

Abstract
Tumors are theoretically capable of eliciting an antitumor immune response, but are often poorly immunogenic. Oncolytic viruses (OVs) have recently emerged as a promising strategy for the immunogenic delivery of tumor-associated antigens (TAAs) to cancer patients. However, safe and effective OV/TAA therapies have not yet been established. We have previously demonstrated that vectors based on Sindbis virus (SV) can inhibit tumor growth and activate the innate immune system in mice. Here, we demonstrate that SV vectors carrying a TAA generate a dramatically enhanced therapeutic effect in mice bearing subcutaneous, intraperitoneal, and lung cancers. Notably, SV/TAA efficacy was not dependent on tumor cell targeting, but was characterized by the transient expression of TAAs in lymph nodes draining the injection site. Early T-cell activation at this site was followed by a robust influx of NKG2D expressing antigen-specific cytotoxic CD8+ T cells into the tumor site, subsequently leading to the generation of long-lasting memory T cells which conferred protection against rechallenge with TAA-positive as well as TAA-negative tumor cells. By combining in vivo imaging, flow cytometry, cytotoxicity/cytokine assays, and tetramer analysis, we investigated the relationship between these events and propose a model for CD8+ T-cell activation during SV/TAA therapy.
Introduction
Oncolytic viruses (OV) are viruses that specifically target and replicate in tumor cells.1 Owing to their selectivity and oncolytic properties, OVs have generated considerable interest as an alternative or adjunct to conventional cancer therapies.2 However, a major limitation of OV therapy is inadequate replication and propagation at the tumor site.3,4 Moreover, for safety reasons, many OVs are designed to be replication deficient in order to prevent them from spreading to healthy tissues, further limiting their oncolytic potential.5
One possible solution to this problem is to supplement direct viral oncolysis with a bystander effect, in which tumor cells not directly infected by the OV will also be destroyed. This can be achieved, for example, by inserting a therapeutic or cytotoxic gene into the OV genome for delivery to the tumor site.6,7 Endowed with natural immunogenicity, some OVs are capable of effective stimulation of the immune system, raising the possibility of using OVs to induce an immunological anticancer bystander effect.8 This idea gained further impetus with the identification9,10 and recent prioritization11 of a variety of clinically relevant tumor-associated antigens (TAA), which can be delivered by the OV (OV/TAA) to the tumor site.12 In their natural state, TAAs are often poorly immunogenic.13 However, by redirecting the antiviral immune response toward the TAA, an immunogenic OV/TAA could potentially break this immunological tolerance. A major goal of OV research should therefore be the development of safe and effective OV/TAA agents.
Sindbis virus (SV), an alphavirus with a positive single-stranded RNA genome,14 represents one of a select number of viruses that have demonstrated exceptional potential both as an OV15,16 and as a viral vaccine.17 We have previously shown that replication-deficient SV vectors target and inhibit the growth of xenograft, syngeneic, and spontaneous tumors in mice.16,18 Recently, we also found that SV induces the activation of natural killer (NK) cells and macrophages in tumor-bearing mice.19 In addition, SV vectors expressing immune-modulating genes such as interleukin 12 (IL-12) have enhanced antitumor16 and immunostimulatory19 effects. Nevertheless, these approaches have not generally led to complete tumor remission.19 Moreover, some tumor cells may not be efficiently targeted by SV,20 underscoring the need to develop new ways of enhancing SV anticancer therapy.
We hypothesized that the unique characteristics of SV vectors, that make them effective oncolytic agents and gene delivery systems (e.g., the ability to disseminate through the bloodstream15 and to deliver high levels of heterologous proteins)21 could also be useful for efficient TAA delivery. Moreover, the SV life cycle, which is characterized by the absence of a DNA phase, rendering the vectors safer, also involves the production of high levels of double-stranded RNA, a potent immunological ‘danger signal',22 and the subsequent activation of the type I interferon pathway.23 The combination of safety, immunogenicity, efficient dissemination, and high TAA expression make SV/TAA an attractive OV/TAA candidate.
In this study, we used the BALB/c CT26 colon carcinoma tumor model to investigate the use of SV as an OV/TAA agent. We found that unlike other tumor models tested, CT26 cells are not targeted by SV in vivo. Nevertheless, SV vectors carrying β galactosidase (LacZ) had a remarkable therapeutic effect in mice bearing LacZ-expressing CT26 tumors. Using the in vivo imaging system (IVIS) for sensitive in vivo detection of luciferase activity,24 we identified the mediastinal lymph nodes (MLN) as a site of early transient heterologous protein expression after intraperitoneal (i.p.) injection of SV vectors carrying firefly luciferase (SV/Fluc). TAA delivery into the MLN marked the starting point of a potent immune response that culminated in the generation of effector and memory CD8+ T cells displaying robust cytotoxicity against LacZ-positive and LacZ-negative tumor cells. This latter phenomenon, known as epitope spreading, has recently been suggested to be an important component of effective cancer immunotherapy in patients.25 Taken together, these findings demonstrate the unique therapeutic potential of SV/TAA, with important implications for the design of clinical trials using oncolytic SV vectors. On the basis of our observations, we suggest a four-step model for the activation of CD8+ T-cell–mediated antitumor immunity during SV/TAA therapy (induction, cytotoxicity, epitope spreading, and memory), providing a framework for future studies involving SV/TAA and similar therapeutic approaches.
Results
SV/LacZ inhibits the growth of LacZ-expressing tumors in immunocompetent mice
In order to evaluate the use of SV vectors carrying TAAs for cancer therapy, we used a LacZ-expressing mouse colon cancer cell line (CT26.CL25) as a model tumor-TAA system. Initially, we tested SV/TAA (SV/LacZ) efficacy in mice bearing subcutaneous (s.c.) tumors. As seen in Figure 1a, SV/LacZ significantly inhibited the growth of LacZ-expressing CT26.CL25 tumors, whereas the control vector SV/Fluc had no observable therapeutic effect (Figure 1a, left panel). On the other hand, both SV/LacZ and SV/Fluc had little effect on the growth of LacZ-negative CT26.WT tumors (Figure 1a, right panel). These results demonstrate that SV/LacZ has a powerful antigen-dependent therapeutic effect in mice bearing s.c. CT26 tumors.
Figure 1.

SV/LacZ inhibits the growth of LacZ-expressing CT26.CL25 tumors. (a) 0.5 × 106 LacZ-expressing CT26.CL25 (left panel) or LacZ-negative CT26.WT (right panel) cells were injected subcutaneously into the right flank of BALB/c mice. Starting on day 9 after tumor inoculation, mice were treated intraperitoneally with SV/LacZ, control SV/Fluc vectors, or media (Mock). Tumor volume (mm3) was measured and plotted (N = 3–4). Data are representative of at least two independent experiments. (b) Kaplan–Meier survival plots of mice bearing peritoneal CT26.CL25 tumors. 2.5 × 104 CT26.CL25 cells were injected intraperitoneally, and treatment started on day 4 (n = 5). Data for the SV/LacZ and mock groups are representatives of two independent experiments. (c) Representative bioluminescence images of SV/LacZ- and control-treated mice bearing lung CT26.CL25.Fluc tumors are shown (left panel). Relative tumor growth (top right panel) was determined by normalizing the luminescence to the first image (day 2) for each individual mouse, and survival rates were plotted (bottom right panel) (n = 5–8). Data are representative of two independent experiments. Data in a and c are expressed as mean ± SEM. *P < 0.05 **P < 0.01. SV, Sindbis viral vector.
In order to investigate SV/LacZ efficacy in a physiologically relevant model of colon cancer, we injected CT26.CL25 cells i.p. to mimic peritoneal carcinomatosis.26 Therapeutic efficacy in this model was assessed by monitoring mouse survival. As in the s.c. model, SV/LacZ was found to have a potent therapeutic effect against these tumors, whereas the control vector (SV/Fluc) had only a minor therapeutic effect (Figure 1b). Next, we evaluated SV/LacZ efficacy against tumors growing in the lung. To supplement our survival data in this model, we constructed Fluc-expressing CT26 cell lines (CT26.CL25.Fluc and CT26.WT.Fluc), which can be used to monitor tumor growth noninvasively using the IVIS imaging system.16 Intravenous injection of Fluc-expressing CT26.CL25 cells produced lung tumors, and we found that SV/LacZ induced complete tumor remission and long-term survival in this model, whereas the control vector, SV/green fluorescent protein (GFP), only slightly delayed tumor growth and did not result in long-term survival (Figure 1c). As in the s.c. tumor model, the enhanced therapeutic effect obtained from SV/LacZ in the lung tumor model was dependent on the expression of the TAA (LacZ) from both the vector and the tumor cells, as LacZ-negative CT26.WT tumor growth was only slightly inhibited by SV/LacZ (Supplementary Figure S1). Notably, the same SV/LacZ vector preparation was used in mice bearing CT26.CL25 and CT26.WT tumors, demonstrating that any differences between vector preparations are not responsible for the enhanced therapeutic effect of SV/LacZ. Taken together, these results demonstrate that SV vectors carrying a TAA induce a potent therapeutic effect in mice bearing TAA-expressing CT26 tumors, regardless of the site of tumor growth.
Mediastinal lymph nodes transiently express antigens delivered by SV vectors and are a site of early T-cell activation during SV therapy
We have previously shown that SV vectors have oncolytic potential and can target certain tumors in vivo.16 In order to evaluate the role of tumor cell targeting in the therapeutic effect observed in the CT26 tumor model, we treated tumor-bearing mice with SV/Fluc vectors, which can be used to monitor vector localization in mice.16 We found that even after multiple injections, SV vectors did not target s.c. growing CT26.CL25 tumors (Figure 2a, left panel). Similarly, the vectors did not target lung tumors; instead, SV/Fluc was seen in the peritoneal fat of tumor-bearing mice 24 hours after the first injection and in the liver, 5 days later (Supplementary Figure S2). This general pattern was not dependent on the presence of tumor cells and occurred in tumor-free mice as well (Supplementary Figure S2). These results are consistent with other studies demonstrating that CT26 cells are not infected by SV in vitro,20 and suggest that the powerful therapeutic effect obtained from SV/LacZ is not dependent on tumor cell targeting.
Figure 2.

TAA expression and T-cell activation occur in the mediastinal lymph nodes. (a) Subcutaneous CT26.CL25 tumor-bearing mice (left panel) or tumor-free mice (right panel) were treated intraperitoneally with SV/Fluc. Three hours after the fifth (left panel) or first (right panel) treatment, bioluminescent images were taken to monitor Fluc expression from the vector. To determine the source of the upper body signal, the MLN was extracted and imaged separately (right panel). Red circles in the left panel indicate the location of the subcutaneous tumor in each mouse. (b) Mice bearing lung CT26.CL25.Fluc tumors were treated with SV/LacZ. Twenty-four hours later, mediastinal and inguinal lymph nodes were extracted and stained to determine the percentage of T cells (CD3 positive, MHC class II negative) in the lymph nodes. Representative plots (left panel) and their quantification (right panel; n = 3) are shown. (c) The expression of CD69 on CD8+ T cells extracted from mediastinal and inguinal lymph nodes of lung tumor-bearing mice 24 hours after intraperitoneal SV/LacZ injection was analyzed. Representative flow cytometry plots (left panel) and bar graphs showing the percentage of CD69-high cells (right panel; n = 3) are shown. Data in b and c are representative of two independent experiments (the second experiment was done in mice bearing intraperitoneal tumors) and are expressed as mean ± SEM. *P < 0.05; **P < 0.01. Fluc, firefly luciferase; ILN, inguinal lymph node; MLN, mediastinal lymph node; S/L, SV/LacZ (SV, Sindbis viral vector); TAA, tumor-associated antigen.
Interestingly, by focusing on very early time points after SV/Fluc injection, we noticed that a transient Fluc signal can be seen in the upper body as early as 3 hours after i.p. SV/Fluc injection (Figure 2a and Supplementary Figure S2). By extracting the MLN and imaging them separately, we determined that the upper body signal originated from these lymph nodes (Figure 2a, right panel). Notably, transient Fluc expression in the MLN occurred in both tumor-bearing and tumor-free mice (Supplementary Figure S2). The MLN has previously been shown to drain the peritoneum27,28 and represents an environment in which antigens delivered by SV vectors (such as Fluc, LacZ, or other TAAs) can potentially be processed and presented to T cells by antigen-presenting cells in the context of SV viral danger signals such as double-stranded RNA.22 The MLN therefore provides a possible location for the induction of an immune response to SV/TAA. Consistent with this hypothesis, the number of T cells in the MLN significantly increased 24 hours after SV/LacZ treatment (Figure 2b). As a control lymph node, we used the inguinal lymph nodes (ILN), which do not directly drain the peritoneal cavity,29 and were not targeted by i.p. injection of SV/Fluc (Figure 2a and additional data not shown). Unlike the MLN, there was no increase in T cells in the inguinal lymph nodes 24 hours after SV/LacZ injection (Figure 2b). In addition to the apparent influx of T cells into the MLN, the expression of CD69, which is an early activation marker of T cells, was highly induced on CD8+ T cells in the MLN 24 hours after SV/LacZ treatment (Figure 2c). By contrast, CD8+ T cells from the control inguinal lymph nodes were significantly less activated, though a slight increase in CD69 expression was observed in these cells (Figure 2c). Taken together, Figure 2 demonstrates that tumor cell targeting is not required for effective SV/LacZ therapy, and suggests that immune cell activation during SV/LacZ therapy may occur far away from the tumor site, e.g. in lymph nodes that drain the SV injection site.
SV/LacZ treatment induces a robust activation of CD8+ T cells
Because we observed the activation of CD8+ T cells in lymph nodes draining the SV injection site, we anticipated that activated CD8+ T cells might subsequently migrate into the injection site in the peritoneum. Using flow cytometry, we confirmed that a large number of CD8+ T cells influx into the peritoneum by 7 days after the first SV/LacZ injection (Figure 3a). These peritoneal CD8+ T cells were activated, as evidenced by their upregulation of NKG2D30 and downregulation of lymph node–homing receptor L-selectin31 (Figure 3b). In addition to the robust influx of activated CD8+ T cells into the peritoneum, a small number of NKG2D-high, L-selectin–low CD8+ T cells could also be seen in the lungs of mice bearing lung CT26.CL25 tumors that were treated with SV/LacZ (Figure 3b).
Figure 3.

SV/LacZ induces potent CD8+ T-cell response. (a) Lung CT26.CL25.Fluc tumor-bearing mice were treated with SV/LacZ or media (Mock). Seven days later, peritoneal cells were analyzed. Representative flow cytometry plots (left panel) and the calculated number of CD8+ T cells (right panel) are shown (mean ± SEM; N = 3). (b) CD8+ T cells from the peritoneum and the lungs were further analyzed to determine their activation state, using NKG2D and L-selectin as activation markers. Representative flow cytometry plots (left panel) and the calculated percentage of activated (NKG2D high, L-selectin low) cells (right panel) are shown (mean ± SEM; N = 3). *P < 0.05; **P < 0.01. SV, Sindbis viral vector.
SV/LacZ treatment induces LacZ-specific effector and memory CD8+ T cells
The fact that SV therapeutic efficacy depends on the expression of LacZ from both the vector and the tumor cells (Figure 1 and Supplementary Figure S1), in conjunction with the robust activation of CD8+ T cells observed during SV/LacZ therapy (Figures 2 and 3) suggest that CD8+ T cells may be involved in the anticancer effect of SV/LacZ in this model. Nevertheless, CD8+ T-cell activation also occurred during SV/GFP and SV/Fluc therapy (Supplementary Figure S3), even though these vectors had significantly lower therapeutic efficacy (Figure 1). We hypothesized that what distinguishes SV/LacZ from the other vectors is its ability to directly stimulate LacZ-specific CD8+ T cells that can subsequently target LacZ-expressing tumors. To demonstrate this concept, we injected SV/LacZ into a LacZ-naive tumor-free mouse, and observed a robust LacZ-specific CD8+ T-cell response in the peritoneum 4 days later (Supplementary Figure S4a). An increase in LacZ-specific CD8+ T cells was also observed in the spleens of s.c. tumor-bearing mice (Figure 4a,b), in the peritoneum of i.p. and lung tumor-bearing mice (Figure 4c), and in the lungs of lung tumor-bearing mice (Figure 4d, left panel) treated with SV/LacZ. Fewer LacZ-specific CD8+ T cells were seen in mice treated with control vectors (Supplementary Figure S4b). As expected, LacZ-specific CD8+ T cells from SV/LacZ-treated mice were characterized by an activated (NKG2D high, L-selectin low) phenotype (Figure 4d, right panel and Supplementary Figure S4a, right panel). Taken together, these results demonstrate that SV/LacZ treatment leads to the potent activation of LacZ-specific CD8+ T cells, providing a possible mechanism for the LacZ-dependent efficacy seen in Figure 1.
Figure 4.

SV/LacZ induces LacZ-specific CD8+ T-cell response. (a,b) Splenocytes from CT26.CL25 subcutaneous tumor-bearing mice were collected and analyzed 2 weeks after SV/LacZ or mock treatment started. (a) Representative tetramer plots and (b) the percentage of tetramer-positive cells are shown (n = 5). (c) Cells from the peritoneal cavity of intraperitoneal and lung tumor-bearing mice 7 days after therapy started were collected, stained, and analyzed (n = 4–5). (d) Lungs from lung tumor-bearing mice 7 days after treatment started were analyzed, and the percentage of activated (NKG2D high, L-selectin low) cells in the subsets of LacZ tetramer positive (tet(+)) and negative (tet(–)) CD8+ T cells in the lungs were analyzed and plotted in Figure 3b (n = 3). Data in b–d are expressed as mean ± SEM. *P < 0.05; **P < 0.01. SV, Sindbis viral vector.
In order to determine if a subset of the LacZ-specific CD8+ T cells generated during SV/LacZ therapy eventually develop into memory T cells, we analyzed splenocytes from SV/LacZ-treated long-term–surviving mice that bore i.p. CT26.CL25 tumors. Using LacZ tetramers in combination with the memory marker CD127, we identified a population (~1% of the CD8+ T-cell splenocyte population) of LacZ-specific, CD127+ memory CD8+ T cells in these mice, more than 3 months after the last SV/LacZ injection. Control splenocytes from naive mice had only background levels of this population (under 0.1%) (Supplementary Figure S4c).
SV/LacZ treatment induces lymphocyte cytotoxicity against CT26.CL25 tumor cells
As shown in Figure 4d, LacZ-specific CD8+ T cells in the lungs of lung tumor-bearing mice treated with SV/LacZ appeared to be activated. In order to investigate the function of these cells, we performed an ex vivo cytotoxicity assay using lung lymphocytes obtained from lung tumor (CT26.CL25)-bearing mice receiving SV/LacZ (or SV/GFP) therapy. As shown in Figure 5a, the viability of CT26.CL25 tumor cells was significantly lower when they were cocultured with lung lymphocytes from SV/LacZ-treated mice compared with when they were cocultured with lymphocytes from mock- or SV/GFP-treated mice. Notably, lung lymphocytes from SV/LacZ-treated mice did not affect the viability of LacZ-negative CT26.WT tumor cells, demonstrating the antigen-specific nature of the immune response in the lung. Consistent with this result, only lung lymphocytes from SV/LacZ-treated mice that were cocultured with LacZ-expressing CT26.CL25 tumor cells showed an increase in interferon (IFN)-γ production (Figure 5b).
Figure 5.

Lymphocytes acquire LacZ-specific cytotoxicity during SV/LacZ therapy. Lung lymphocytes were extracted from CT26.CL25.Fluc lung tumor-bearing mice 7 days after mock (−), SV/GFP (G), or SV/LacZ (L) treatment started. Extracted lung lymphocytes were cocultured with CT26.CL25.Fluc cells (CT26.CL25) or CT26.WT.Fluc cells (CT26.WT) for 2 days to determine (a) the cytotoxicity of lung lymphocytes against each tumor cell population and (b) IFN-γ secretion from the lung lymphocytes in response to coculture with each tumor cell population, as described in materials and methods (data in a and b are expressed as mean ± SD, n = 3). **P < 0.01 (significantly different from mock). ND, not detected; SV, Sindbis viral vector.
CD8+ T cells are required for the antigen-specific enhanced therapeutic effect of SV/LacZ
The results of the cytotoxicity and IFN-γ secretion assays (Figure 5) are consistent with the in vivo observation that SV/LacZ has a significantly stronger therapeutic effect against CT26.CL25 tumors than control vectors (Figure 1), and with the observation that SV/LacZ induces a powerful LacZ-specific CD8+ T-cell response in tumor-bearing mice (Figure 4). Taken together, these results strongly suggest the involvement of CD8+ T cells in the antigen-specific benefits of SV/TAA therapy. In order to directly determine the role of CD8+ T cells in the therapeutic effects observed, we depleted the CD8+ T-cell population in mice bearing s.c. (Figure 6a), peritoneal (Figure 6b), and lung (Figure 6c) tumors, and confirmed that in the absence of CD8+ T cells, the therapeutic efficacy of SV/LacZ was greatly reduced in all three models.
Figure 6.
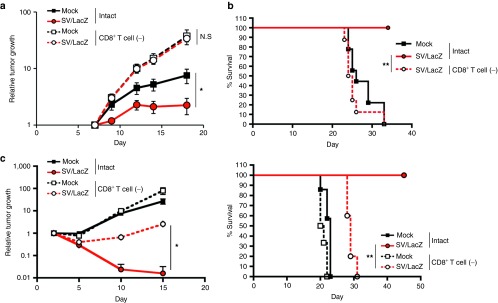
CD8+ T cells are required for the enhanced therapeutic effect of SV/LacZ. The therapeutic effect of SV/LacZ was compared between intact and CD8+ T cell depleted (CD8+ T cell (−)) mice in the (a) subcutaneous, (b) intraperitoneal, and (c) lung tumor models. (a) The size of CT26.CL25 subcutaneous tumors at indicated time points was measured and plotted for each group (n = 5). (b) Survival rates in CT26.CL25 intraperitoneal tumor-bearing mice were monitored and plotted as Kaplan–Meier survival plots (n = 8–9). (c) Tumor growth (left panel) and survival rates (right panel) in CT26.CL25.Fluc lung tumor-bearing mice were analyzed. Relative tumor growth was quantified as in Figure 1c, and survival rates are shown as Kaplan–Meier survival plots (n = 5). Data in a and c are expressed as mean ± SEM. *P < 0.05; **P < 0.01. NS, not significant; SV, Sindbis viral vector.
SV/LacZ therapy induces epitope spreading
Surprisingly, we found that, unlike lung lymphocytes, splenocytes from SV/LacZ-treated tumor-cured mice acquired cytotoxicity against not only CT26.CL25 cells, but also LacZ-negative CT26.WT cells (Figure 7a). Consistently, an increase in IFN-γ production was observed when these splenocytes were cocultured with CT26.WT cells, although the extent of the production was lower than when they were cocultured with CT26.CL25 cells (Figure 7b). On the basis of these results, we hypothesized that SV/LacZ-treated tumor-cured mice might have acquired resistance to LacZ-negative CT26.WT tumors. To determine if this was the case, we injected CT26.WT cells i.v. (Figure 7c) or i.p. (data not shown) into SV/LacZ-treated tumor-cured mice, and found that the tumors did not grow. By contrast, tumor growth was readily observed in control (naive) mice. These results suggest that an immune response to endogenous CT26 tumor antigens might have developed as a consequence of SV/LacZ therapy, a concept known alternatively as epitope spreading, antigen spreading, determinant spreading, or antigen cascade.32 To confirm that epitope spreading occurred during SV/LacZ therapy, we focused on gp70, which is an endogenous CT26 TAA. As shown in Figure 7d, an increase in IFN-γ secretion from splenocytes taken from SV/LacZ-treated tumor-cured mice was observed after culturing these cells with either gp70 or LacZ peptides, whereas, no peptide induced IFN-γ secretion from naive splenocytes. These results indicate that splenocytes from SV/LacZ-treated tumor-cured mice could respond to endogenous CT26 TAAs such as gp70 in addition to LacZ. Consistent with this observation, flow cytometry analysis using gp70 tetramers demonstrated that the number of gp70-specific CD8+ T cells was increased in the spleens of SV/LacZ-treated tumor-cured mice compared with naive mice (Figure 7e). Taken together, these results indicate that SV/LacZ therapy against CT26.CL25 tumors induced epitope spreading, which led to the development of immunity against other antigen(s) expressed on the CT26 tumors.
Figure 7.

Immunity against endogenous CT26 TAAs develops during SV/LacZ therapy. (a,b) Splenocytes were extracted from CT26.CL25.Fluc lung tumor-bearing mice at 7 days after mock (−), SV/GFP (G), or SV/LacZ (L) treatment started. Extracted splenocytes were cocultured with CT26.CL25.Fluc (CT26.CL25) or CT26.WT.Fluc (CT26.WT) cells for 2 days to determine (a) the cytotoxicity of the splenocytes toward each tumor cell population and (b) IFN-γ secretion from the splenocytes in response to coculture with each tumor cell population, as described in materials and methods (mean ± SD, n = 3). (c) CT26.WT.Fluc tumor was inoculated intravenously into naive and CT26.CL25 SV/LacZ-treated tumor-cured mice at more than 60 days after the last SV/LacZ treatment, and tumor growth in the lung was analyzed at the indicated time points by bioluminescent imaging. The left panel shows representative bioluminescence images of two independent experiments. The right panel shows the quantification of tumor bioluminescence at the indicated time points (mean ± SEM; n = 8. (d) CT26.WT.Fluc tumors were inoculated intravenously into naive (N) and SV/LacZ-treated tumor-cured mice (S) at more than 30 days after the last SV/LacZ treatment. Eight days after tumor inoculation, splenocytes were extracted from each mouse and incubated with LacZ, gp70, or control peptides for 3 days. After the incubation, LacZ- or gp70-specific induction of IFN-γ secretion was analyzed as described in materials and methods (mean ± SEM, n = 3). (e) The number of gp70-specific CD8+ T cells in splenocytes extracted in (d) was quantified by flow cytometry using gp70 tetramers (mean ± SD; n = 3). *P < 0.05, **P < 0.01 (significantly different from mock or naive). ND, not detected; SV, Sindbis viral vector.
Discussion
In this study, we used a mouse cancer-TAA system to investigate the use of SV vectors carrying TAAs for cancer therapy and made the following key observations: (i) SV represents a potentially powerful therapeutic platform for the immunogenic delivery of TAAs, (ii) the therapeutic benefit obtained from SV/TAA does not necessarily require the direct targeting of tumor cells, (iii) SV/TAA therapy involves transient early delivery of the TAA to lymph nodes draining the injection site, in particular, the MLN in the case of i.p. SV injection, (iv) SV/TAA therapy induces a potent TAA-specific CD8+ T-cell response, that is subsequently redirected against tumor cells expressing the cognate TAA, (v) SV/TAA therapy leads to epitope spreading, providing a possible solution to the problem of tumor escape by TAA loss or modification, and (vi) SV/TAA therapy ultimately leads to long-term survival of tumor-bearing mice and to the generation of long-lasting memory CD8+ T cells against multiple TAAs.
Over the past few decades, a variety of methods have been developed for the immunogenic delivery of TAAs, including the employment of vectors that target antigen-presenting cells33 or are directly injected into lymph nodes.34 Here, we demonstrate that a single i.p. injection of SV/TAA leads to the rapid immunogenic delivery of TAAs to the MLN. TAA expression in the MLN is transient, and probably would have remained unnoticed without the use of the sensitive IVIS imaging system. I.p. injections are frequently used in animal studies and are becoming increasingly common in the clinic.35 Our observation of transient TAA expression and subsequent T-cell activation at this site (Figure 2) may therefore have broad implications for the development of cancer immunotherapies. In this context, Hsu et al. have recently demonstrated that i.p.-injected cytomegalovirus resulted in the productive infection of CD169+ macrophages in the MLN.28 Consistent with this, depletion of macrophages substantially reduced the expression of SV-derived heterologous protein in the MLN (unpublished data). Notably, however, the induction of anti-TAA CD8+ T-cell immunity was not significantly inhibited in macrophage-depleted mice that were treated with SV/LacZ (unpublished data). This discrepancy may be resolved by the observation that while both macrophages and dendritic cells (DC) express viral antigens in draining lymph nodes, only DC efficiently present these antigens to naive CD8+ T cells.36 Another possible explanation is the fact that additional lymph nodes, besides the MLN, drain the peritoneal cavity. Indeed, transient heterologous protein expression was also observed in the abdominal cavity of SV-treated mice (Supplementary Figure S2, top panel). Further studies are needed, and are underway, to clarify the role of TAA delivery to the MLN during SV/TAA therapy.
One possible future application of SV targeting of the MLN is the development of dual expression SV vectors that deliver TAAs in conjunction with appropriate immune-stimulating cytokines to create optimal lymph node conditions for T-cell stimulation. Moreover, because transient MLN TAA expression can be induced multiple times by injecting SV/TAA every 3–4 days (unpublished data), different cytokines can be delivered at different stages of SV/TAA therapy to further tailor the antitumor immune response. We have previously demonstrated that SV vectors carrying IL-12 have an enhanced therapeutic effect in tumor-bearing mice16 and promote the IFNγ-dependent activation of M1 type macrophages.19 However, the effects of IL-12 delivery to the MLN have not specifically been investigated. Another potential future application of MLN targeting is the use of SV vectors to target and/or to deliver payloads to mediastinal masses such as those derived from certain neurogenic tumors.37
Besides the activation of T cells in the MLN, there appears to be a systemic redistribution of CD8+ T cells early after SV/TAA injection. Various tissues, including the peritoneum (Supplementary Figure S3a) and the lung (Supplementary Figure S5) show a reduction in CD8+ T cells in the first 1–2 days after SV/TAA injection. The apparent efflux of T cells from these tissues coincides with their influx into the MLN (Figure 2b). It is interesting to note that during this early phase, lung tumors in SV/TAA-treated mice already appear to shrink (Figure 1c). Moreover, this early therapeutic effect was also observed in mice treated with control vectors that do not express the TAA (Figure 1c), in SV-treated mice bearing tumors that do not express the TAA (Supplementary Figure S1), and in SV/TAA-treated mice that were depleted of CD8+ T cells (Figure 6c). One possible explanation for this is the activation of NK cells by SV. We have previously shown that SV therapy induces a robust NK cell response in tumor-bearing mice.19 In the CT26 lung model, a rapid influx of NKG2D-expressing NK cells into the lung was observed as early as 2 days after SV injection, several days before the maximum influx of TAA-specific CD8+ T cells (Supplementary Figure S5). A detailed analysis of the NK-cell response in the lung and other tissues will be the subject of future investigations.
One of the limitations of cancer vaccine strategies is that the inherent heterogeneity and genomic instability of tumor cell populations, coupled with the selective pressure induced by the treatment, can lead to tumor evasion by loss or modification of the TAA used in the vaccine.38,39 In this context, an interesting and therapeutically significant observation is the induction of epitope spreading, i.e., the expansion of the antitumor T-cell response to incorporate novel TAAs that are endogenous to the tumor but not delivered by the vector32 during SV/TAA therapy (Figure 7). The precise mechanism underlying epitope spreading in our model is not yet known. For example, it is not clear why epitope spreading was observed in splenocytes (Figure 7), but not lung lymphocytes (Figure 5), 7 days after the first SV injection. One possible explanation for these results is that the influx of new CD8+ T cells into the lungs was limited by day 7, because the tumors were already largely eliminated from the lungs by this point (Figure 1c and Supplementary Figure S1a). Epitope spreading is currently being evaluated in clinical trials, and a positive correlation between this phenomenon and therapeutic efficacy has been observed.25,40 These developments may signify a paradigm shift in the design of cancer vaccines, whereby an emphasis would be placed on the induction of a strong diversified T-cell response that could potentially be effective even against tumors with heterogeneous antigen expression.
In summary, this study represents a proof of concept for the use of SV/TAA for cancer therapy and provides valuable insight into the mechanisms underlying SV/TAA efficacy. A simplified four-step model for the activation of CD8+ T cells during SV/TAA therapy is depicted in Figure 8, which can be used to guide future studies. Notably, using SV vectors that carry a TAA not only greatly enhances SV efficacy, but also abrogates the need for tumor cell targeting—a hitherto prerequisite for effective oncolytic SV therapy—thereby paving the way for a much broader application of SV anticancer therapy. The current findings, which stem from our previous investigation into the oncolytic potential of SV,15,16 compliment and expand upon earlier studies on the use of SV nucleic acid41 and replicon particle42 vaccines and illustrate the versatility of SV anticancer therapy. Future work will focus on protocol optimization (e.g., by altering the route and frequency of injection) and provide a more detailed mechanistic analysis of SV/TAA efficacy.
Figure 8.

Four-step model for the activation of CD8+ T cells during SV/TAA therapy. Step 1: Intraperitoneal injection of SV/LacZ results in transient immunogenic expression of LacZ in the mediastinal lymph nodes (dark blue arrow), followed by the induction of T-cell activation at this site and/or in alternative locations (light blue arrow). Natural killer cells are also activated against the tumor cells (brown arrow). Step 2 (red arrow): LacZ-specific CD8+ T-cell cytotoxicity results in the destruction of tumor cells and the subsequent release of tumor associated antigens such as LacZ and gp70. Step 3 (green arrows): Antigen-presenting cells capture and present these antigens to CD8+ T cells in the tumor-draining lymph nodes, resulting in epitope spreading, including the induction of gp70-specific CD8+ T cells that can potentially target LacZ(−) tumor cell escape variants. Step 4 (purple arrows): memory CD8+ T cells against a variety of tumor-associated antigens are generated. APC, antigen-presenting cell; LN, lymph node; MLN, mediastinal lymph node; NK, Natural killer cell; SV, Sindbis viral vector; TAA, tumor-associated antigen; Tc, cytotoxic CD8+ T cell; Tm, memory CD8+ T cell.
Materials and Methods
Cell lines. Baby hamster kidney (BHK), CT26.WT, and LacZ-expressing CT26.CL25 cells were obtained from the American type culture collection. Firefly luciferase (Fluc)-expressing CT26 cells (CT26.WT.Fluc and CT26.CL25.Fluc) for noninvasive bioluminescent imaging were generated by stable transfection of a Fluc-expressing plasmid into CT26.WT and CT26.CL25 cells. The Fluc-expressing plasmid was constructed by introducing a SV40 promoter sequence into the multicloning site of pGL4.20 vector (Promega, Madison, WI).
Cell culture. BHK cells were maintained in minimum essential a-modified media (a-MEM) (Mediatech, VA) with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, GA). CT26.WT, CT26.CL25, CT26.WT.Fluc, and CT26.CL25.Fluc cells were maintained in Dulbecco-modified essential media (DMEM) containing 4.5 g/l glucose (Mediatech) supplemented with 10% FBS. All basal media were supplemented with 100 mg/ml of penicillin–streptomycin (Mediatech) and 0.5 mg/ml of amphotericin B (Mediatech). For culturing CT26.CL25 and CT26.CL25.Fluc cells, 0.4 mg/ml of G418 sulfate (Mediatech, Manassas, VA) was added to the basal media. For culturing CT26.WT.Fluc and CT26.CL25.Fluc cells, 5 μg/ml of puromycin (Sigma-Aldrich, St Louis, MO) was added to the basal media.
SV/TAA production. SV/LacZ was used as an immunogenic SV/TAA agent, and SV/Fluc and SV/GFP were used as control vectors. SV/Fluc was also used for imaging experiments (see below). Vectors were produced as previously described.16 Briefly, plasmids carrying the replicon (SinRep5-LacZ, SinRep5-GFP, or SinRep5-Fluc) or DHBB helper RNAs (SinRep5-tBB) were linearized with XhoI (for SinRep5-LacZ, SinRep5-GFP, and SinRep5-tBB) or PacI (for SinRep5-Fluc). In vitro transcription was performed using the mMessage mMachine RNA transcription kit (Ambion, Austin, TX). Helper and replicon RNAs were then electroporated into BHK cells and incubated at 37 °C in α-MEM supplemented with 10% FBS. After 12 hours, the media was replaced with OPTI-MEM I (Invitrogen, Carlsbad, CA), supplemented with CaCl2 (100 µg/ml), and cells were incubated at 37 °C. After 24 hours, the supernatant was collected, centrifuged to remove cellular debris, and frozen at −80 °C. Vector titers were determined as previously described15 and were similar in all three vectors (SV/LacZ, SV/Fluc, and SV/GFP).
Mice and tumor inoculation. Four- to eight-week-old female BALB/c mice were purchased from Taconic (Germantown, NY). For the s.c. tumor model, 0.5 × 106 or 1 × 106 CT26.WT or CT26.CL25 cells in 0.2 ml phosphate-buffered saline (PBS) were injected s.c. into the right flank of each mouse. For the i.p. tumor model, 2.5 × 104 or 5 × 104 CT26.CL25 cells in 0.2 ml PBS were injected i.p. into each mouse. For the lung tumor model, 0.3 × 106 CT26.WT.Fluc or CT26.CL25.Fluc cells in 0.2 ml PBS were injected i.v. into each mouse.
Therapeutic efficacy. In the s.c. tumor model, treatment started after tumor volume was more than 40 mm3 (volume = width × width × length/2). In the i.p. tumor model, treatment started on day 4 after tumor cell inoculation. In the lung tumor model, treatment started on day 3 after tumor cell inoculation. SV/LacZ, SV/GFP, or SV/Fluc (~107 plaque-forming units in 0.5 ml of OPTI-MEM I) and mock treatments (0.5 ml of OPTI-MEM I supplemented with 100 mg/l CaCl2) were administered i.p. four times a week for 2 weeks, for a total of eight treatments. Therapeutic efficacy was monitored in three ways: tumor volume (for s.c. tumors, measured with mechanical calipers), tumor luminescence (for lung tumors), and survival (for i.p. and lung tumors). Noninvasive bioluminescent imaging was done using the IVIS Spectrum imaging system (Caliper Life Sciences, Alameda, CA), and tumor growth was quantified using the Living Image 3.0 software (Caliper Life Sciences), as previously described.16 Survival was monitored and recorded daily.
Bioluminescent imaging of SV/Fluc. Tumor-bearing and tumor-free mice were injected with SV/Fluc (~107 plaque-forming units in 0.5 ml of OPTI-MEM I 0.5ml) i.p. After the treatment, bioluminescence signal was detected by IVIS at the indicated time points as previously described.16
Ex vivo cytotoxicity assay. Lung lymphocytes or splenocytes from tumor-bearing mice were collected 7 days after SV treatment started. Lung lymphocytes (1 × 105/ml) or splenocytes (2 × 106/ml) were cocultured with CT26.WT.Fluc cells (2 × 104/ml) or CT26.CL25.Fluc cells (2 × 104/ml) in a 24-well plate for 2 days in 1 ml RPMI 1640 supplemented with 10% FBS. Culture media were then collected for IFN-γ secretion assays, and the remaining cells in each well were washed twice with PBS. Cells were then lysed with 100 µl of M-PER Mammalian Protein Extraction Reagent (Pierce, Rockford, IL) per well. Cytotoxicity was assessed based on the viability of the CT26 cells, which was determined by measuring the luciferase activity in each well. Luciferase activity was analyzed by adding 100 µl of Steady-Glo reagent (Promega) to each cell lysate and measuring the luminescence using a GLOMAX portable luminometer (Promega).
IFN-γ secretion assay. Lung lymphocytes (1 × 105/ml) or splenocytes (2 × 106/ml) were stimulated by CT26 tumor cells (2 × 104/ml) or immunogenic peptides (5 µg/ml) in a 24-well plate in 1 ml RPMI 1640 (Mediatech) supplemented with 10% FBS. The peptides used were the LacZ peptide TPHPARIGL,43 the gp70 peptide SPSYVYHQF,44 or the P1A peptide LPYLGWLVF as a negative control.45 After stimulation, IFN-γ levels in the media were measured using a mouse IFN-γ Quantikine ELISA kit (R&D systems, Minneapolis, MN). TPHPARIGL and SPSYVYHQF-mediated increase in IFN-γ secretion was calculated by subtracting the IFN-γ levels in the control (LPYLGWLVF stimulated) samples from the IFN-γ levels in the TPHPARIGL- and SPSYVYHQF-stimulated samples.
Flow cytometry. Antimouse antibodies anti-CD8a eFluor 450 and eFluor 650NC, anti-CD4 PE-Cyanine7, anti-CD69 PE, anti-CD314 (NKG2D) PE-Cyanine7, anti-CD62L (L-selectin) FITC and Alexa Fluor 700, and anti-CD45 eFluor 450 were purchased from eBioscience (San Diego, CA). PE-labeled LacZ tetramers were obtained from the NYU Vaccine and Cell Therapy Core (New York, NY), and allophycocyanin-labeled gp70 tetramers were obtained from the NIH Tetramer Core Facility (Atlanta, Georgia). For flow cytometry analysis of lung lymphocytes and splenocytes, mice were killed and their lungs and spleens were extracted. The extracted lungs were chopped into small pieces and incubated with a digestion mix (collagenase I (50 µg/ml), collagenase IV (50 µg/ml), hyaluronidase V (25 µg/ml), and DNAse I (20 U/ml)) for 30 minutes at 37 °C. Extracted spleens and digested lungs were then mashed through 70–100 µm cell strainers, followed by a treatment with 1× RBC lysis buffer (eBioscience) to eliminate red blood cells. Peritoneal cells were collected from peritoneal exudates as previously described.19 Cells were then stained with various Abs, washed twice with HBSS (Mediatech), and analyzed using an LSR II machine (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo (Tree Star, San Carlos, CA).
CD8+ T-cell depletion. CD8+ T cells were depleted using anti-CD8 antibody (clone 2.43) (Bio X cell, Lebanon, NH). 0.4 mg antibody in 0.2 ml PBS was injected into each mouse, starting 1 day before the first SV treatment, and then every 2–3 days for 2 weeks. Control mice were injected with PBS.
Ethics statement. All mice experiments were approved by the institute of Animal Care and Use Committee at the New York University Medical Center (protocol number 090413-03).
Statistics. For flow cytometry, IVIS imaging, ELISA, tumor growth, and survival experiments, Student's t tests (two-tailed), analysis of variance (ANOVA) followed by Dunnett's test, or Kaplan–Meier log-rank test were done using Prism 4 for Macintosh (GraphPad Software, La Jolla, CA).
SUPPLEMENTARY MATERIAL Figure S1. The enhanced therapeutic effect of SV/LacZ in mice bearing lung tumors is dependent on LacZ expression on the tumors. Figure S2. SV does not target CT26 tumors in the lung. Figure S3. SV/Fluc and SV/GFP induce CD8+ T-cell response. Figure S4. SV/TAA induces the activation of effector and memory LacZ-specific CD8+ T cells. Figure S5. NK cells are activated at an early stage of SV therapy.
Acknowledgments
We thank Dr. Christine Pampeno and Vincent DiGiacomo for critical reading of this manuscript. This work was supported by the US Public Health grants CA100687 from the National Cancer Institute, National Institutes of Health, and Department of Health and Human Services. The contents of this paper are being utilized for a patent. According to the rules and regulations of New York University School of Medicine, if this patent is licensed by a third party, the authors may receive benefits in the form of royalties or equity participation.
Supplementary Material
References
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- Wein LM, Wu JT, Kirn DH. Validation and analysis of a mathematical model of a replication-competent oncolytic virus for cancer treatment: implications for virus design and delivery. Cancer Res. 2003;63:1317–1324. [PubMed] [Google Scholar]
- Vähä-Koskela MJ, Le Boeuf F, Lemay C, De Silva N, Diallo JS, Cox J, et al. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J Virol. 2013;87:2363–2366. doi: 10.1128/JVI.01609-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildner O, Blaese RM, Morris JC. Therapy of colon cancer with oncolytic adenovirus is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res. 1999;59:410–413. [PubMed] [Google Scholar]
- Hermiston TW, Kuhn I. Armed therapeutic viruses: strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002;9:1022–1035. doi: 10.1038/sj.cgt.7700542. [DOI] [PubMed] [Google Scholar]
- Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng JC, Levin B, Hirano T, Yee H, Pampeno C, Meruelo D. In vivo antitumor activity of Sindbis viral vectors. J Natl Cancer Inst. 2002;94:1790–1802. doi: 10.1093/jnci/94.23.1790. [DOI] [PubMed] [Google Scholar]
- Tseng JC, Levin B, Hurtado A, Yee H, Perez de Castro I, Jimenez M, et al. Systemic tumor targeting and killing by Sindbis viral vectors. Nat Biotechnol. 2004;22:70–77. doi: 10.1038/nbt917. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Bergmann CC, Takita-Sonoda Y, Murata K, Rodrigues EG, Nussenzweig RS, et al. Recombinant Sindbis viruses expressing a cytotoxic T-lymphocyte epitope of a malaria parasite or of influenza virus elicit protection against the corresponding pathogen in mice. J Virol. 1998;72:6907–6910. doi: 10.1128/jvi.72.8.6907-6910.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng JC, Hurtado A, Yee H, Levin B, Boivin C, Benet M, et al. Using sindbis viral vectors for specific detection and suppression of advanced ovarian cancer in animal models. Cancer Res. 2004;64:6684–6692. doi: 10.1158/0008-5472.CAN-04-1924. [DOI] [PubMed] [Google Scholar]
- Granot T, Venticinque L, Tseng JC, Meruelo D. Activation of cytotoxic and regulatory functions of NK cells by Sindbis viral vectors. PLoS ONE. 2011;6:e20598. doi: 10.1371/journal.pone.0020598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PY, Guo JH, Hwang LH. Oncolytic Sindbis virus targets tumors defective in the interferon response and induces significant bystander antitumor immunity in vivo. Mol Ther. 2012;20:298–305. doi: 10.1038/mt.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenbeek PJ, Frolov I, Rice CM, Schlesinger S. Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J Virol. 1993;67:6439–6446. doi: 10.1128/jvi.67.11.6439-6446.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Leitner WW, Hwang LN, deVeer MJ, Zhou A, Silverman RH, Williams BR, et al. Alphavirus-based DNA vaccine breaks immunological tolerance by activating innate antiviral pathways. Nat Med. 2003;9:33–39. doi: 10.1038/nmxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TC, Burns SM, Contag CH. In vivo bioluminescence imaging for integrated studies of infection. Cell Microbiol. 2004;6:303–317. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- Corbière V, Chapiro J, Stroobant V, Ma W, Lurquin C, Lethé B, et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res. 2011;71:1253–1262. doi: 10.1158/0008-5472.CAN-10-2693. [DOI] [PubMed] [Google Scholar]
- Lopes Cardozo AM, Gupta A, Koppe MJ, Meijer S, van Leeuwen PA, Beelen RJ, et al. Metastatic pattern of CC531 colon carcinoma cells in the abdominal cavity: an experimental model of peritoneal carcinomatosis in rats. Eur J Surg Oncol. 2001;27:359–363. doi: 10.1053/ejso.2001.1117. [DOI] [PubMed] [Google Scholar]
- Tilney NL. Patterns of lymphatic drainage in the adult laboratory rat. J Anat. 1971;109 Pt 3:369–383. [PMC free article] [PubMed] [Google Scholar]
- Hsu KM, Pratt JR, Akers WJ, Achilefu SI, Yokoyama WM. Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J Gen Virol. 2009;90 Pt 1:33–43. doi: 10.1099/vir.0.006668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- Arbonés ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Frolov I, Perri S, Ji Y, MacKichan ML, zur Megede J, et al. Infection of human dendritic cells by a sindbis virus replicon vector is determined by a single amino acid substitution in the E2 glycoprotein. J Virol. 2000;74:11849–11857. doi: 10.1128/jvi.74.24.11849-11857.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70:9031–9040. doi: 10.1158/0008-5472.CAN-10-0699. [DOI] [PubMed] [Google Scholar]
- Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- Duwe BV, Sterman DH, Musani AI. Tumors of the mediastinum. Chest. 2005;128:2893–2909. doi: 10.1378/chest.128.4.2893. [DOI] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergati M, Intrivici C, Huen NY, Schlom J, Tsang KY.2010Strategies for cancer vaccine development. J Biomed Biotechnol 2010Article ID: 596432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael MG, Benavides LC, Holmes JP, Gates JD, Mittendorf EA, Ponniah S, et al. Results of the first phase 1 clinical trial of the HER-2/neu peptide (GP2) vaccine in disease-free breast cancer patients: United States Military Cancer Institute Clinical Trials Group Study I-04. Cancer. 2010;116:292–301. doi: 10.1002/cncr.24756. [DOI] [PubMed] [Google Scholar]
- Leitner WW, Ying H, Driver DA, Dubensky TW, Restifo NP. Enhancement of tumor-specific immune response with plasmid DNA replicon vectors. Cancer Res. 2000;60:51–55. [PMC free article] [PubMed] [Google Scholar]
- Cheng WF, Hung CF, Hsu KF, Chai CY, He L, Polo JM, et al. Cancer immunotherapy using Sindbis virus replicon particles encoding a VP22-antigen fusion. Hum Gene Ther. 2002;13:553–568. doi: 10.1089/10430340252809847. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971–3980. [PubMed] [Google Scholar]
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci USA. 1996;93:9730–9735. doi: 10.1073/pnas.93.18.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Bacík I, Irvine KR, Yewdell JW, McCabe BJ, Anderson RW, et al. Antigen processing in vivo and the elicitation of primary CTL responses. J Immunol. 1995;154:4414–4422. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.