by qPCR using DNA purified from white blood cells. The average copy number of 2bF9 cassette per cell in the group of recipients conditioned with 1,100 cGy total body irradiation (TBI) was 0.36 ± 0.16 (n = 9), which was not significantly different from the 660 cGy group (0.38 ± 0.12; n = 7; P = 0.78; Figure 1b). These results demonstrate that engraftment of 2bF9 LV genetically modified HSCs are viable in FIXnull mice preconditioned with the myeloablative regimen 1,100 cGy TBI or, more importantly, a clinically relevant nonmyeloablative regimen of 660 cGy TBI.
Figure 1.

Genetic analysis of 2bF9 lentivirus (LV)-tranduced BMT recipients. (a) PCR analysis of the 2bF9 transgene. Genomic DNA was purified from peripheral white blood cells from primary (1°) and secondary (2°) 2bF9 LV-transduced hematopoietic stem cells recipients. FIXnull and C57BL/6 wild-type (WT) mice were used as controls. 2bF9 transgenic mouse DNA was used as a positive control for the 2bF9 transgene. Absence of the WT mFIX and presence of the disrupted mFIX gene confirmed that these mice were in the FIXnull background. Shown is one representative experiment that was performed three times. (b) Real-time quantitative PCR determined the average copy number of the 2bF9 transgene per cell in BMT recipients. Genomic DNA was purified from peripheral white blood cells, and 150 ng of DNA was analyzed for the 2bF9 transgene with normalization to ApoB. Bars represent mean ± SD. There were two groups of 2bF9 LV-transduced recipients. One was preconditioned with lethal total body irradiation, 1,100 cGy, the other with sublethal 660 cGy. For individual mice analyzed more than once over the course of study, the average copy number was calculated. Genomic DNA from FIXnull and 2bF9 homozygous transgenic mice was used as controls.
hFIX expression in 2bF9 LV-transduced recipients
To measure FIX expression, we used an enzyme-linked immunosorbent assay (ELISA) assay to quantitate hFIX antigen (hFIX:Ag) and a chromogenic assay to determine the functional FIX activity (FIX:C). Both FIX:Ag and FIX:C were detected in platelet lysates of FIXnull mice that received 2bF9 LV-transduced HSCs. The expression levels of both FIX:Ag and FIX:C decreased at 8 weeks after transplantation but remained steady under either lethal irradiation of 1,100 cGy or sublethal 660 cGy irradiation for the rest of the time points during the study course (Figure 2a,c). The average antigen levels of FIX were 2.89 ± 1.75 mU/108 platelets (ranging from 1.10 to 7.02 mU/108 platelets; n = 9) in the recipients preconditioned with 1,100 cGy and 1.87 ± 1.30 mU/108 platelets (ranging from 0.69 to 4.43 mU/108 platelets; n = 7) in the 660 cGy group (Figure 2b), whereas the average FIX: C levels were 1.67 ± 1.15 (ranging from 0.39 to 4.41 mU/108 platelets) and 1.13 ± 0.85 mU/108 platelets (ranging from 0.39 to 2.92 mU/108 platelets), respectively (Figure 2d). The average levels of the platelet-FIX expression including both FIX:Ag and FIX:C in the 1,100 cGy group appeared to be higher than those obtained from the 660 cGy group, but there were no significant differences between the two groups (Figures 2b,d). There were no significant differences in the ratio of platelet FIX:C to FIX:Ag between the 1,100 cGy and the 660 cGy groups at any of the time points. At 3 weeks after transplantation, the ratio of platelet FIX: C to FIX: Ag was at the lowest level of about 0.4 and increased over time with a peak of around 1 at 17–21 weeks. Although the ratio in the 1,100 cGy group appears slightly lower than those obtained from the 660 cGy group for most of the time points, there is no significant difference between the two groups (Figures 2e,f).
Figure 2.
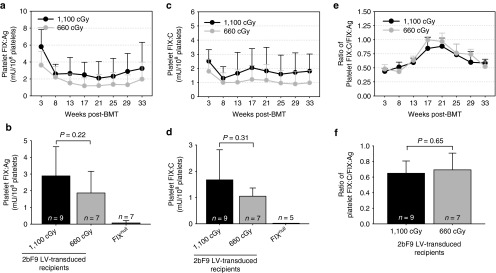
Analysis of platelet FIX expression in 2bF9 lentivirus (LV)-transduced recipients. Platelet-FIX expression in 2bF9 LV-transduced primary recipients was monitored for 33 weeks. Isolated platelets were lysed in 0.5% CHAPS. The levels of FIX:Ag in platelet lysates were determined by ELISA assay and FIX:C were determined by chromogenic assay. Platelets from FIXnull mice were used as a negative control. Data shown were summarized from 9 and 7 recipients in the 1100 cGy and the 660 cGy group respectively and expressed as the mean ± SD. (a) Average expression levels of platelet-FIX:Ag at each time point. (b) Average expression levels of platelet-FIX:Ag over the study period. (c) Average expression levels of platelet-FIX:C at each time point. (d) Average expression levels of platelet-FIX:C over the study period. (e) Ratio of platelet-FIX: C to FIX: Ag in 2bF9 LV-transduced mice at each time point. (f) Average ratio of platelet-FIX:C to FIX:Ag over the study period.
A small amount of FIX was detected in the 2bF9 LV-transduced recipient plasma with the average levels of 1.83 ± 1.67 mU/ml (n = 9) in the 1,100 cGy group and 1.09 ± 1.06 mU/ml (n = 7) in the 660 cGy group. There was no significant difference between the two groups (P = 0.74; Figure 3a). To analyze the distribution of the FIX between platelets and plasma, we normalized FIX levels to total whole-blood FIX content in each group. The results demonstrated that 94.59 ± 2.13% (n = 9) of whole-blood FIX was stored in platelets in the group of recipients preconditioned with 1,100 cGy TBI, which was not significantly different from the 660 cGy group (95.11 ± 2.68%, n = 7; P = 0.65; Figure 3b). The distribution of FIX in platelets and plasma is consistent with our previous findings in the animals that received bone marrow cells from 2bF9 transgenic mice.19
Figure 3.

Analysis of FIX expression in the plasma of 2bF9 lentivirus (LV)-transduced recipients. (a) Quantification of the levels of FIX expression in mouse plasma. Plasmas were collected from 2bF9 LV-transduced recipients and hFIX:Ag were quantified by the human FIX-specific ELISA assay. Normal pool of human plasma was used as the standards. Plasmas from FIXnull and wild-type C57BL6 (WT) mice were used as controls. (b) The distribution of the FIX transgene product in mouse whole blood. To analyze the distribution of the FIX between platelets and plasma, we normalized FIX levels to total whole-blood hFIX content in individual mice. 1° BMT*: data presented in this group were summarized from the primary recipients from which bone marrow mononuclear cells were collected and transplanted into the secondary recipients.
The cellular location of 2bF9 transgene protein was determined by immunofluorescent confocal microscopy, demonstrating that the 2bF9 transgene protein was detected in 2bF9 LV-transduced platelets as shown in green in Figure 4d,g and colocalized with endogenous mouse von Willebrand factor as shown in yellow in merged images (Figure 4f,i). As expected, no FIX was detected in FIXnull controls (Figure 4a). The percentage of platelets that expressed hFIX protein in 2bF9 LV-transduced recipients was analyzed by flow cytometry (Figure 4j). Results showed 20.8 ± 12.1% (ranging from 5.9 to 38.5%) and 14.8 ± 10.7% (ranging from 7.5 to 35.8%) of platelets expressing hFIX respectively in the recipients preconditioned with 1,100 cGy and 660 cGy at 32 weeks after transplantation, with no significant difference between the two groups (Figure 4k).
Figure 4.

Immunofluorescent staining of hFIX transgene protein in platelets of 2bF9 lentivirus (LV)-transduced recipients. (a–i) Confocal microscopy determined the localization of transgene protein expression. Isolated platelets from untransduced FIXnull (a–c) and 2bF9 LV-transduced mice were immunostained for both human FIX (hFIX; a, d, and g) and mouse VWF (b, e, and h). The two images were merged in c, f, and i. hFIX is shown in green, VWF in red, and colocalization in yellow. Shown is one representative experiment that was performed three times. (j–k) Flow cytometry analysis of hFIX expression. Isolated platelets were stained with anti-mouse CD41/integin αIIb mAb and anti-hFIX PoAb. Platelets from FIXnull and 2bF9 transgenic (2bF9tg+) mice were used as controls. Representative flow cytometry plots (j) and graph (k) show the percentage of hFIX transgene expression in 2bF9 LV-transduced recipients.
Collectively, these data demonstrated that sustained platelet-FIX expression was attained in 2bF9 LV-transduced HSC recipients under either myeloablative 1,100 cGy or nonmyeloablative 660 cGy TBI.
Phenotypic correction assessment
To assess whether 2bF9 LV-mediated HSC transduction and syngeneic transplantation can restore hemostasis in hemophilia B mice, tail-clipping tests and rotational thromboelastometry analysis were used in this study. As shown in Figure 5a, eight of nine 2bF9 LV-transduced recipients survived tail clipping in the 1,100 cGy group and all seven recipients in 660 cGy group survived tail clipping. The one that did not survive in clipping had the lowest FIX expression (0.39 mU/108 platelets) in that group. By contrast, only 2 out of 10 FIXnull control mice survived tail clipping.
Figure 5.
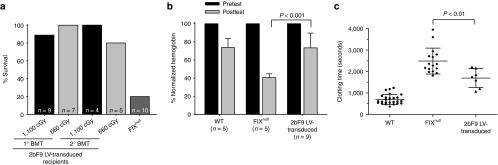
Phenotypic correction assessment. (a) Tail-clipping test assessing phenotypic correction of FIXnull mice. Tail clipping tests were performed on the recipients at least 12 weeks after transplantation. Mice surviving beyond 24 hours were considered to have achieved phenotypic correction. (b) Tail-bleeding test grading phenotypic correction of FIXnull mice. Tail tips were clipped using a 1.6-mm diameter template. Blood samples were collected and hemoglobin levels were measured before and 6 hours after tail transection. Hemoglobin was normalized by defining the pretest levels of hemoglobin as 100%. (c) Rotational thromboelastometry analysis of whole-blood clotting time. Blood samples were collected from vena cava. Wild-type and FIXnull mice were used as controls.
To grade phenotypic correction in the transduced recipients, the tail-bleeding test was used. Hemoglobin (Hb) levels were measured before and 6 hours after tail transection. As shown in Figure 5b, after tail clipping, the Hb levels in 2bF9 LV-transduced recipients remained 73.41 ± 16.31% (n = 9), which were significantly higher than in FIXnull controls (40.52 ± 4.28%; n = 5; P < 0.001). There was no difference between 2bF9 LV-transduced recipients and wild-type mice (73.70 ± 10.18%; n = 5; P = 0.97).
To further assess the clinical efficacy of 2bF9 gene therapy, the whole-blood clotting time in 2bF9 LV-transduced recipients was determined by rotational thromboelastometry analysis. The whole-blood clotting time was 1697 ± 437 seconds (n = 7) in 2bF9 LV-transduced recipients, which was significantly shorter than FIXnull control (2490 ± 603 seconds; n = 16; P < 0.01; Figure 5c). Collectively, these results confirm that hemostasis is improved in hemophilia B mice after platelet gene therapy.
Sustained expression of FIX in sequential transplantation recipients
To confirm that long-term engrafting HSCs were successfully genetically modified by 2bF9 LV, some of the primary (1°) recipients were killed at 9 months after primary transplantation. BM mononuclear cells were collected and sequential transplantation was performed. As shown in Figure 1a,2b, F9 proviral DNA was detected in all secondary (2°) recipients. The average copy numbers of 2bF9 proviral DNA per cell were 0.55 ± 0.42 (n = 4) and 0.15 ± 0.12 (n = 5) in the recipients preconditioned with 1,100 cGy and 660 cGy, respectively, which were not significantly different from those obtained from their donors (0.55 ± 0.28; n = 3, in the 1,100 cGy group and 0.30 ± 0.06; n = 4, in the 660 cGy group; Figure 6a).
Figure 6.
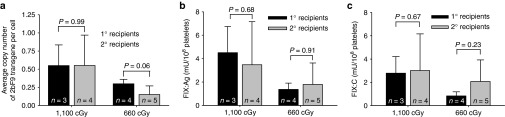
Sustained platelet-FIX expression in sequential transplantation recipients. To ascertain that long-term repopulating hematopoietic stem cells were transduced by 2bF9 LV, BM mononuclear cells isolated from some primary (1°) recipients (n = 3 in the 1,100 cGy group and n = 4 in the 660 cGy group) were transplanted into secondary (2°) recipients preconditioned with 1,100 cGy (n = 4) or 660 cGy (n = 5). Animals were analyzed for 2bF9 transgene expression 4 weeks after transplantation. Bars represent mean ± SD. (a) Average copy numbers of 2bF9 proviral DNA per cell determined by qPCR. For individual mice analyzed more than once over the study, the average copy number was calculated. (b) The expression levels of platelet-FIX:Ag in 2bF9 LV-transduced recipients determined by ELISA assay. For individual mice analyzed more than once over the study, the average platelet-FIX:Ag was calculated. (c) The expression levels of platelet-FIX:C in 2bF9 LV-transduced recipients determined by chromogenic assay. For individual mice analyzed more than once over the study, the average platelet-FIX:C was calculated.
The expression of platelet FIX was sustained in the 2° recipients. The average levels of platelet FIX:Ag were 4.42 ± 3.71 mU/108 platelets in the 1,100 cGy group and 1.40 ± 0.62 mU/108 platelets in the 660 cGy group, whereas the platelet FIX:C levels were 3.01 ± 3.15 and 2.07 ± 1.85 mU/108 platelets, respectively. The levels of both FIX:Ag and FIX:C in the 2° recipients were not significantly different from those obtained from their donors (Figure 6b,c). A small amount of FIX:Ag was also detected in the plasma of the 2° recipients with an average level of 1.22 ± 1.32 mU/ml (n = 9), which was not significantly different from the level obtained from their donors (0.86 ± 0.83, n = 7; P = 0.57; Figure 3a). The distribution of FIX in platelets and plasma in the 2° recipients was similar to those in the 1° recipients with 93.61 ± 3.63% of whole-blood FIX stored in platelets (Figure 3b). The tail clip survival test demonstrated that all four recipients in the 1100 cGy group and four of the five animals in the 660 cGy group survived tail clipping (Figure 5a). Collectively, these data demonstrate that platelet-FIX expression in the secondary recipients was sustained, leading to phenotypic correction.
Immune tolerance developed in 2bF9 LV-transduced recipients
To explore the immune response after 2bF9 gene therapy in the hemophilia B mouse model, a modified chromogenic-based Bethesda assay was used to determine the levels of anti-FIX inhibitory antibodies and the ELISA assay was used to quantitate the levels of total anti-FIX antibodies. Similar to the naive control FIXnull mice (preimmunization), neither inhibitory nor noninhibitory antibodies were detected in the recipients that received 2bF9 LV-transduced HSCs (Figure 7a,b). To investigate whether immune tolerance was induced in 2bF9 LV-transduced recipients, we challenged the recipients with rhFIX in the presence of adjuvant. After two immunizations, only one of nine 2bF9 LV-transduced recipients developed a low titer of anti-FIX inhibitors (1.6 BU/ml) as determined by the Bethesda assay. By contrast, all of the FIXnull mice produced inhibitory antibodies under the same immunization, ranging from 17 to 43 BU/ml (n = 8; Figure 7a). Similar results were found when using the ELISA assay for quantification of total anti-FIX antibodies. The amounts of total anti-FIX antibodies in 2bF9 LV-transduced recipients were barely detectable in four of the nine recipients (0.36 ± 0.43 µg/ml, ranging from 0.19 to 1.07 µg/ml). By contrast, all of the FIXnull mice produced high levels of anti-FIX antibodies with an average of 332.85 ± 212.89 µg/ml (ranging from 90.85 to 690.73 µg/ml) after rhFIX immunization (Figure 7b). These results demonstrate that immune tolerance was induced in the 2bF9 LV-transduced recipients.
Figure 7.
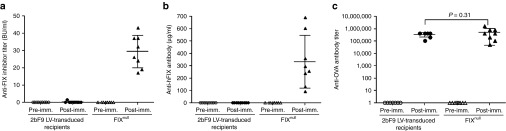
The development of immune tolerance in 2bF9 lentivirus (LV)-transduced recipients. (a) Quantification of inhibitory antibodies in mouse plasma by Bethesda assay. Plasmas were collected from 2bF9 LV-transduced recipients for Bethesda assay to determine the titers of anti-FIX inhibitory antibodies. None of the recipients developed anti-FIX inhibitory antibodies after gene therapy. To explore whether immune tolerance was induced in 2bF9 LV-transduced recipients, animals were challenged with recombinant human FIX (rhFIX) in the presence of adjuvant and plasmas were collected for Bethesda assay. FIXnull mice were immunized with the same protocol as a control. (b) Quantification of total anti-FIX antibodies in mouse plasma by ELISA assay. (c) Quantification of anti-ovalbumin (OVA) antibody titers in mouse serum by ELISA assay. To ensure that the immune systems in 2bF9 LV-transduced recipients were not defective and that the immune tolerance is FIX-specific, animals were immunized with OVA absorbed on Alum by i.p. injection and serums were collected for ELISA assay to determine the titers of anti-OVA antibodies. FIXnull mice were immunized with the same protocol as a control.
To investigate whether the immune tolerance induction in the 2bF9 LV-transduced recipients is FIX antigen specific, we immunized recipients with a nonspecific antigen, ovalbumin (OVA), which is a well-characterized immunogen commonly used in immunology studies. As expected, after the FIX-unrelated antigen OVA immunization, high titers of anti-OVA IgG was found in both the 2bF9 LV-transduced recipients and FIXnull control mice with no statistically significance between the two groups (P = 0.31; Figure 7c). Taken together, these data strongly suggest that 2bF9 lentiviral gene delivery to HSCs following HSC transduction and syngeneic transplantation induces FIX-specific immune tolerance in hemophilia B mice.
Discussion
Although data from clinical trials using AAV-mediated liver-restricted gene therapy of hemophilia B in humans are very encouraging, for individuals with severe liver disease or neutralizing antibodies to AAV, an alternative gene therapy approach might be desired.
Our previous studies using a transgenic mouse model have demonstrated that FIX can be ectopically expressed and stored in platelet α-granules and that platelet-derived FIX can rescue the bleeding diathesis in hemophilia B mice.19 This provides proof-of-principal that platelet-targeted FIX expression could be successful for gene therapy of hemophilia B. To apply this to a clinically translatable gene therapy, we use LV-mediated 2bF9 gene delivery to HSCs to introduce FIX expression in platelets. Our data from the current study have demonstrated that the platelet-specific expression of FIX induced by LV-mediated HSC transduction and syngeneic transplantation improves bleeding diathesis and induces immune tolerance in hemophilia B mice.
Sustained therapeutic levels of platelet-FIX can be achieved in hemophilia B mice following ex vivo HSC transduction and syngeneic transplantation. The transgene expression was sustained not only in the primary recipients but also in the sequential BMT recipients, demonstrating that long-term engrafting HSCs were successfully genetically modified by 2bF9 LV. Since 95% of FIX in the whole blood in 2bF9 LV-transduced recipients was stored in platelets, the questions that need to be addressed are what fraction of platelets would need to be transduced and what the optimal level of FIX expression per platelet is needed in order to restore hemostasis to FIXnull mice. Our studies do not address the second question, but our results demonstrated as little as 6–10% of platelets needed to be transduced was sufficient to rescue the bleeding diathesis in hemophilia B mice in our tail clip survival test model.
Since FIX is a vitamin K-dependent protein, it is important to assure that the newly synthesized FIX protein precursor is functionally carboxylated in the transduced megakaryocytes/platelets, becoming completely functional FIX protein in the platelets after 2bF9 gene therapy. The ratio of functional platelet-FIX:C to FIX:Ag reflects the degree to which FIX is completely carboxylated. We found that ~44 and 48% platelet-FIX was functionally carboxylated in the groups of animals preconditioned with the lethal 1,100 cGy TBI and the sublethal 660 cGy TBI, respectively, at 3 weeks after HSC transplantation. The ratio of platelet-FIX:C to FIX:Ag increased with time after transplantation, reaching a peak of almost 100% carboxylated platelet-FIX in the 660 cGy group at week 17, but gradually declined after and then to ~52% at week 33. A similar trend was found in the group preconditioned with the 1,100 cGy TBI. The lower ratio in the early phase after transplantation could be due to the deficiency of vitamin K. It has been reported20,21,22 that vitamin K deficiency is one of the complications of the intensive preconditioning in patients undergoing BM transplantation, which may be prevented by weekly intravenous administration of vitamin K.
Although we still do not yet know why the percentage of γ-carboxylated platelet-FIX in 2bF9 LV-transduced recipients declined in later time points, it might be due to an age-related effect. It has been reported that some vitamin K-dependent proteins, e.g., osteocalcin and matrix Gla protein, are less carboxylated in the elderly, which is interpreted as age-dependent impairment of γ-carboxylation.23,24,25,26,27 One strategy to increase the functional platelet-FIX activity is to incorporate the gain-of-function mutation FIX Padua (R338L) that has been shown to exhibit normal antigen levels, but up to ninefold higher specific activity.28 Recent studies have demonstrated that utilizing the codon-optimized FIX transgene significantly increases FIX expression in both preclinical and clinical hepatocyte-targeted gene therapy trials.12,29 Cantore and coworkers have reported that the combined effect of codon optimization and hyperfunctional FIX Padua targeted to hepatocytes resulted in a 15-fold increase in the efficacy of FIX gene therapy in hemophilia B mice.30 Whether using the codon-optimized FIX Padua will improve the efficacy of platelet-targeted FIX gene therapy is the subject of further investigation by our group.
One challenge in hemophilia B gene therapy is the development of immune responses to the components of the gene transfer vector and/or the transgene product, which is one of the main causes of failure of gene therapy. Several factors, including the type of gene delivery vector, the route of administration of vector, the targeted cell type, and the transgene expression cassette, may impact the immune response in gene therapy of hemophilia B.31,32,33,34,35 In AAV-mediated gene therapy, humoral and cellular immunity to AAV is an obstacle which has resulted in a substantial fraction of patients being excluded from ongoing clinical trials because the AAV vectors used in gene therapy trials are derived from a wild-type parvovirus, which naturally infects humans.14 Lentivirus-mediated in vitro transduction followed by transplantation of the transduced HSCs may circumvent the immune response to the viral vectors used for gene transfer because in vivo systemic delivery of vector is avoided.
The transgene product may be seen as a neoantigen by the host immune system, which may elicit a humoral immune response, resulting in anti-FIX neutralizing antibody development and/or a cellular immune response that will cause immune-mediated destruction of transduced cells, ultimately resulting in the loss of transduced cells and transgene expression. The immune response against the transgene FIX protein may be avoided using tissue-specific promoters. Indeed, no anti-FIX inhibitory antibodies developed in gene therapy of hemophilia B when FIX expression was targeted to hepatocytes or erythrocytes using tissue-specific promoters.31,36 In our current study, a platelet-specific αIIb promoter was used to direct FIX expression to platelets, resulting in the storage of FIX in platelet α-granules. Notably, we found that none of the transduced animals developed inhibitory or noninhibitory antibodies during the entire study course. Although the distributions of FIX are very different between platelet- versus hepatocyte/or erythrocyte-restricted FIX expression, no anti-FIX antibodies developed in any of the three models. It has been shown that immune responses to transgene products are primed by professional antigen-presenting cells (APC) that are transduced and express antigen.37,38,39 Using tissue-specific promoters to direct transgene expression may avoid transgene expression in APC and thereby abrogate undesired immune responses against transgene product.
The ideal gene therapy protocol should not only introduce sustained therapeutic levels of FIX protein but also induce immune tolerance to the transgene protein. This would be particularly beneficial because protein infusion could then be administered in some special situations (e.g., surgery in which a greater level of FIX may be required) with minimized risk of inhibitor development. In the current study, we found that LV-mediated platelet-targeted gene therapy can induce FIX-specific immune tolerance, which was defined as the specific reduction or elimination of inhibitor responses,40 in FIXnull mice under either a lethal or sublethal TBI-conditioning regimen. Whether 2bF9 lentiviral gene delivery to HSCs will induce immune tolerance in FIXnull mice with preexisting anti-FIX immunity will be explored by our group in the future.
In summary, our studies have demonstrated that 2bF9 LV-mediated HSC transduction followed by syngeneic hematopoietic stem cell transplantation can efficiently introduce sustained therapeutic levels of platelet-derived FIX in hemophilia B mice and that platelet-targeted FIX gene therapy can induce antigen-specific immune tolerance. The combination of HSCT and platelet-restricted gene therapy may be a promising approach for gene therapy to achieve phenotypic correction and immune tolerance induction in hemophilia patients. Further assessment of the efficacy and safety of such an approach will need to be pursued in a large animal model.
Materials and Methods
Mice. All the animals used in this study were in a C57BL/6 genetic background. FIX-deficient (FIXnull) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in our animal facility. Isoflurane or xylazine/ketamine was used for anesthesia. All mice were kept in pathogen-free microisolator cages at the animal facilities operated by the Medical College of Wisconsin. Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Vector construction, production, and titering. The 2bF9 lentiviral vector (pWPT-2bF9) harboring the human FIX expression cassette under the control of the platelet-specific glycoprotein IIb gene (αIIb) promoter was constructed as described in our previous report.19 Recombinant LV (2bF9 LV) production and titering were performed using the protocols as described in our previous report.17
Hematopoietic stem cells (HSCs) transduction and transplantation. Bone marrow (BM) cells from FIXnull mice were collected from femurs and tibia as previously described.17 Murine HSCs were isolated using the anti-Sca-1 MicroBead Kit (Miltenyl Biotec, Auburn, CA). The transduction of Sca-1+ cells with 2bF9 LV were performed in accordance with the procedures described in our previous reports.17,18 Briefly, the Sca-1+ cells were cultured in completed X-VIVO 10 media (Lonza Walkersville, Walkersville, MD) containing a cytokine cocktail for 48 hours at 37 °C in 5% CO2. The cells were transduced on RetroNectin-coated plates with 2bF9 LV at a multiplicity of infection of ~10. Six- to eight-week-old FIXnull mice were conditioned with 660 or 1,100 cGy TBI using a Gammacell 40 Exactor cesium irradiator (Best Theratronics, Ottawa, Ontario, Canada). Twenty-four hours after irradiation, 1 × 106 2bF9 LV-transduced Sca-1+ cells were infused into each mouse by retro-orbital vein injection. Animals were analyzed starting at 3 weeks after transplantation. At 9 months after transplantation, some animals were euthanized, BM cells were harvested, and sequential transplantation was carried out by transferring BM mononuclear cells into irradiated FIXnull secondary recipients. Blood samples were collected from tail or eye bleeds monthly as described in our previous report.41
PCR and qPCR. Genomic DNA was purified from peripheral white blood cells using QIAamp DNA blood mini kit (Qiagen, Hilden, Germany). A 350-bp fragment from the 2bF9 cassette was amplified as previously described.19 To confirm the FIXnull background, primers (5′-TCCTGTCATCTCACCTTGCTC-3′ and 5′-AACAGGGATAGTAAGATTGTTCC-3′) amplifying a 550-bp fragment of the disrupted mouse FIX were used. Primers (5′-TGGAAGCAGTATGTTGGTAAGC-3′ and 5′-AACAGGGATAGTAAGATTGTTCC-3′) were used to amplify a 320-bp fragment of wild-type mouse FIX. Amplification was performed using GoTaq Green Master Mix (Promega, Madison, WI). DNA from 2bF9 transgenic (2bF9tg+) mice19 was used as a positive control. Water was used as a negative control. For qPCR, 150 ng of genomic DNA was analyzed for the quantification of the 2bF9 proviral DNA sequence, with normalization to ApoB using iQ Supermix (Bio-Rad Laboratories, Hercules, CA) as previously described.17 DNA from 2bF9tg+/+ and FIXnull mice were used as controls.
FIX assays. FIX expression was quantitated by both FIX antigen (FIX: Ag) and functional FIX activity (FIX:C) assays. Platelets or plasma were isolated from 2bF9 LV-transduced recipients as previously described.41 Platelet lysate were prepared by lysing platelet pellets in 25 µl of 0.5% CHAPS (the zwitterionic detergent, 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, MP Biomedicals, Solon, OH) per 107 platelets. FIX: Ag was measured using a human FIX-specific ELISA assay. A 96-well plate was coated with 50 µl of 2 µg/ml of mouse anti-hFIX monoclonal antibody (AHIX-5041, Haematologic Technologies, Essex Junction, VT) at 4 °C overnight. Fifty microliters of series diluted platelet lysate or plasma was applied to the coated wells and incubated at room temperature for 2 hours. Goat anti-hFIX antibody (Affinity Biologicals, Ancaster, ON, Canada) was used as the primary, and a biotin-labeled mouse anti-goat antibody (Pierce, Rockford, IL) was used as the secondary antibody for detection. Human reference plasma was used as the standard. Platelets or plasmas collected from FIXnull mice were analyzed in parallel for each assay as negative controls. The levels of FIX:C in platelet lysates were quantitated by a chromogenic assay using the BIOPHEN Factor IX kit (Hyphen BioMed, Neuville-sur-Oise, France). A standard curve was established using serially diluted normal human pooled plasma. Platelets from FIXnull mice were processed in parallel for each assay as a negative control.
Immunofluorescent confocal microscopy. Intracellular location of the 2bF9 transgene protein was determined by immunofluorescent confocal microscopy as previously described.19,42 Briefly, platelets isolated from 2bF9 LV-transduced recipients were spun, fixed, and permeabilized on glass slides. The slides were blocked with 2.5% normal donkey serum for 1 hour, and incubated with a primary antibody goat anti-hFIX (Affinity Biologicals) at 4 °C overnight, then stained with rabbit anti-human VWF polyclonal antibody that crossreacts with mouse VWF (DAKO, Carpinteria, CA) for 2 hours and detected by AlexaFluor-488- or AlexaFluor-568-conjugated anti-goat and anti-rabbit secondary antibodies (Invitrogen, Eugene, OR) for 30 minutes. Platelets isolated from FIXnull mice were processed in parallel for each experiment. Nonspecific isotype IgG control antibodies served as the negative controls. Immunofluorescent detection was performed by confocal microscopy using an Olympus FV1000-MPE Multiphoton Microscope (Olympus, Tokyo, Japan).
Flow cytometry analysis. Approximately 2 × 107 platelets isolated from 2bF9 LV-transduced recipients were fixed with 0.5 ml of fixation buffer (BD Cytofix, BD Biosciences, San Diego, CA) at 4 °C for 30 minutes and permeabilized with 0.5% Triton X-100 (Thermo Scientific, Rockford, IL) for 20 minutes. Cells were blocked with PBS/2% BSA/2.5% normal donkey serum for 30 minutes and then stained with 5 µg/ml of goat anti-human FIX antibody (Affinity Biologicals) in PBS/2% BSA for 1 hour. After being washed, samples were stained with a donkey anti-goat antibody conjugated with AlexaFluor-488 (Invitrogen) and a rat anti-mouse Integrin αIIb antibody directly conjugated with PE (MWreg30, Santa Cruz Biotechnology, Santa Cruz, CA) at 1:100 in PBS/2% BSA for 1 hour. Platelets collected from FIXnull and 2bF9tg+/+ mice were used as controls. Cells were analyzed using an LSRII Green flow cytometer (BD Biosciences, San Jose, CA) and data were analyzed with Flow Jo software (TreeStar, Ashland, OR).
Phenotypic correction assessment. Tail clip survival tests were performed to access phenotypic correction as previously described.42 Briefly, after transplantation and at least 3 months bone marrow reconstitution, the tails of anesthetized mice were clipped at a diameter of 1.6 mm, without subsequent cauterization. FIXnull mice were tested in parallel as a control. Animals surviving beyond 24 hours were considered to have achieved phenotypic correction. The tail bleeding assay was used to grade the phenotypic correction in our transduced animals. Similar to the tail clip survival test, animals were anesthetized and the tail tip was transected using a 1.6-mm diameter template without subsequent cauterization. Fifty microliters of blood was collected before and 6 hours after the tail was transected, and Hb was measured using the Scil Vet ABC Plus blood counter (Scil Animal Care Company, Gurnee, IL). The Hb was normalized by defining the pretest levels of Hb as 100%. Whole-blood clotting time was determined by Rotational thromboelastometry analysis using a rotational thromboelastometry delta analyzer (Munich, Germany) as described in our previous report.19
Immunization of mice. At 5 months or later after transplantation, animals were immunized with recombinant human FIX (rhFIX; BeneFIX, Wyeth Pharmaceuticals, Cambridge, MA) at a dose of 200 U/kg in the presence of adjuvant (Ribi) (Sigma-Aldrich, St Louis, MO) by intraperitoneal injection twice with a 3-week interval. Two weeks after the last immunization, plasmas were collected to determine the titer of anti-FIX antibodies. As an antigen nonspecific immunization control, some animals were subsequently sensitized with OVA (Sigma-Aldrich) at a dose of 25 μg per mouse absorbed onto Imject Alum (Thermo, Rockford, IL) by intraperitoneal injection weekly for a total of three immunizations. One week after the last immunization, blood samples were collected for the ELISA assay to determine the titers of anti-OVA antibodies. FIXnull mice were immunized in parallel as a control.
Detection of antibodies. The titers of anti-hFIX inhibitory antibodies were determined by a modified Bethesda assay43 and the total anti-hFIX antibodies were quantitated by the ELISA assay. For the Bethesda assay, sequential dilutions of mouse plasma were incubated with an equal volume of 1-U/ml rhFIX at 37 °C for 2 hours, and residual FIX:C was subsequently measured using the chromogenic assay. The assay has a sensitivity of 1 BU/ml. For the ELISA assay, a 96-well plate was coated with 100 µl of 1-U/ml rhFIX at 4 °C overnight. Serial dilutions of mouse plasma were applied to the coated wells in duplicate and incubated at room temperature for 2 hours. Horseradish peroxidase-conjugated goat anti-mouse IgG (Pierce) at 1:3,000 was used as the detecting antibody, followed by incubation with orthophenylenediamine substrate. A standard curve was established using a serial dilution of anti-hFIX monoclonal antibody AHIX (Haematologic Technologies). The assay has a sensitivity of 0.16 µg/ml.
The titers of anti-OVA antibodies were determined by ELISA assay. A 96-well plate was coated with 100 µl of 10-µg/ml OVA at 4 °C overnight. Serial dilutions of mouse plasma were added to coated wells in duplicate and incubated at room temperature for 2 hours. Bound antibodies were detected by horseradish peroxidase-conjugated goat-anti-mouse IgG antibody (Pierce). The plate was developed with orthophenylenediamine substrate. Plasmas from FIXnull mice were processed in parallel. Antibody titers were expressed as the highest dilution of plasma samples showing a positive result (optical density >0.3) in the ELISA assay.44
Statistical analysis. All data are presented as the mean ± SD, and statistical comparisons of experimental groups were evaluated by unpaired Student's t-test. A value of P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by the National Institutes of Health grant HL-102035, the Hemophilia Association of New York grant, the Children's Hospital Foundation, the MACC Fund, and the Rebecca Slye Endowment Fund to Q. S. Y. C. was supported by the National Hemophilia Foundation JGP Postdoctoral Research Fellowship. G. Z. was supported by the National Natural Science Foundation of China (No.81170531) and the Special Medical Project of Hangzhou (20110833B30).
References
- Benson G, Auerswald G, Elezovic I, Lambert T, Ljung R, Morfini M, et al. Immune tolerance induction in patients with severe hemophilia with inhibitors: expert panel views and recommendations for clinical practice. Eur J Haematol. 2012;88:371–379. doi: 10.1111/j.1600-0609.2012.01754.x. [DOI] [PubMed] [Google Scholar]
- Franchini M.Lippi G.Montagnana M.Targher G.Zaffanello M.Salvagno GL.et al.2009Anaphylaxis in patients with congenital bleeding disorders and inhibitors. Blood Coagul Fibrinolysis 20225–229. [DOI] [PubMed] [Google Scholar]
- Jadhav M, Warrier I. Anaphylaxis in patients with hemophilia. Semin Thromb Hemost. 2000;26:205–208. doi: 10.1055/s-2000-9824. [DOI] [PubMed] [Google Scholar]
- Lusher JM. Inhibitor antibodies to factor VIII and factor IX: management. Semin Thromb Hemost. 2000;26:179–188. doi: 10.1055/s-2000-9821. [DOI] [PubMed] [Google Scholar]
- Shibata M, Shima M, Misu H, Okimoto Y, Giddings JC, Yoshioka A. Management of haemophilia B inhibitor patients with anaphylactic reactions to FIX concentrates. Haemophilia. 2003;9:269–271. doi: 10.1046/j.1365-2516.2003.00772.x. [DOI] [PubMed] [Google Scholar]
- Warrier I, Lusher JM. Development of anaphylactic shock in haemophilia B patients with inhibitors. Blood Coagul Fibrinolysis. 1998;9 suppl. 1:S125–S128. [PubMed] [Google Scholar]
- Warrier I. Management of haemophilia B patients with inhibitors and anaphylaxis. Haemophilia. 1998;4:574–576. doi: 10.1046/j.1365-2516.1998.440574.x. [DOI] [PubMed] [Google Scholar]
- Niemeyer GP, Herzog RW, Mount J, Arruda VR, Tillson DM, Hathcock J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. 2009;113:797–806. doi: 10.1182/blood-2008-10-181479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda VR, Stedman HH, Haurigot V, Buchlis G, Baila S, Favaro P, et al. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood. 2010;115:4678–4688. doi: 10.1182/blood-2009-12-261156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurigot V, Mingozzi F, Buchlis G, Hui DJ, Chen Y, Basner-Tschakarjan E, et al. Safety of AAV factor IX peripheral transvenular gene delivery to muscle in hemophilia B dogs. Mol Ther. 2010;18:1318–1329. doi: 10.1038/mt.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Louboutin JP, Bell P, Greig JA, Li Y, Wu D, et al. Muscle-directed gene therapy for hemophilia B with more efficient and less immunogenic AAV vectors. J Thromb Haemost. 2011;9:2009–2019. doi: 10.1111/j.1538-7836.2011.04491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuddenham E. Gene therapy for haemophilia B. Haemophilia. 2012;18 suppl. 4:13–17. doi: 10.1111/j.1365-2516.2012.02823.x. [DOI] [PubMed] [Google Scholar]
- High KA. The gene therapy journey for hemophilia: are we there yet. Hematology Am Soc Hematol Educ Program. 2012;2012:375–381. doi: 10.1182/asheducation-2012.1.375. [DOI] [PubMed] [Google Scholar]
- Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcedo R, Morizono H, Wang L, McCarter R, He J, Jones D, et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol. 2011;18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Wilcox DA, Fahs SA, Fang J, Johnson BD, DU LM, et al. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5:352–361. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- Kuether EL, Schroeder JA, Fahs SA, Cooley BC, Chen Y, Montgomery RR, et al. Lentivirus-mediated platelet gene therapy of murine hemophilia A with pre-existing anti-factor VIII immunity. J Thromb Haemost. 2012;10:1570–1580. doi: 10.1111/j.1538-7836.2012.04791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Shi Q, Fahs SA, Kuether EL, Walsh CE, Montgomery RR. Factor IX ectopically expressed in platelets can be stored in alpha-granules and corrects the phenotype of hemophilia B mice. Blood. 2010;116:1235–1243. doi: 10.1182/blood-2009-11-255612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston TN, Dudley JM, Shearer MJ, Schey SA. Vitamin K prophylaxis in high-dose chemotherapy. Lancet. 1995;345:1245. doi: 10.1016/s0140-6736(95)92031-5. [DOI] [PubMed] [Google Scholar]
- Barron MA, Doyle J, Zlotkin S. Vitamin K deficiency in children pre-bone marrow transplantation. Bone Marrow Transplant. 2006;37:151–154. doi: 10.1038/sj.bmt.1705215. [DOI] [PubMed] [Google Scholar]
- Toor AA, Slungaard A, Hedner U, Weisdorf DJ, Key NS. Acquired factor VII deficiency in hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2002;29:403–408. doi: 10.1038/sj.bmt.1703381. [DOI] [PubMed] [Google Scholar]
- Liu G, Peacock M. Age-related changes in serum undercarboxylated osteocalcin and its relationships with bone density, bone quality, and hip fracture. Calcif Tissue Int. 1998;62:286–289. doi: 10.1007/s002239900432. [DOI] [PubMed] [Google Scholar]
- Szulc P, Chapuy MC, Meunier PJ, Delmas PD. Serum undercarboxylated osteocalcin is a marker of the risk of hip fracture in elderly women. J Clin Invest. 1993;91:1769–1774. doi: 10.1172/JCI116387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa N, Shiraki M, Suhara Y, Kamao M, Tanaka K, Okano T. Vitamin K status of healthy Japanese women: age-related vitamin K requirement for gamma-carboxylation of osteocalcin. Am J Clin Nutr. 2006;83:380–386. doi: 10.1093/ajcn/83.2.380. [DOI] [PubMed] [Google Scholar]
- Shea MK, O'Donnell CJ, Vermeer C, Magdeleyns EJ, Crosier MD, Gundberg CM, et al. Circulating uncarboxylated matrix gla protein is associated with vitamin K nutritional status, but not coronary artery calcium, in older adults. J Nutr. 2011;141:1529–1534. doi: 10.3945/jn.111.139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann JC, Ames BN. Vitamin K, an example of triage theory: is micronutrient inadequacy linked to diseases of aging. Am J Clin Nutr. 2009;90:889–907. doi: 10.3945/ajcn.2009.27930. [DOI] [PubMed] [Google Scholar]
- Finn JD, Nichols TC, Svoronos N, Merricks EP, Bellenger DA, Zhou S, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. 2012;120:4521–4523. doi: 10.1182/blood-2012-06-440123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantore A, Nair N, Della Valle P, Di Matteo M, Màtrai J, Sanvito F, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. 2012;120:4517–4520. doi: 10.1182/blood-2012-05-432591. [DOI] [PubMed] [Google Scholar]
- Nakai H, Herzog RW, Hagstrom JN, Walter J, Kung SH, Yang EY, et al. Adeno-associated viral vector-mediated gene transfer of human blood coagulation factor IX into mouse liver. Blood. 1998;91:4600–4607. [PubMed] [Google Scholar]
- Ge Y, Powell S, Van Roey M, McArthur JG. Factors influencing the development of an anti-factor IX (FIX) immune response following administration of adeno-associated virus-FIX. Blood. 2001;97:3733–3737. doi: 10.1182/blood.v97.12.3733. [DOI] [PubMed] [Google Scholar]
- Cohn EF, Zhuo J, Kelly ME, Chao HJ. Efficient induction of immune tolerance to coagulation factor IX following direct intramuscular gene transfer. J Thromb Haemost. 2007;5:1227–1236. doi: 10.1111/j.1538-7836.2007.02522.x. [DOI] [PubMed] [Google Scholar]
- Vandendriessche T, Thorrez L, Acosta-Sanchez A, Petrus I, Wang L, Ma L, et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost. 2007;5:16–24. doi: 10.1111/j.1538-7836.2006.02220.x. [DOI] [PubMed] [Google Scholar]
- Zhang TP, Jin DY, Wardrop RM, 3rd, Gui T, Maile R, Frelinger JA, et al. Transgene expression levels and kinetics determine risk of humoral immune response modeled in factor IX knockout and missense mutant mice. Gene Ther. 2007;14:429–440. doi: 10.1038/sj.gt.3302881. [DOI] [PubMed] [Google Scholar]
- Chang AH, Stephan MT, Sadelain M. Stem cell-derived erythroid cells mediate long-term systemic protein delivery. Nat Biotechnol. 2006;24:1017–1021. doi: 10.1038/nbt1227. [DOI] [PubMed] [Google Scholar]
- Corr M, Lee DJ, Carson DA, Tighe H. Gene vaccination with naked plasmid DNA: mechanism of CTL priming. J Exp Med. 1996;184:1555–1560. doi: 10.1084/jem.184.4.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe B, Selby M, Barnett S, Baenziger J, Walker CM. Induction of cytotoxic T lymphocytes by intramuscular immunization with plasmid DNA is facilitated by bone marrow-derived cells. Proc Natl Acad Sci USA. 1996;93:8578–8583. doi: 10.1073/pnas.93.16.8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesielska A, Hadaczek P, Mittermeyer G, Zhou S, Wright JF, Bankiewicz KS, et al. Cerebral infusion of AAV9 vector-encoding non-self proteins can elicit cell-mediated immune responses. Mol Ther. 2013;21:158–166. doi: 10.1038/mt.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, Pratt KP, Miao CH. Progress toward inducing immunologic tolerance to factor VIII. Blood. 2013;121:4449–4456. doi: 10.1182/blood-2013-01-478669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, et al. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112:2713–2721. doi: 10.1182/blood-2008-02-138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, Cooley BC, et al. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116:1974–1982. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadarowski JJ, Czapek EE, Ontiveros JD, Pedraza JL. Modification of the Bethesda assay for factor VIII or IX inhibitors to improve efficiency. Acta Haematol. 1988;80:134–138. doi: 10.1159/000205619. [DOI] [PubMed] [Google Scholar]
- Hausl C, Maier E, Schwarz HP, Ahmad RU, Turecek PL, Dorner F, et al. Long-term persistence of anti-factor VIII antibody-secreting cells in hemophilic mice after treatment with human factor VIII. Thromb Haemost. 2002;87:840–845. [PubMed] [Google Scholar]