

Abstract
Stem cell survival and retention in myocardium after injury following adoptive transfer is low. Elevated catecholamine levels coinciding with myocardial injury adversely affect cardiac progenitor cell (CPC) survival. The G protein-coupled receptor kinase 2 (GRK2)-derived inhibitory peptide, βARKct, enhance myocyte contractility, survival, and normalize the neurohormonal axis in failing heart, however salutary effects of βARKct on CPC survival and proliferation are unknown. Herein, we investigated whether the protective effects of βARKct expression seen in the failing heart relate to CPCs. Modified CPCs expressing βARKct enhanced AKT/eNOS signaling through protective β2-adrenergic receptors (β2-ARs). In addition, to the actions of βARKct expression on β2- AR signaling, pharmacologic inhibition of GRK2 also increased β2-AR signaling in nonengineered CPCs (lacking βARKct) but had limited effects in βARKct engineered CPCs providing evidence for the strength of the βARKct in inhibiting GRK2 in these cells. Increased proliferation and metabolic activity were observed in βARKct-engineered CPCs following catecholamine stimulation indicating improved adrenergic tolerance. βARKct modification of CPCs increased survival and proliferation following adoptive transfer in an acute myocardial infarction model concomitant with increased expression of β-AR. Thus, βARKct engineering of CPCs promotes survival and proliferation of injected cells following myocardial infarction, which includes improved β-adrenergic tolerance essential for stem cell survival.
Introduction
The β-adrenergic receptor (β-AR) system regulates cardiac contractility1,2 that is an integral part of cardiac homeostasis, with elevated adrenergic drive occurring during postnatal development concurrent with extensive cellular proliferation3 as well as in the failing heart.1,4 Loss of contractile performance in the pathologically challenged heart can be initially compensated for by increased adrenergic drive but chronic elevation of catecholamine levels leads to deleterious consequences for cardiac structure and function.4,5 Although the adult myocardium possesses limited reparative potential in the form of endogenous stem cells, the functional regenerative activity of cardiac progenitor cells (CPCs) is compromised by adrenergic stimulation.6 Therefore, the ability of CPCs to mediate repair in the myocardium appears to be influenced by adrenergic stimulation that is normally present in response to myocardial injury, with differential effects in acute vs. chronic states.
The β-adrenergic system in the heart mediates effects primarily through β1- and β2-AR subtypes with β1-ARs regulating cardiac contractility via coupling exclusively to the G protein, Gs7. Myocardial injury coincides with activation of β1-AR mediated signaling with increased catecholamine levels that in the chronic condition prompts myocyte death and necrosis.5,8 In contrast, β2-AR signaling can antagonize cardiomyocyte death9,10 and promotes proliferation, mobilization, and differentiation of stem and progenitor cells.11,12,13,14 Thus, the β2-AR has been classified as a protective AR and this occurs through its properties of coupling not only to Gs but also Gi.9,10 Consistent with this and in the context of myocardial repair, we have recently found that β2-AR signaling promotes CPC survival and proliferation.6 CPCs in a lineage “uncommitted” state express β2-ARs almost exclusively while the β1-AR is acquired in the process of cardiac lineage commitment.6 β1-AR expression on “committed” CPCs leads to sensitization to apoptotic signaling in response to increased catecholamine stimulation, thereby limiting cardiac repair and regeneration in response to pathologic injury.
When the heart is injured or stressed there is activation of the sympathetic nervous system and adrenergic overdrive via excessive catecholamines that leads to uncoupling and desensitization of β-ARs in the heart, which is orchestrated through the upregulation of G protein-coupled receptor kinase 2 (GRK2).15,16 Enhanced GRK2 activity has been shown to contribute to cardiac decompensation,17 and even independent of its uncoupling properties on β-ARs, it appears to be a prodeath kinase especially in stressed myocytes.18,19 Importantly, inhibition of GRK2 or its genetic ablation its has been shown to improve cardiac function in myocardial injury and heart failure animal models via improving inotropic reserve, improving myocyte survival and also via normalization of the neurohormonal axis that can occur through improved β-AR signaling.18,20,21 GRK2 signaling inhibition can be achieved using βARKct, a small peptide isolated from the Gβγ binding site of GRK2, a process required for GRK2's actions on activated G protein-coupled receptors.22 βARKct gene therapy for the treatment of pathologic myocardial injury has been successfully implemented in experimental animal models20,21 with the focus specifically on cardiomyocytes. In this study, we studied the actions of GRK2 inhibition specifically on CPC biology and reparative processes in the failing heart to target improved adrenergic signaling and tolerance to combat against sympathetic overdrive. We find that βARKct engineering of human CPCs (hCPCs) isolated from heart failure patients increases survival, proliferation, and enhances β-adrenergic regulation in cells following cardiac commitment. Collectively, these studies point toward the β-adrenergic system as an important modulator of CPC function, providing a rationale for strategies to enhance “adrenergic tolerance” via GRK2 inhibition and βARKct treatment as an interventional approach to improve outcomes in cellular therapy for the treatment of heart disease.
Results
βARKct overexpression in hCPCs
hCPCs were isolated from patients undergoing left ventricular assist device implantation as described previously.23 hCPCs overexpressing green fluorescent protein eGFP (hCPCe) or βARKct (hCPCeβct) were modified using lentiviral vectors Lv-egfp or Lv-egfp + βARKct (Supplementary Figure S1a). Modification efficiency after lentiviral transduction was 77.5% for hCPCe and 80.6% for hCPCeβct as measured by GFP expression using flow cytometric analysis (Supplementary Figure S1b). Expression of eGFP or βARKct in the modified hCPCs was confirmed by immunoblot analysis (Supplementary Figure S1c). To verify that the βARKct was indeed inhibiting GRK2 and blocking desensitization of resident β2-ARs, cAMP responses in control CPCs and βARKct-engineered CPCs was measured acutely over time after isoproterenol (ISO) treatment and indeed, we found increased cAMP responses with βARKct expression (Supplementary Figure S1d). Additional molecular characterization revealed no change in the expression of β-ARs after βARKct modification in hCPCs (Supplementary Figure S1e). These results indicate successful modification of the hCPC to express βARKct and the eGFP tag for subsequent functional studies.
Molecular signaling in βARKct-engineered hCPCs after catecholamine stimulation
Consequences of βARKct modification for molecular signal transduction were characterized in βARKct-engineered hCPC after catecholamine stimulation. Effect of βARKct modification was assessed as readout for GRK2, a known βARKct target and AKT, which has been previously shown to be activated because of increased β2-AR signaling. GRK2 phosphorylation at the known extracellular signal-regulated kinase/mitogen-activated protein kinase site (Ser 670) increased 2.1- and 2.6-fold in hCPCeβct under acute (2 hours) or chronic (6 hours) catecholamine stimulation relative to 1.4- and 0.9-fold in hCPCe, respectively (Figure 1a). Furthermore, AKT phosphorylation significantly increased 2.1- and 1.7-fold in hCPCeβct after 2 or 6 hours of catecholamine stimulation compared with 1.3 and 1.2 fold in hCPCe (Figure 1a). There was an increase in basal activation of AKT in CPCs with βARKct expression (0 hour), however the fold changes histogram represents normalized values for each cell line. Treatment of hCPCeβct or hCPCe cells with a GRK2 pharmacological inhibitor (Methyl 5-[2-(5-nitro-furyl)vinyl]-2-furoate) was done before catecholamine stimulation and this decreased GRK2 expression by 73% in hCPCe at 6 hours in comparison with only a 39% decrease in hCPCeβct, indicating that pharmacological GRK2 inhibition is also effective in blocking desensitization of the β-AR mediated signaling analogous to the impact of βARKct expression in hCPCeβct (Figure 1c). Since CPCs express only β2-AR and we have shown previously that catecholamine stimulation can enhance AKT, effect of GRK2 inhibitor treatment on hCPCe and hCPCeβct was assessed in terms of AKT activity. AKT phosphorylation was increased as a result of transgene modification and cell selection in either hCPCeβct or hCPCe cells by 2.1- or 1.8-fold, respectively, but catecholamine stimulation for 6 hours did not reveal significant differences in AKT phosphorylation between hCPCe and hCPCeβct (Figure 1c).
Figure 1.
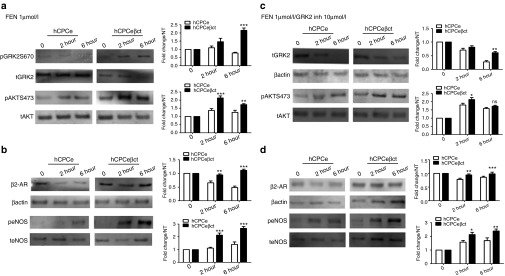
Characterization of molecular signaling in βARKct-engineered hCPCs. (a) Increased GRK2 phosphorylation in human cardiac progenitor cells overexpressing βARKct (hCPCeβct) after 2 and 6 hours of catecholamine stimulation compared with GFP expressing hCPCs (hCPCe; n = 3). Furthermore, increased AKT phosphorylation in hCPCeβct compared with hCPCe after catecholamine stimulation (n = 3). (b) Increased expression of β2-adrenergic receptor in hCPCeβct compared with hCPCe in response to catecholamine treatment. hCPCeβct maintain β2-adrenergic receptor even after 6 hours of catecholamine treatment compared with hCPCe (n = 3). Increased eNOS phosphorylation in hCPCeβct after catecholamine stimulation compared with hCPCe indicating activation of the prosurvival signaling (n = 3). (c) Decreased GRK2 levels in hCPCe compared with hCPCeβct after treatment with GRK2 inhibitor in the presence of catecholamine stimulation. Increased AKT phosphorylation in hCPCeβct compared with hCPCe after 2 hours of catecholamine stimulation in the presence of GRK2 inhibitor but the difference in not significant after 6 hours. (d) Increased β2-AR expression in hCPCeβct after 2 hours of catecholamine stimulation compared with hCPCe but not significant at 6 hours. Similarly, increased eNOS phosphorylation in hCPCeβct compared with hCPCe at 2 and 6 hours, respectively. NS, nonsignificant, hCPCe vs. hCPCeβct *P < 0.05, **P < 0.01, ***P < 0.001. GFP, green fluorescent protein; GRK2, G protein-coupled receptor kinase 2; hCPC, human cardiac progenitor cell.
Activation of β2-AR increases survival signaling in CPCs6 and cardiomyocytes9 in response to catecholamine stimulation. Importantly, β2-AR expression was preserved in hCPCeβct after catecholamine stimulation for 2 or 6 hours, whereas catecholamine stimulation in control cCPCe cells caused β2-AR expression to decrease 35 and 52% at 2 or 6 hours, respectively (Figure 1b). Furthermore, β2-AR expression after GRK2 inhibitor treatment decreased by 22% in hCPCe compared with hCPCeβct after 2 hours but does not change significantly between hCPCe and hCPCeβct after 6 hours of GRK2 inhibition in conjunction with catecholamine stimulation (Figure 1d) indicating that the beneficial effect of βARKct depends, in part, upon GRK2 activity that correlates with preservation of β2-AR expression in nonengineered CPCs lacking βARKct. Activation of β2-AR signaling increases AKT phosphorylation that, in turn, activates downstream eNOS activity.18 Indeed, eNOS phosphorylation increases of 2.1- or 2.6-fold was observed in hCPCeβct cells after 2 or 6 hours of catecholamine stimulation as compared with increases of only 1.1- or 1.4-fold in control hCPCe cells (Figure 1b). Similarly, following treatment with the pharmacologic GRK2 inhibitor, eNOS phosphorylation increased higher in control cells (1.5-fold at 2 hours and 1.6-fold at 6 hours) while this inhibitor did not alter improved eNOS activation in βARKct containing CPCs (Figure 1d). Collectively, these results indicate that βARKct prevents β2-AR desensitization and promotes signaling related to survival and proliferation mediated by AKT/eNOS signaling in response to catecholamine stimulation. Consistent with βARKct results, pharmacologic GRK2 inhibition also increases β2-AR/AKT/eNOS-mediated signaling in hCPCe after catecholamine stimulation but does impart any change when GRK2 is already inhibited by the βARKct.
Cell Survival is increased in βARKct-engineered hCPCs subjected to oxidative stress
hCPCeβct cells show increased viability relative to hCPCe cells when in culture for 5 days (P < 0.001; Supplementary Figure S2a). Oxidative stress induced by a H2O2 challenge led to a significant reduction of cell death in hCPCeβct cells relative to hCPCe cells (P < 0.01; Supplementary Figure S2b). Concordant with the cytoprotective effect of βARKct,18 significant phosphorylation increases for both AKT (P < 0.01) and eNOS (P < 0.001) were observed in hCPCeβct compared with hCPCe cells after oxidative stress (Supplementary Figure S2 c–d).
Proliferation enhanced in βARKct-engineered hCPCs
βARKct modification increases AKT activation in CPCs (Figure 1) that may lead to increased proliferation in the presence of catecholamines. Catecholamine-driven proliferation was increased in hCPCeβct compared with hCPCe cells at day 3 (P < 0.05) and day 5 (P < 0.05; Figure 2a). Consistent with these βARKct results, pharmacologic GRK2 inhibition together with catecholamine stimulation prompted proliferation of hCPCeβct relative to hCPCe that was significantly increased at day 3 (P < 0.05), but this differential proliferative response was lost by day 5 in culture (Figure 2c) suggesting that long-term pharmacologic inhibition of GRK2 achieves effects similar to constitutive βARKct expression. Concordantly, metabolic activity was significantly increased in hCPCeβct relative to hCPCe cells at day 3 (P < 0.05) and day 5 (P < 0.01; Figure 2b). Effects on metabolic activity resulting from pharmacologic GRK2 inhibition together with catecholamine stimulation mirrored proliferation findings, with increased metabolic activity at day 3 (P < 0.01) that becomes comparable by day 5 between hCPCeβct relative to hCPCe (Figure 2d). Proliferation or metabolic activity in either hCPCeβct or hCPCe was not affected by pharmacologic GRK2 inhibition alone. Therefore, βARKct increases proliferation and metabolic activity in hCPCeβct relative to hCPCe in response to catecholamine stimulation and the potentiating effect of βARKct is mimicked by pharmacologic GRK2 inhibition in control cells.
Figure 2.

Increased proliferation and metabolic activity in hCPCeβct. (a) CyQuant assay; hCPCeβct exhibited enhanced proliferation compared with hCPCe at 3 and 5 days after catecholamine stimulation (n = 3). (b) Metabolic activity measured by using MTT reagent in hCPCeβct demonstrated improved metabolic activity relative to hCPCe (n = 3). NS, nonsignificant, hCPCe FEN vs. hCPCeβct FEN *P < 0.05, **P < 0.01, ***P < 0.001. hCPCeβct treated with GRK2 inhibitor in the presence of catecholamine stimulation have increased proliferation (c) and metabolic activity (d) compared with hCPCe at day 3 (n = 3). However, there is no significant change in proliferation and metabolic activity between hCPCe and hCPCeβct at 5 days in the presence of GRK2 inhibitor and catecholamine stimulation. hCPCe and hCPCeβct treated with GRK2 inhibitor alone does not show any effect on proliferation and metabolic activity. NS, nonsignificant, hCPCe FEN/inh vs. hCPCeβct FEN/inh *P < 0.05, **P < 0.01, ***P < 0.001. GRK2, G protein-coupled receptor kinase 2; hCPC, human cardiac progenitor cell.
Increased survival of hCPCeβct cells with β1-AR overexpression and catecholamine challenge
CPCs express β2-AR in an uncommitted state but acquire β1-AR in the process of cardiac lineage commitment leading to sensitization to catecholamine-induced cell death.6 Therefore, effect of βARKct modification in promoting CPC survival in the presence of catecholamine-induced β1-AR stimulation was analyzed. hCPCeβct or hCPCe cells were infected with a β1-AR adenovirus and subsequently challenged with ISO to analyze ISO-β1-AR mediated cell death.8 Consistent with the proapoptotic role of β1-AR overexpression, cell death was increased at 2 hours (23.9%) and 6 hours (37.1%) in ISO-treated hCPCe cells compared with control nontreated hCPCe cells (7.6%) (Figure 3a). The presence of hCPCeβct conferred significant protection at 2 (14.8%, P < 0.01) and 6 hours (20.7%, P < 0.001) under these β1-AR mediated proapoptotic conditions (Figure 3a). The involvement of functional β1-AR overexpression was confirmed by inclusion of metoprolol, a β1-AR specific antagonist, which abrogated ISO-induced cell death in hCPCeβct and hCPCe (Figure 3a). The proapoptotic effect of β1-AR signaling in cardiomyocytes is mediated, at least in part, by PKA and cAMP activation24 that leads to a variety of homologous and heterologous desensitization effects25,26,27 in response to chronic catecholamine stimulation. Increased cAMP levels were observed in β1-AR overexpressing hCPCe cells after 2 and 6 hours of catecholamine stimulation (Figure 3b) indicating activation of β1-AR mediated signaling. In contrast, less chronic cAMP levels were observed in hCPCeβct cells (Figure 3b). These experiments support the postulate that βARKct expression confers protection against ISO-induced cell death, even when the hCPCs acquire more proapoptotic β1-ARs.
Figure 3.

Increased survival in hCPCeβct overexpressing β1-adrenergic receptor in response to catecholamine stimulation. (a) Increased survival in hCPCeβct compared with hCPCe overexpressing β1-adrenergic receptor and in the presence of isoproterenol as evidenced by FACS based cell death analysis (n = 3). No change after isoproterenol induced cell death in metoprolol treated hCPCeβct and hCPCe. (b) Enhanced cAMP activity in hCPCe overexpressing β1-adrenergic receptor stimulated with isoproterenol at 2 and 6 hours compared with hCPCeβct as measured by cAMP assay (n = 3). hCPCe + β1-AR vs. hCPCeβct + β1-AR *P < 0.05, **P < 0.01, ***P < 0.001. hCPC, human cardiac progenitor cell.
Increased survival of hCPCeβct cells in situ in the heart following myocardial infarction
Superiority of hCPCeβct cells in the face of adrenergic challenge relative to hCPCe cells in terms of proliferation and survival, as we found in vitro above (Figure 2; Supplementary Figure S2) was validated in vivo using adoptively transfer of syngeneic murine CPCs into acutely infarcted mouse myocardium characterized by high circulating catecholamine levels as previously reported.1 Indeed, significantly increased epinephrine (P < 0.001) and norepinephrine (P < 0.001) serum levels were observed 3 days after myocardial infarction compared with normal mice (Supplementary Figure S3). Murine CPCs engineered to express βARKct (mCPCeβct) show typical characteristics of increased proliferation and metabolic activity in vitro (Supplementary Figure S4a–c) as previously observed for hCPCeβct cells (Figure 2). Survival and proliferation of mCPCeβct vs. mCPCe cells was assessed at day 3 after myocardial infarction and adoptive transfer. Adoptively transferred cells were identified by coincident immunolabeling for c-kit+/GFP+ in myocardial sections. The number of c-kit+/GFP+ cells was a significant 2.1-fold higher in hearts receiving mCPCeβct compared with mCPCe (Figure 4 a–c). Similarly, proliferating cell nuclear antigen (PCNA)+/GFP+ cells were also significantly increased (25.1%) in mCPCeβct compared with mCPCe (10.2%) indicating increased proliferation of the donated CPC population (Figure 4d–f). Lastly, apoptotic cell death as indicated by TUNEL+/GFP+ coincidence was reduced (3.7%) in hearts receiving mCPCeβct compared with mCPCe (10.5%) at 3 days (Figure 4g–i). Collectively these findings support the beneficial effect of engineered mCPCeβct cell transfer in the context of acute myocardial infarction injury over normal CPCs.
Figure 4.

CPC survival and proliferation is enhanced with βARKct engineering in hearts after acute myocardial infarction. (a–b) Increased c-kit+/GFP+ cells within hearts transplanted with hCPCeβct compared with hCPCe transplanted hearts after 3 days of infarction stained with GFP (green) c-kit (red), sarcomeric actin (blue), and nuclei (white) Scale bar = 40 µm. (c) Quantitation of c-kit+/GFP+ cells in hCPCeβct and hCPCe transplanted hearts. (d–f) Increased PCNA+/GFP+ cells in hCPCeβct hearts compared with hCPCe stained with PCNA (red), GFP (green), sarcomeric actin (blue), and nuclei (white) along with corresponding quantitation. (g–i) Reduced TUNEL+/GFP+ in hCPCeβct hearts compared with hCPCe stained with TUNEL (red), GFP (green), sarcomeric actin (blue), and nuclei (white) along with corresponding quantitation. hCPCe vs. hCPCeβct *P < 0.05, **P < 0.01, ***P < 0.001. GFP, green fluorescent protein; hCPC, human cardiac progenitor cell.
Increased frequency of β1-AR+ mCPCeβct in situ following myocardial infarction
As shown above, βARKct engineering increases CPC survival and proliferation in hearts after acute ischemic injury supporting expansion of the mCPCeβct cells following adoptive transfer. We have shown previously that lineage unspecified β2-AR+ CPCs acquire β1-AR expression (the major myocyte β-AR sub-type) upon cardiogenic lineage commitment and sensitization to catecholamine-induced cell death.6 The βARKct modified mCPCeβct group yields an increased number of GFP CPCs colocalized with β1-AR (2.3%) compared with mCPCe group (0.3%; Figure 5a–c). Similarly, increased β2-AR+/GFP+ cells were observed in mCPCeβct (20.1%) group compared with mCPCe (11.5%; Figure 5d–f) in the infarcted myocardium at day 3 compared with mCPCe. Collectively, these results indicate that βARKct engineering increases survival of cardiogenic CPCs and allow for improved viability of the adoptively transferred mCPCeβct cells in repair and regenerative processes and since β1-ARs are present there appears to be improved catecholamine and adrenergic tolerance of these cells in the infarcted myocytes due to GRK2 inhibition.
Figure 5.
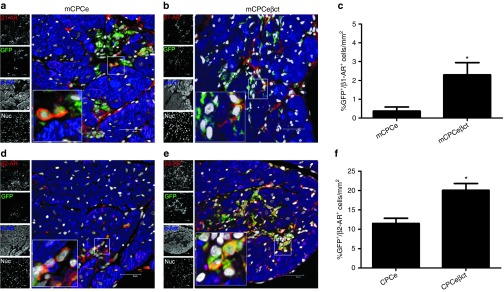
Increased survival of β1-adrenergic receptor expressing cells in hearts after acute infarction. (a–b) Increased survival of β1-AR+6 cells colocalized with GFP (green) in the hearts with CPCeβct compared with CPCe. (c) Quantitation of β1-AR+/GFP+ CPCs. (d–f) Increased survival of β2-AR+/GFP+ cells in CPCeβct transplanted hearts compared with CPCe along with corresponding quantitation. Scale bar = 40 µm. hCPCe vs. hCPCeβct *P < 0.05, **P < 0.01, ***P < 0.001. GFP, green fluorescent protein; hCPC, human cardiac progenitor cell.
Discussion
Adoptive transfer of CPCs into pathologically challenged myocardium improves cardiac function in a variety of animal models.23,28 However, poor survival, engraftment, and lineage commitment of adoptively transferred CPCs in damaged myocardium is a significant limitation for efficacy of cellular therapy.29,30 The stressed/injured myocardial environment is characterized by increased circulating catecholamine levels and subsequent adrenergic over-drive1 that can alter survival and proliferation characteristics of adoptively transferred CPCs. Although β2-AR expression favors CPC survival and proliferation in an uncommitted state, the acquisition of β1-AR during lineage commitment increases sensitization to catecholamine-induced cell death.6 Findings in this study support the postulate that survival and proliferation of adoptively transferred CPCs can be enhanced by genetic engineering of CPCs with βARKct to enhance adrenergic tolerance in a damaged heart by inhibiting GRK2, which has previously been shown to be a prodeath kinase in myocytes.18,19
Catecholamine dysregulation in pathologic injury or chronic stress leads to adrenergic hyperactivity and induces the upregulation of GRK2, a critical regulator of cardiac G protein-coupled receptors including β-ARs.31 Silencing GRK2 activity can prevent cardiomyocyte loss, improve heart function, and normalize the neurohormonal axis including lowering of circulating catecholamines due to improved signaling through β-ARs as well as improved function of the heart.16,18,19,20 Salutary effects of βARKct upon survival and proliferation in CPCs in the face of adrenergic challenge were indeed due to GRK2 inhibition as a pharmacological inhibitor of this GRK imparted similar in vitro properties on proliferation and survival after β-AR stimulation and the inhibitor did very little on top of βARKct showing that this peptide is inhibiting GRK2 in these CPCs. Furthermore, βARKct increased cAMP production acutely and imparted protection against chronic catecholamine stimulation by reducing cAMP levels (Figure 3) as a consequence of PKA activation25,27 and increased β1-AR26 phosphorylation, reinforcing the long-term beneficial effect of βARKct expression on hCPCe cells despite an acute increase in cAMP production. The βARKct, a synthetic peptide developed from the carboxyl terminus of GRK2 that binds to membrane-associated and disassociated Gβγ subunits, which is a requirement of GRK2 activation and its actions in the heart include improved adrenergic signaling and cardiac contractility and function.32,33,34 The milieu of acutely infarcted myocardium and associated catecholamine storm provides an appropriate experimental setting to demonstrate this robust βARKct-mediated protection of CPCs. Indeed, βARKct-engineered CPCs displayed enhanced survival, proliferation and also increased β1-AR expression that would mark increased adrenergic tolerance as the CPCs commit to a cardiomyocyte lineage (Figures 4 and 5). Salutary effects of βARKct coupled together with observations of adrenergic signaling effects in CPCs prompted this study, which supports use of βARKct engineering to enhance adrenergic tolerance, survival, and proliferation of CPCs in the damaged myocardium.
Future studies will be needed to determine if this engineering of CPCs can improve cardiac function and outcomes in ischemic heart failure. However, we had shown that modifying CPCs with the βARKct confers superior ability to withstand the catecholamine storm immediately after transplantation in an acute pathological setting promoting survival, proliferation and commitment. Moreover, lentiviral modification of CPCs with βARKct provides sustained expression of the transgene in CPCs in contrast to pretreatment of CPCs with pharmacological GRK2 inhibitor before transplantation. Previously it has been shown that CPCs express low levels of both β-ARs35, which is in contrast to to CPCs used in this study that only express β2-AR and acquire β1-AR upon cardiac commitment and maybe attributed to different CPC isolation and expansion procedures. The molecular basis of βARKct-mediated improvement in survival and proliferation in CPCs rests with inhibition of GRK2 activity that has been recently shown to be a prodeath kinase in the heart that can limit prosurvival pathways including AKT and eNOS downstream, at least in part, through the β2-AR18 and Gi protein activation, the β-AR sub-type known to protect against cateholamine-induced cell death. Since the cardioprotective effects of βARKct seem to be in part due to AKT activation suggesting AKT gene transfer as a possible alternative. However, recently it has been shown that AKT modification of CPCs enhances proliferation but prevents CPCs from cardiac commitment.36 Furthermore, βARKct works in a delineate fashion; it does not only regulate adrenergic overdrive but at the same time enhance AKT mediate prosurvial signaling.
βARKct and subsequent GRK2 inhibition has shown to be cardioprotective in the acute setting,18 and also has been shown to reverse heart failure in several animal models including a recent study showing significant cardiac improvement in a large animal preclinical efficacy study in heart failure pigs.21
Importantly, the βARKct inhibits GRK2 through blocking Gβγ binding and activation of this kinase, and in addition to its palliative effects on heart function, the βARKct has been shown to limit vascular smooth muscle restenosis and also limit hypertension in an animal model.37,38 Therefore, future studies will continue to explore exact mechanisms for βARKct-mediated improvement of CPC survival and engraftment in the ischemic myocardium as it is possible that effects are cell-specific and different than the heart failure rescuing mechanisms that have been seen in myocytes. For example, it has been shown that in addition of the βARKct's ability to block Gβγ-mediated G protein-coupled receptor kinase activity, this peptide can inhibit the prodeath activities of GRK2 localized to mitochondrial via Hsp90 binding.19 Finally, studies will need to delve further into delineating the adrenergic signaling network in CPCs with the goal of defining molecular interventional targets to promote enhancement of reparative and regenerative processes. Increased surviving CPCs because of βARKct engineering will enable enhanced CPC participation in repair and regeneration processes potentially providing a novel therapeutic modality for the treatment of heart failure.
Materials and Methods
Cell culture and differentiation. Human CPCs were isolated from ventricular samples obtained as discarded tissue of patients receiving left ventricular assist device implantation.23 The samples were provided without patient identifiers and generated as a normal by-product of a surgical procedure, so therefore the protocol was deemed nonhuman subjects research by IRB review. Human CPC were maintained in as described in Supplementary Materials and Methods. Mouse CPCs are isolated from syngeneic male FVB mice and cultured for 3 weeks in cardiac stem cell media as described in Supplementary Materials and Methods.28,39
Lentiviral transduction. CPCs were genetically modified through a bicistronic lentiviral vector to deliver enhanced GFP transcribed from an internal ribosomal entry site in conjunction with the βARK-ct gene (CPCeβct). Additional details are provided in Supplementary Materials and Methods.
Pharmacological treatments. CPCs were serum starved overnight and treated with Fenoterol (FEN; 1 µmol/l, Sigma-Aldrich, St. Louis, MO, USA) for 2 or 6 hours in serum free medium before CyQuant labeling, metabolic assays and protein analysis. ISO (1 μmol/l) treatment of CPCs to induce catecholamine stress was performed for 2 or 6 hours before flow cytometric analysis. β1-antagonist Metoprolol (1 μmol/l; Sigma-Aldrich) was used for manipulation of adrenergic stimulation. CPCs were treated with Methyl 5-[2-(5-nitro-2-furyl)vinyl]-2-furoate as a pharmacological inhibitor of GRK2 activity (10 µmol/l, Millipore) for 1 hour in serum free medium before catecholamine stimulation. CPCs were treated with H2O2 for 3 hours in low serum medium to induce oxidative stress before FACS based cell death analysis.
CyQuant and metabolic assay. CyQuant and 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay of CPC were performed with plating of cells in quadruplicate (2,000 cells/well) in 96-well plates, followed by incubation with CyQuant (Invitrogen) or MTT reagent (Sigma, St Louis, MO) as previously described.6,28,40
Immunoblots. Immunoblotting was performed as described.40,41 Details regarding sample preparation and antibodies are provided in Supplementary Materials and Methods and Table S1.
Flow cytometry. Cell death was measured by Annexin V staining (BD Biosciences, San Jose, CA, USA) according to manufacturer instructions. Cytometry is performed by using a BD FACSAria Flow Cytometer (BD Biosciences).
Animal studies. Institutional Animal Care and Use Committee approval was obtained for all animal studies. Detailed experimental procedures are provided in Supplementary Materials and Methods.
Immunohistochemistry. Immunocytochemistry, TUNEL assays, and immunohistochemistry were performed as previously described6,28,40 with additional information in Supplementary Materials and Methods.
cAMP assay. cAMP levels were measured by competitive enzyme immunoassay from cell lysates of hCPCs treated with catecholamine in combination with metoprolol by using the cAMP parameter assay kit (R&D systems).
Catecholamine assay. Plasma epinephrine (Epi) and norepinephrine (NEpi) levels were determined by enzyme-linked immunosorbent assay performed on mouse plasma samples using the BI-CAT enzyme-linked immunosorbent assay kit from ALPCO Diagnostics (Windham, NH) with detailed methods provided in Supplementary Materials and Methods.
Statistics. Statistical analysis was performed using Student's t-test. Comparison of more than two groups was performed by one-way ANOVA or two-way ANOVA with Bonferroni's post hoc test. P value less than 0.05 is considered statistically significant. Error bars represent SEM. Statistical analysis is performed using Graph Pad prism v 5.0 software.
SUPPLEMENTARY MATERIAL Figure S1. Characterization of βARKct overexpression in human CPCs. Figure S2. Enhanced viability and survival in hCPCeβct in response to oxidative stress. Figure S3. Serum plasma levels of epinephrine and norepinephrine. Figure S4. βARKct engineering in mouse CPCs. Table S1. Antibodies. Materials and Methods.
Acknowledgments
We thank all members of the Sussman Lab for their critical review of the manuscript. There are no conflicts of interest. M.A.S. is supported by National Heart, Lung, and Blood Institute awards R21-HL102714, R01-HL067245, R37-HL091102, P01-HL085577, RC1-HL100891, R21-HL102613, R21 HL104544 and R01 HL105759. W.J.K. is supported by R01HL085503, R37-HL061690, P01-HL108806 and P01-HL075443. D.G.T. is supported by HL105414. M.K. is supported by American Heart Association postdoctoral fellowship (11POST7370097).
Supplementary Material
References
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999;51:651–690. [PubMed] [Google Scholar]
- Collis LP, Srivastava S, Coetzee WA, Artman M. beta2-Adrenergic receptor agonists stimulate L-type calcium current independent of PKA in newborn rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H2826–H2835. doi: 10.1152/ajpheart.00101.2007. [DOI] [PubMed] [Google Scholar]
- Vatner DE, Asai K, Iwase M, Ishikawa Y, Shannon RP, Homcy CJ, et al. Beta-adrenergic receptor-G protein-adenylyl cyclase signal transduction in the failing heart. Am J Cardiol. 1999;83 12A:80H–85H. doi: 10.1016/s0002-9149(99)00266-0. [DOI] [PubMed] [Google Scholar]
- Iwai-Kanai E, Hasegawa K, Araki M, Kakita T, Morimoto T, Sasayama S. alpha- and beta-adrenergic pathways differentially regulate cell type-specific apoptosis in rat cardiac myocytes. Circulation. 1999;100:305–311. doi: 10.1161/01.cir.100.3.305. [DOI] [PubMed] [Google Scholar]
- Khan M, Mohsin S, Avitabile D, Siddiqi S, Nguyen J, Wallach K, et al. ß-Adrenergic regulation of cardiac progenitor cell death versus survival and proliferation. Circ Res. 2013;112:476–486. doi: 10.1161/CIRCRESAHA.112.280735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hébert TE, Allen BG. Nuclear ß-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011;23:89–98. doi: 10.1016/j.cellsig.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, et al. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, et al. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–337. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Foerster K, Groner F, Matthes J, Koch WJ, Birnbaumer L, Herzig S. Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through beta 2-adrenoceptors. Proc Natl Acad Sci USA. 2003;100:14475–14480. doi: 10.1073/pnas.1936026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri DJ, Mackay EW, Hamlin AS, Marathe SV, Nandam LS, Vaidya VA, et al. Norepinephrine directly activates adult hippocampal precursors via beta3-adrenergic receptors. J Neurosci. 2010;30:2795–2806. doi: 10.1523/JNEUROSCI.3780-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider JE, Polster SP, Lee S, Charles NJ, Adhikari N, Mariash A, et al. Chronic treatment with clenbuterol modulates endothelial progenitor cells and circulating factors in a murine model of cardiomyopathy. J Cardiovasc Transl Res. 2009;2:182–190. doi: 10.1007/s12265-009-9089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel A, Shivtiel S, Kalinkovich A, Ludin A, Netzer N, Goichberg P, et al. Catecholaminergic neurotransmitters regulate migration and repopulation of immature human CD34+ cells through Wnt signaling. Nat Immunol. 2007;8:1123–1131. doi: 10.1038/ni1509. [DOI] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–144. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theilade J, Strøm C, Christiansen T, Haunsø S, Sheikh SP. Differential G protein receptor kinase 2 expression in compensated hypertrophy and heart failure after myocardial infarction in the rat. Basic Res Cardiol. 2003;98:97–103. doi: 10.1007/s00395-003-0395-x. [DOI] [PubMed] [Google Scholar]
- Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrofski JA, Koch WJ. The beta-adrenergic receptor kinase in heart failure. J Mol Cell Cardiol. 2003;35:1167–1174. doi: 10.1016/s0022-2828(03)00243-8. [DOI] [PubMed] [Google Scholar]
- Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, et al. Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res. 2010;107:1140–1149. doi: 10.1161/CIRCRESAHA.110.221010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Sato PY, Chuprun JK, Peroutka RJ, Otis NJ, Ibetti J, et al. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ Res. 2013;112:1121–1134. doi: 10.1161/CIRCRESAHA.112.300754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, et al. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raake PW, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, et al. AAV6.ßARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J. 2013;34:1437–1447. doi: 10.1093/eurheartj/ehr447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, et al. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J Am Coll Cardiol. 2012;60:1278–1287. doi: 10.1016/j.jacc.2012.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, et al. Cardiotoxic and cardioprotective features of chronic ß-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, et al. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal. 2004;16:1397–1403. doi: 10.1016/j.cellsig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Maurice DH. Vascular smooth muscle cell phenotype-dependent phosphodiesterase 4D short form expression: role of differential histone acetylation on cAMP-regulated function. Mol Pharmacol. 2005;68:596–605. doi: 10.1124/mol.105.014126. [DOI] [PubMed] [Google Scholar]
- Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, et al. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation. 2009;120:2077–2087. doi: 10.1161/CIRCULATIONAHA.109.884403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- Müller-Ehmsen J, Whittaker P, Kloner RA, Dow JS, Sakoda T, Long TI, et al. Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol. 2002;34:107–116. doi: 10.1006/jmcc.2001.1491. [DOI] [PubMed] [Google Scholar]
- White DC, Hata JA, Shah AS, Glower DD, Lefkowitz RJ, Koch WJ. Preservation of myocardial beta-adrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proc Natl Acad Sci USA. 2000;97:5428–5433. doi: 10.1073/pnas.090091197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tevaearai HT, Eckhart AD, Shotwell KF, Wilson K, Koch WJ. Ventricular dysfunction after cardioplegic arrest is improved after myocardial gene transfer of a beta-adrenergic receptor kinase inhibitor. Circulation. 2001;104:2069–2074. doi: 10.1161/hc4201.097188. [DOI] [PubMed] [Google Scholar]
- Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA. 2001;98:5809–5814. doi: 10.1073/pnas.091102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AS, White DC, Emani S, Kypson AP, Lilly RE, Wilson K, et al. In vivo ventricular gene delivery of a beta-adrenergic receptor kinase inhibitor to the failing heart reverses cardiac dysfunction. Circulation. 2001;103:1311–1316. doi: 10.1161/01.cir.103.9.1311. [DOI] [PubMed] [Google Scholar]
- Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, et al. Acute beta-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem. 2007;282:11397–11409. doi: 10.1074/jbc.M607391200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KM, Din S, Gude N, Konstandin MH, Wu W, Quijada P, et al. Cardiac progenitor cell commitment is inhibited by nuclear Akt expression. Circ Res. 2011;108:960–970. doi: 10.1161/CIRCRESAHA.110.237156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Smithwick LA, Lefkowitz RJ, Koch WJ. Targeting Gbeta gamma signaling in arterial vascular smooth muscle proliferation: a novel strategy to limit restenosis. Proc Natl Acad Sci USA. 1999;96:3945–3950. doi: 10.1073/pnas.96.7.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn HI, Xi Y, Pesant S, Harris DM, Hyslop T, Falkner B, et al. G protein-coupled receptor kinase 2 expression and activity are associated with blood pressure in black Americans. Hypertension. 2009;54:71–76. doi: 10.1161/HYPERTENSIONAHA.108.125955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransioli J, Bailey B, Gude NA, Cottage CT, Muraski JA, Emmanuel G, et al. Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells. 2008;26:1315–1324. doi: 10.1634/stemcells.2007-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitabile D, Bailey B, Cottage CT, Sundararaman B, Joyo A, McGregor M, et al. Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin. Proc Natl Acad Sci USA. 2011;108:6145–6150. doi: 10.1073/pnas.1017935108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Völkers M, Din S, Avitabile D, Khan M, Gude N, et al. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J. 2011;32:2179–2188. doi: 10.1093/eurheartj/ehq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.