

Abstract
Self-complementary adeno-associated viral (AAV) vectors expressing human factor IX (hF.IX) have achieved transient or sustained correction of hemophilia B in human volunteers. High doses of AAV2 or AAV8 vectors delivered to the liver caused in several patients an increase in transaminases accompanied by a rise in AAV capsid-specific T cells and a decrease in circulating hF.IX levels suggesting immune-mediated destruction of vector-transduced cells. Kinetics of these adverse events differed in patients receiving AAV2 or AAV8 vectors causing rise in transaminases at 3 versus 8 weeks after vector injection, respectively. To test if CD8+ T cells to AAV8 vectors, which are similar to AAV2 vectors are fully-gutted vectors and thereby fail to encode structural viral proteins, could cause damage at this late time point, we tested in a series of mouse studies how long major histocompatibility (MHC) class I epitopes within AAV8 capsid can be presented to CD8+ T cells. Our results clearly show that depending on the vectors' genome, CD8+ T cells can detect such epitopes on AAV8's capsid for up to 6 months indicating that the capsid of AAV8 degrades slowly in mice.
Introduction
Vectors based on adeno-associated virus (AAV) expressing human factor IX (F.IX) achieved in animals including mice, dogs, and nonhuman primates sustained gene transfer and correction of disease in models of hemophilia B.1,2,3 In a phase 1 clinical trial, human subjects with severe hemophilia B were infused with recombinant AAV vectors derived from the human serotype 2 (AAV2) expressing human factor IX (AAV2-hF.IX) for intrahepatic expression. One of the two patients in the highest dose group of 2 × 1012 vector genomes (vg)/kg developed therapeutic levels of F.IX by week 2. By 4 weeks after vector infusion, levels of F.IX started to decrease and within a few weeks returned to pregene therapy levels. At the time when F.IX levels started to decrease, the patient showed an asymptomatic increase in transaminases, which eventually resolved spontaneously in a time course that paralleled the decline in F.IX levels.4 Overall, the patient's clinical course was suggestive of immune-mediated destruction of AAV-transduced hepatocytes; no such findings had been observed in animal models. The trial was continued with a reduced dose of vector. The next patient did not develop detectable levels of F.IX upon gene transfer. Nevertheless, this patient also presented with asymptomatic transaminitis, with a time course relative to therapy identical to that seen in the previous one. The second patient's T cell responses to the capsid antigen of the AAV2 vector and the transgene product were assessed carefully before and after gene transfer.4 He had no detectable AAV2 capsid-specific T cells in his peripheral blood before gene transfer. Such a response developed after gene transfer and then eventually subsided. The trial was halted as it was felt that its design was unsuited to circumvent the postulated CD8+ T cell-mediated destruction of AAV2-transduced hepatocytes. A subsequent trial for AAV-mediated correction of hemophilia used a self-complementary (sc)AAV8 vector that preclinically had achieved therapeutic levels of F.IX at lower vector doses.5 Additional data had indicated that capsid antigens of AAV8, which uses a different receptor than AAV2, might be less susceptible to recognition by CD8+ T cells,6,7 although in vitro stimulation studies suggested otherwise.8 In the scAAV8-hF.IX trial, all subjects, who received doses of vector ranging from 2 × 1010 to 2 × 1012 vg/kg, developed therapeutic levels of F.IX. At week 8, one of the subjects in the high-dose cohort presented with a marked increase in transaminases accompanied by a modest drop in F.IX and an increase in circulating AAV capsid-specific T cells as measured by IFN-γ ELISpot assays.5 The patient was treated with steroids, which resulted in a rapid decline in transaminases and a stabilization of F.IX levels. The second patient developed a very small increase in transaminases around week 9 after gene transfer and was immediately treated with steroids. Transaminases returned to baseline. In this patient, T cell responses were not informative due to low cell viability.5 In both subjects, the onset of transaminitis was markedly delayed compared to that observed in the earlier AAV2 trial where patients showed evidence of liver cell destruction after 3–4 weeks.4 This raises questions regarding the hypothesis that AAV capsid-specific CD8+ T cells were causative for the liver cell destruction, especially as earlier work had suggested that AAV8 uncoats more rapidly,9 which implies that its capsid degrades faster thus offering targets for specific CD8+ T cells for a comparatively shorter time.
We previously reported on a mouse model that allows us to track CD8+ T cell recognition of AAV capsid epitopes over time.10 These data showed that CD8+ T cells respond to the endogenous AAV2 capsid epitope within virus protein (VP)3 for at least 3 weeks after intravenous (i.v.) vector injection while responses to a foreign epitope placed into the less abundant VP2 could be observed for at least 10 weeks. Using this model, we show here that although the endogenous AAV8 epitope fails to elicit strong or sustained recall responses of CD8+ T cells, a foreign epitope within AAV capsid VP2 is recognized and elicits CD8+ T cell expansion for months. The extent and duration of the CD8+ T cell response was influenced by the vectors' genome; AAV8 vector with single-stranded genomes (ssAAV8) elicited a longer and more pronounced CD8+ T cell response compared to scAAV8 vectors. Empty AAV8 vectors that are generated during vector production and that by some but not all investigators are removed during vector purification were relatively poor stimulators of CD8+ T cells suggesting a role for innate immune responses in vector-driven expansion of specific CD8+ T cells.
Results
Quality control of AAV vectors
Insertion of sequences into the capsid of AAV may affect assembly or infectivity of virions. We routinely conduct silver stains and western blots of all AAV vectors and these results (Figure 1a,b) showed the expected banding patterns for AAV8 with wild-type or modified capsids. We incorporated 10 copies of the SIINFEKL including spacers of two amino acids in lengths between each SIINFEKL sequence. VP2 has a molecular mass of ~72 kDa and upon incorporating an additional 100 amino acids, modified VP2 would be expected to have a mass of ~85 kDa. A band of this size was detected by silver stain (Figure 1a) or western blot (Figure 1b) of the modified vectors. Some other bands not corresponding to the expected sizes of VP1-3 were seen including a fairly pronounced band in the lanes containing modified vectors that comigrated with the VP2 band of the wild-type vector. These bands may reflect unmodified VP2 or VP2 variants arising due to low-level translation from alternative start codons or low frequency occurrence of alternative splicing.11 Alternatively, the additional bands may be due to impurities unrelated to capsid.12 The banding pattern of empty vectors mirrored that of vectors containing genome. Relative ratios of capsid VP1, VP2, and VP3 were comparable for WT AAV8, ssAAV8(MOVA)-hF.IX, and ssAAV8(MOVA)-hF.IX empty particles. To examine AAV capsid quality and confirm genome-containing and empty vectors were employed, optical density measurements were performed and UV absorbance was determined. The predicted 260/280 absorbance measurement for genome-containing and empty AAV vectors is 1.50 and 0.75 nm. As expected, the ssAAV8(MOVA)-hF.IX and WT AAV8 vector samples had a UV absorbance of 1.52 and 1.50 nm, respectively, while the ssAAV8(MOVA)-hF.IX empty vector had an absorbance of 0.65 nm. To ensure that the incorporation of 10 copies of the SIINFEKL did not change vector tropism, we i.v. injected male C57Bl/6 mice with 2 × 1011 viral particles of a Gag expressing ssAAV8 or ssAAV8(MOVA)-hF.IX and isolated DNA from the liver, lung, and spleen 24 hours postadministration. Vector genomes were quantified by real-time TaqMan (Figure 2a). Vector genome levels in the different tissues were comparable in the mice administered with either vector. To ensure that capsid-modified AAV8 vectors remained functional, groups of male C57Bl/6 mice were injected i.v. with AAV8 or AAV8(MOVA) expressing hF.IX under a hepatocyte-specific promoter. Levels of hF.IX measured from plasma at different times thereafter were comparable in magnitude (Figure 2b) and upon transfer of either vector were equally sustained. These results show that the capsid modification did not affect vectors assembly or infectivity and therefore most likely had limited effects on the structure of the vectors as reported previously for another VP2-modified vectors.13 They furthermore demonstrate that the preparation of empty AAV vector that was used for our studies was composed of intact virions.
Figure 1.

Silver stain and western blots of purified AAV8 vectors with wild-type or SIINFEKL-modified capsid. (a) Silver staining of an SDS-PAGE gel of empty rAAV8, ssAAV8(MOVA)-hF.IX, and ssAAV8(MOVA)-hF.IX vectors. (b) Western blot of rAAV8, ssAAV8(MOVA)-hF.IX, and empty ssAAV8(MOVA)-h.FIX vectors.
Figure 2.

Vector tropism and in vivo transgene product expression. (a) Graphs show the levels of AAV vector genome 24 hours after intravenous transfer in liver, lungs and spleen of mice injected with an AAV8 vector with wild-type capsid (closed bars) or a SIINFEKL-modified capsid (open bars). Differences between the two vectors were not statistically significant by t test. (b) Graphs show average levels of h.FIX ± SD in groups of five male BALB/c mice on day 14 following intravenous injections with AAV8 vectors with wild-type (left) or SIINFEKL-modified (right) capsid. Differences between the groups were not statistically significant (Mann–Whitney test).
Response to the endogenous immunodominant CD8+ T cell epitope within AAV8 capsid
To assess kinetics of CD8+ T cell recognition of epitopes within AAV8 capsid, groups of Thy1.2+ BALB/c mice were injected intramuscularly with 1 × 1011 virus particles of an adenovirus human serotype 5 (Ad) vector expressing the capsid antigen of AAV8 under a cytomegalovirus promoter (Ad-AAV8cap). Splenocytes were harvested 3–4 months later and labeled with carboxyfluorescein succinimidyl ester (CFSE). Labeled splenocytes were transferred i.v. into Thy1.1+ BALB/c mice, which had been injected 2 days, or 1, 3, or 7 weeks earlier i.v. with 2 × 1011 vg of ssAAV8-hF.IX. Mice that had not been injected with ssAAV8-hF.IX but were transfused with AAV capsid-specific memory T cells served as controls. To measure recognition of the endogenous AAV8 capsid epitopes by the memory CD8+ T cells within the donor splenocyte population, recipient mice were euthanized 10 days later. Splenocytes were isolated and stained with an antibody to Thy1.2 to distinguish between donor and recipient T cells, an antibody to CD8, and a tetramer specific for one of the AAV8 capsid epitopes. Cells were analyzed by flow cytometry. Plots were gated on tetramer+CD8+Thy1.2+ cells, and loss of CFSE staining in this population was used as a measure for proliferation of AAV capsid-specific memory CD8+ T cells. The experimental scheme is shown in Figure 3. AAV capsid-specific CD8+ T cells showed marginal proliferation at the early time points for up to week 3 compared to control cells. The difference in proliferation was statistically significant after correction for type I errors as determined both by % of cells within the population that divided only in mice that received cells one week after AAV8 transfer. By week 3 after vector transfer, AAV capsid-specific CD8+ T cells no longer proliferated. Overall, even at early time points, the level of proliferation was marginal suggesting that the endogenous AAV8 capsid epitope was only poorly recognized. Figure 4a shows the summary of the results and Figure 4b shows representative histograms for the different time points.
Figure 3.

Experimental design. To test for responses to endogenous AAV capsid epitopes, splenocytes from mice immunized 3–4 months earlier with an Ad vector expressing AAV8cap were labeled with carboxyfluorescein succinimidyl ester (CFSE) and then transferred intravenously into sThy-1 congenic recipient mice that had been injected intravenously with an AAV8 vector. Recipient mice were euthanized 10 days later and CFSE-expression on donor-derived AAV capsid-specific CD8+ T cells was determined. Cells were gated according to CFSE expression to assess those that had not proliferated (generation 0, those that had proliferated once (generation 1), twice (generation 2), etc. To discount cells that had undergone homeostatic proliferation during the 10 days in recipient mice, cells that proliferated once were viewed as being part of generation 0. Cells with extensive loss of CFSE were grouped together as generation 6. Events in each gated generation were divided by the log 2 of the generation. The sum reflects total cells at the onset (before they proliferated). Events of generation 0 were subtracted from number of total cells at onset to determine number of cells that proliferated. This number divided by total cells at onset (×100) reflects percent of cells that proliferated. Events of generations 1–6 were divided by the number of the generation. Events of cells divided by the later number was used to determine the proliferation index (PI). (http://www.flowjo.com/v9/html/proliferation.html).
Figure 4.

Proliferation of AAV8 capsid-specific memory CD8+ T cells in AAV8-hF.IX-transduced mice. Thy1.1 BALB/c mice were intravenously injected with 2 × 1011 vg of AAV8-hF.IX. Mice then received 5 × 107 CFSE-labeled splenocytes from the Thy1.2 BALB/c mice, which had been immunized with 1 × 1011 vp of Ad-AAV8cap 3–4 months earlier, at day 2, weeks 1, 3, or 7 following AAV8 transfer. The proliferation of Thy1.2+CD8+AAV8-tet+ cells was tested 10 days later from the spleens of the recipient mice. Control mice did not receive AAV8-hF.IX before the adoptive transfer of splenocytes. (a) Average proliferation indices (average number of division cycles) +SD are shown on the left, average % cells proliferating + SD are shown on the right. (b) Shows representative histograms from a mouse for each time point. Numbers of mice/group were as follows: day 2 = 3 mice, week 1 = 5 mice, week 3 = 3 mice, week 7 = 3 mice, controls (combined from the different time points of this series of experiments) = 12 mice. Significance was determined by multiple Student's t test with Holm–Sidak correction for type 1 errors comparing experimental to control mice. For the proliferation indices, P values were not significant. For % cells proliferating, P values were significant for week 1 (P = 0.01). Significant differences to controls are indicated by (*).
Response to a foreign epitope placed into the capsid of a ssAAV8 vector
Lack of recognition of a single AAV8 epitope in laboratory mice may reflect destruction of this particular epitope during capsid processing and may therefore not faithfully recapitulate T cell responses in humans, which being outbred can recognize a wide array of different AAV capsid epitopes.8,14 We therefore in the next set of experiments injected Thy1.1+ C57BL/6 mice with a ssAAV8-hF.IX vector that had been capsid-modified to express 10 copies of SIINFEKL within VP2. This vector was termed ssAAV8(MOVA)-hF.IX. SIINFEKL is the immunodominant epitope of ovalbumin in H-2b mice. CFSE-labeled splenocytes from OT-1 mice,15 which are transgenic for the T cell receptor (TcR) to SIINFEKL bound to major histocompatibility class (MHC) I complexes of H-2b mice, were transferred into C57BL/6 mice that had been injected at various previous time points with ssAAV8(MOVA)-hF.IX. As in the previous experiment, recipient mice were euthanized 10 days after OT-1 splenocyte transfer and lymphocytes were isolated from spleens and liver. In this set of experiments, cells were stained with fluorochrome-labeled antibodies to Thy1.2, CD8, and Vα2; the latter is the V region used by the SIINFEKL-specific TcR of OT-1 mice. Splenocytes from individual mice and liver-derived lymphocytes that depending on recovered cell numbers were either pooled or tested from individual mice and were analyzed by flow cytometry. Cells were gated onto live Thy1.2+CD8+Vα2+ cells. Expression levels of CFSE within this cell population were used as a read-out for proliferation of SIINFEKL-specific CD8+ T cells in response to the ssAAV8(MOVA)-hF.IX vector. As shown in Figure 5a,b, SIINFEKL as expressed on the capsid of the ssAAV8(MOVA)-hF.IX vector was readily recognized by OT-1 CD8+ T cells. In all mice transferred with OT-1 splenocytes 2 days or 2 weeks after ssAAV8(MOVA)-hF.IX vector injection, the majority of splenic SIINFEKL-specific donor CD8+ T cells had proliferated so extensively that CFSE staining was no longer detected. Transfer of CFSE-labeled OT-1 spenocytes during weeks 10–12 after ssAAV8(MOVA)-hF.IX vector injection resulted in continued very high proliferation of SIINFEKL-specific donor CD8+ T cells in two out of seven mice with all of the transferred OT-1 CD8+ T cells showing complete loss of CFSE. The remaining five mice showed reduced but still pronounced proliferation. By 16–17 weeks after ssAAV8(MOVA)-hF.IX vector injection, results continued to be variable with some mice still showing pronounced proliferation while in others proliferation was modest. By weeks 20–22, splenic donor OT-1 cells of only one of the mice showed complete loss of CFSE stain and average results for the proliferation index remained highly significant for all mice. SIINFEKL-specific hepatic donor CD8+ T cells continued to proliferate in most mice although the % cells dividing declined by weeks 16–17 as compared to the earlier time points. Liver lymphocytes and splenocytes were isolated from the same mice. The continued high loss of CFSE in liver-derived SIINFEKL-specific donor CD8+ T cells—which had to be pooled in some experiments prior to analysis due to paucity of CD8+ cells— indicates that even in mice that no longer showed strong proliferation of SIINFEKL-specific donor CD8+ T cells in spleens, such T cells continued to proliferate extensively in the liver. Considering AAV8's high tropism for hepatocytes, this is likely due to persistence of capsid antigen.
Figure 5.
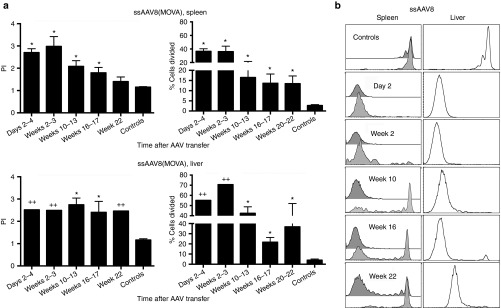
Proliferation of CD8+ T cells from OT-1 mice in ssAAV8(MOVA)-hF.IX transduced mice. Thy1.1 C57BL/6 mice were injected intravenously with 5 × 1011 vg of AAV8(MOVA)-hF.IX. Mice then received 5 × 106 CFSE-labeled splenocytes from OT-1 mice at days 2–4, weeks 2–3, 10–12, 16–17, or 20–22 following AAV8 transfer. The proliferation of Thy1.2+CD8+Vα2+ cells in spleens of individual mice and lymphocytes isolated from pooled or individual livers was tested 10 days later. Control mice did not receive AAV8(MOVA)-hF.IX before splenocytes were adoptively transferred. The proliferation of cells from liver was tested in some experiments with pooled samples; thus P values were not calculated for all liver samples. (a) Average proliferation indices (average number of division cycles) ± SD are shown on the left, average % cells proliferating ± SD are shown on the right. (b) Shows representative histograms for each time point. Numbers of mice/group were as follows: days 2–4: spleens = 5, liver pooled; weeks 2–3: spleens = 5, liver pooled; weeks 10–12: spleens = 7, liver = 3; weeks 16–17: spleens = 8, liver = 8; weeks 20–22: spleen = 6, liver pooled; controls (combined from the different time points of this series of experiments): spleens = 13; liver = 6. Significance was determined by multiple Student's t test with Holm–Sidak correction for type 1 errors comparing experimental to control mice. For the proliferation indices, significant P values were as follows: (spleens) days 2–4 < 0.0001; weeks 2–3 < 0.0001; weeks 10–13 < 0.0001; weeks 16–17 = 0.0004; (livers, for unpooled samples only): weeks 10–12 < 0.0001; weeks 16–17 = 0.014. For % cells proliferating, significant P values were as follows: (spleens) days 2–4 < 0.0001, weeks 2–3 < 0.0001, weeks 10–17 = 0.0006, weeks 16–17 = 0.004, weeks 20–22 = 0.01; (liver) weeks 10–13 < 0.0001; weeks 16–17 < 0.0001. Significant differences to controls are indicated by (*); pooled liver samples are highlighted by (++).
To assess if incorporation of SIINFEKL-encoding sequences into the gene for VP2 changes processing of the AAV8 capsid, we tested if CD8+ T cells to the endogenous AAV8 capsid epitope, which had failed to proliferate strongly to wild-type AAV8 capsid as shown in Figure 4, responded to this epitope as presented by the ssAAV8(MOVA)-hF.IX vector. For this set of experiments, Thy1.1 BALB/c mice were injected i.v. with ssAAV8(MOVA)-hF.IX vector. Two days later, they received CFSE-labeled splenocytes from Thy1.2 BALB/c mice immunized 4 months previously with an Ad-AAV8cap vector. As shown in Figure 6, tetramer+ donor CD8+ T cells failed to strongly proliferate to the endogenous AAV epitope of the capsid-modified vector indicating that incorporation of SIINFEKL was unlikely to affect processing or overall stability of the endogenous epitope of the AAV8 vector.
Figure 6.

The stability of ssAAV8(MOVA)-hF.IX vector. Thy1.1 BALB/c mice were injected intravenously with 2 × 1011 vg of AAV8(MOVA)-hF.IX. On day 2, mice received 5 × 107 CFSE-labeled splenocytes from Thy1.2 BALB/c mice, which had been immunized with 1 × 1011 virus particles of the Ad-AAV8cap vector 4 months earlier. Proliferation of Thy1.2+CD8+AAV8-tet+ cells were tested 10 days later using lymphocytes isolated from spleens. The control mice were intravenously injected with phosphate-buffered saline before splenocytes were adoptively transferred. The average number of division cycle (proliferation index) and the percentage of cells of the original sample which divided (% divided) were calculated. Error bars represent SD for two to three mice per group.
Response to a foreign epitope placed into the capsid of a scAAV8 vector
The structure of the AAV vectors' genome, i.e., single stranded versus self-complementary not only influences onset and magnitude of transgene product expression but also innate immune responses, which are more readily induced by scAAV vectors.16 Increased activation of innate immunity resulting in the production of interferons in turn may increase antigen processing by upregulating proteasomal enzymes and TAP and increasing cell surface expression of MHC molecules. To assess proliferation against scAAV8 vectors, we constructed such vectors expressing hF.IX and carrying 10 copies of SIINFEKL within VP2. Excluding the 10 copies of SIINFEKL, the ss and scAAV8 vector genomes were identical. Using the same adoptive transfer approach as described above, OT-1 cells that homed to the spleens of the recipient mice proliferated when transferred between days 4 to weeks 3 after scAAV8(MOVA)-h.F.IX vector injection. Proliferation then ceased to be statistically significant in spleens (Figure 7a,b). As was seen in mice injected with ssAAV8 vectors, SIINFEKL-specific donor CD8+ T cells that homed to the liver proliferated more extensively than those that homed to spleen but again their proliferation as determined by % cells proliferating ceased to be significant as compared to proliferation of cells transferred into control mice by 12–13 weeks after vector injection. Overall, proliferation to antigens within the capsid of scAAV8 vectors was significantly less pronounced (P < 0.05) and of shorter duration than to those with the capsid of ssAAV8 vectors.
Figure 7.
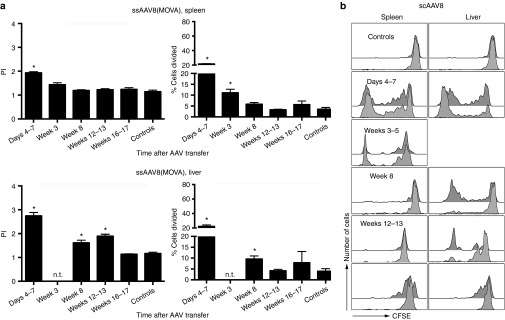
Proliferation of CD8+ T cells from OT-1 mice in scAAV8(MOVA)-hF.IX-transduced mice. The experimental design and the layout of the graphs are identical to those of experiments shown in Figure 2a,b using scAAV8(MOVA)-F.IX vectors for injection of mice. Numbers of mice per group were as follows: (spleens) days 4–7 = 5, weeks 3–5 = 3, week 8 = 5, weeks 12–13 = 2, weeks 16–17 = 3, controls = 8; (liver) days 4–7 = 5, weeks 3–5 = not tested, week 8 = 5, weeks 12–13 = 3; weeks 16–17 = 9, controls = 12. For the proliferation indices, significant P values were as follows: (spleens) days 4–7 < 0.0001, weeks 3–5 = 0.01; (liver) days 4–7 = < 0.0001, week 8 = 0.001, weeks 12–13 = 0.0001. For % cells proliferating, significant P values were as follows: (spleens) days 4–7 < 0.0001, weeks 3–5 = 0.0007; (liver): days 4–7 = 0.01, week 8 = 0.006. Significant differences to controls are indicated by (*).
Response to a foreign epitope placed into the capsid of empty AAV vectors
During AAV vector production, the bulk of the preparation is composed of empty viral capsids that fail to incorporate genomes. Current procedures to remove empty capsid, which have no therapeutic benefits for gene transfer recipients, are inconsistently used so that even vectors used in clinical trials have varied the ratios of full to empty virus particles. To assess if contaminations with empty vector would enhance the risk of AAV capsid-specific T cell responses, we tested T cell proliferation to SIINFEKL displayed by empty AAV8 vectors (Figure 8a,b). Splenic CD8+ T cells that were transferred 4 days after injection of empty AAV8(MOVA)-h.F.IX vectors showed low but significant differences for both the proliferation index and the percentage of dividing cells between experimental and control mice. By week 8, the proliferation index was still marginally higher in experimental animals while the percentage of proliferating cells became comparable in spleens. By week 8, cells ceased to proliferate in spleens. Donor CD8+ T cells isolated from livers showed low statistically significant proliferation only in mice that received OT-1 cells on day 4. Vector transfer donor cells showed loss of CFSE expression (Figure 5b).
Figure 8.
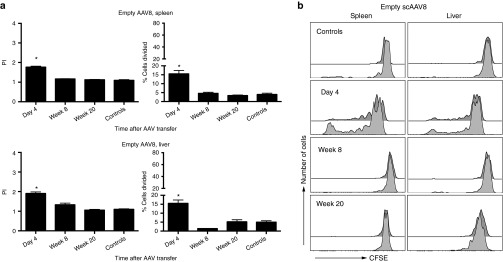
Proliferation of CD8+ T cells from OT-1 mice in mice injected with empty AAV8 vector. The experimental design and the layout of the graphs are identical to those of experiments shown in Figure 2a,b using an empty AAV8(MOVA) vector, derived from a ssAAV preparation for injection of mice. Numbers of mice per group were as follows: (spleen) day 4 = 4, week 8 = 3, week 20 = 2, controls = 3; (liver) day 4 = 4,; week 8 = 5, week 20 = 4, controls = 4. For the proliferation indices, significant P values were as follows: (spleens) day 4 = 0.001; (liver) day 4, 0.001. For % cells proliferating, significant P values were as follows: (spleens) day 4 = 0.004; (liver) day 4 = 0.0009, week 4 = 0.006.
Discussion
After years of setbacks, gene transfer for permanent replacement of missing or faulty genes is finally showing success. In 2008, it was reported that an AAV2 vector carrying the retinal pigment epithelium-specific 65-kDa protein gene that was given into the subretinal space of humans with Leber's congenital amaurosis resulted in recovery of visual/retinal function, most markedly in children.17,18 Vector doses to the confined subretinal space were low, which may have prevented induction of adaptive immune responses. In 2011, another trial heralded success by achieving upon hepatic transfer of a scAAV8 vector-sustained levels of hF.IX in hemophilia B patients.5
In a previous trial, a comparable dose of a ssAAV2-h.FIX vector had accomplished similar levels of hF.IX4 indicating that unlike in preclinical animals,19,20 scAAV8 vectors may not necessarily achieve higher transgene product expression in human liver compared to ssAAV2 vectors. In the ssAAV2 trial, F.IX expression declined to baseline with a concomitant rise and fall in transaminases and the appearance of AAV capsid-specific T cells, as shown in one subject.4 These data were consistent with a hypothesis that in humans, memory CD8+ T cell responses to AAV capsid induced by a natural infection during childhood had been reactivated and eliminated the AAV-transduced hepatocytes. Immune rejections due to AAV capsid antigen-specific T cells have not been observed in experimental animals, such as mice or dogs, which are not naturally infected by AAV.
In the more recent clinical trial, scAAV8 vectors were used for several reasons. In animals, AAV8 vectors have higher tropism for liver compared to AAV2 vectors.21 ScAAV vectors express higher levels of transgene product more rapidly as the rate-limiting step of second-strand DNA synthesis is circumvented22 and it was assumed that this would allow for dose sparing, which in turn may prevent induction of cellular immune responses. In mice and nonhuman primates, AAV2 and related serotypes induce capsid-specific T cell responses unlike vectors from other AAV clades such as AAV8.6,7 Differential induction of capsid-specific T cells was linked to receptor usage; AAV2-like serotypes unlike those related to AAV8 bind to heparin sulfate proteoglycan, which allows for their uptake by human dendritic cells.6 It was thus proposed by some authors that AAV8 would be less likely to elicit capsid-specific T cell responses.
Nevertheless, the results of the recent scAAV8 hemophilia trial share some similarities with those of the previous ssAAV2 trial. At least one patient in the scAAV8 trial developed a 10-fold increase in transaminases and a rise in AAV capsid-specific T cells, which was rapidly controlled by prednisolone. Liver cell destruction did not become evident till 8–9 weeks after gene transfer unlike in the previous ssAAV2 trial where transaminases and T cells increased by 4 weeks after gene transfer. In both trials, AAV vectors were gutted and did not contain genes encoding capsid or regulatory proteins. Capsid antigens present on the virion are thus, but for epitopes of the transgene product, the sole potential targets for T cells.
We reported previously using a mouse model that endogenous epitopes of ssAAV2 capsid stimulate CD8+ T cells for at least 3 weeks, while a foreign T cell epitope placed at multiple copies into VP2 elicits CD8+ T cell proliferation for up to 10 weeks. At this later time point, T cell expansion becomes markedly reduced compared to that observed at 2 weeks after vector transfer, indicating that most of the epitopes had been eliminated. To assess if the late T cell response in human scAAV8 vector recipient could realistically reflect expansion of T cells to residual epitopes of AAV8 capsid, we herein describe similar experiments conducted with an AAV8 serotype vector. In agreement with previous reports, AAV8 vectors with wild-type capsid failed to trigger strong expansion of memory CD8+ T cells in mice. Although this could reflect lack of vector uptake by cells able to induce recall responses, other explanations such as proteasome-mediated destruction of the AAV8 epitope mimicked by the MHC class I tetramer had to be considered. We therefore repeated the experiments using an AAV8 vector that expresses SIINFEKL within capsid VP2. Humans recognize mainly epitopes within VP3 of AAV,8 which is present at a 10-fold higher copy number on AAV capsid compared to VP2. To compensate for this difference, we constructed AAV8 vectors carrying 10 copies of SIINFEKL. These vectors, in contrast to AAV8 vectors carrying wild-type capsid, elicit very robust responses of SIINFEKL-specific CD8+ T cell responses. An unexpected finding here was that the degree and duration of T cell proliferation depended on the vectors' genome: ssAAV8 vector-induced proliferative CD8+ T cell responses for over 22 weeks while responses to scAAV8 capsid ceased after 12–13 weeks. In addition, at the earlier time points when both vectors triggered proliferative responses, responses to epitopes on ssAAV8 vectors were significantly higher than those to epitopes on scAAV8 capsids.
ScAAV vectors differ from ssAAV vectors by lack of a terminal resolution site in one of the ITRs, which allows for folding of the single-stranded complementary genome into a double-stranded DNA.22,23 The capsids of ssAAV and scAAV vector are identical. Nevertheless, differences in genome affect innate responses and as we reported previously, scAAV vectors trigger more vigorous innate responses through the Toll-like receptor-9.16 Although innate cytokine responses such as interleukins and interferons come up in serum very rapidly after AAV gene transfer and then subside, local responses may be maintained for longer through gradual release of genomes from episomally retained AAV vectors. Some cytokines, especially interferons, upregulate the cells' proteolytic machinery, including proteasomes and ubiquitin, while other cytokines such as IL-6 may induce counterbalancing deubiquinating enzymes.24 Accelerated degradation by factors induced by the innate immune system may explain the shortened T cell responses to scAAV vectors when compared to ssAAV vectors. More rapid destruction of capsids cannot explain the lower proliferative T cell responses to scAAV vectors at early time points. We can only speculate that other factors, such as differential induction of immunoinhibitory responses, including regulatory T cells known to suppress transgene product-specific T and B cell responses and expected to control immune responses to the capsid, may play additional yet to be defined roles.
The virtual absence of a proliferative CD8+ T cell response to empty AAV vectors, which would not be expected to induce an inflammatory response beyond that induced by the small injury caused by the injection, may reflect that activation of an innate immune response is essential to allow for T cell proliferation by inducing upregulation of molecules that are key for T cell stimulation such as MHC class I antigens or costimulators. The low proliferative CD8+ T cell response to empty AAV vectors (similar experiments were conducted with AAV2 vectors and gave similar results, data not shown) needs to be interpreted with caution as our experimental setup does not allow us to exclude that contamination of gene transfer AAV vectors with empty capids may, within the setting of an innate immune response to genome containing vectors, result in enhanced T cell responses to AAV capsid epitopes.
The very prolonged proliferation of capsid-specific CD8+ T cells especially in mice that had received ssAAV8 vectors was surprising as it suggests that capsid antigens degrade very slowly. Most AAV particles that are taken up by cells in the end fail to enter the nucleus but rather accumulate in late endosomes, lysosomes, or perinuclear spaces. Presentation of antigen presumably requires degradation of AAV capsid within cytoplasmic compartments rather than the nucleus. Two pathways can lead to extranuclear degradation of AAV capsid. Enzymes, such as cathepsin L and B, which both bind AAV virions,25 could digest capsid antigens within lysosomes, generating peptides for MHC class II presentation and induction of CD4+ T cell responses. Proteasomes can destroy ubiquinated proteins in the cytoplasm. As AAV capsids are first phosphorylated and then ubiquinated in the endosomes,26 proteosomes may lead to processing of AAV particles or fragments once they have reached the cytoplasm for transport of peptides into the endoplasmic reticulum followed by their association with MHC class I molecules, which in turn is required for the stimulation of CD8+ T cell responses.
Alternatively, AAV vectors remain extracellularly as stable antibody-antigen complexes. Such immune complexes can bind to Fc receptors and thereby be deposited onto follicular dendritic cells within lymphatic tissues where they can persist for months in vivo.
We believe whether or not long-term persistence of AAV8 capsid antigens poses sustained risks for human AAV8 vector recipients will depend on the underlying mechanism. If AAV8 capsid antigens are predominantly presented by slow capsid degradation in the targeted cells, such as hepatocytes in our mouse model, patients will remain at risk for immune-mediated hepatocyte destruction and loss of the therapeutic protein for months. If, on the other hand, AAV8 capsid proteins persist in the form of antigen-antibody complexes on Fc-receptor+ cells, they may induce AAV capsid-specific immune responses. But, in the absence of AAV epitopes on vector-targeted hepatocytes, they will do no harm.
Materials and Methods
Mice. Six- to eight-week-old male BALB/c mice (Thy1.1 and Thy1.2) and C57BL/6 mice (Thy1.1) were purchased from the Jackson Laboratory (Bar Harbor, ME) or the National Cancer Institute (Bethesda, DC). OT-1 mice were bred at the Wistar Institute and male mice were used at 6–10 weeks of age. Mice were housed at the Animal Facility of the Wistar Institute and treated according to the institutional rules for animal welfare following approved protocols.
Vectors. The E1- and E3-deleted Ad human serotype 5 vectors expressing capsid antigens of AAV8 under a cytomegalovirus promoter (Ad-AAV8cap) and AAV8-hF.IX were prepared as described before.7
The plasmids and methods used to propagate and purify AAV8(MOVA)-hF.IX were similar to those of AAV8-hF.IX, except that the plasmids encoding the AAV8 capsid genes differed. Two plasmids encoding the AAV8 capsid gene and the sequence of SIINFEKL within VP2 were produced to generate AAV8 vectors expressing SIINFEKL on the viral capsid. The first plasmid has a mutated VP2 start codon and only expresses AAV8 VP1 and VP3 proteins. The second plasmid has mutated VP1 and VP3 start codons and only expresses VP2 protein, in which 10 SIINFEKL-encoding sequences (TCGATCATCAATTTTGAGAAACTG) were inserted right after the ACG start codon into the BglII site (AGATCT). Each SIINFEKL-encoding sequence was linked by 2-alanine-encoding sequences (GCAGCT). Recombinant AAV vectors and empty capsids, including MOVA capsid variants were generated by transient transfection in HEK293 cells and purified by an optimized cesium chloride double-gradient ultracentrifugation process as described by Ayuso et al.,27 with modifications. After AAV particles (vector genome containing particles and empty capsids) recovered after the first CsCl centrifugation were separated in the second CsCl gradient step, two bands were visualized and extracted, the lower band at density ~1.4 gm/ml corresponding to the vector genome containing vector particles, and the upper band at density ~1.3 gm/ml, corresponding to empty capsids.
Quality control of vectors. AAV Vectors and empty capsids were characterized for purity and titer by SDS-PAGE silver stain analysis using 10% acrylamide or 4–12% gradient acrylamide gels and stained using SilverXpress silver staining kit (InVitrogen, Grand Island, NY) and for absence of endotoxin by Limulus amebocyte lysate assay (LAL EndoSafe; Charles River Laboratories, Malvern, PA).28,29 For western blot, AAV vectors and empty capsids were run on a SDS-PAGE gel (4–12% gradient gel, Invitrogen, Carlsbad, CA), and protein bands were transferred to nitrocellulose. Western blot analysis was performed using Monoclonal antibody B1 (American Research Products, Belmont, MA) as primary antibody and HRP-conjugated sheep anti-mouse IgG (Promega, Madison, WI) as secondary antibody to detect AAV capsid proteins (ECL, Amersham, Piscataway, NJ). Optical density measurement of genome-containing and empty AAV viral particles were performed as follows. Spectrophotometry was performed on purified AAV preparations to confirm λ = 260 nm/280 nm; absorbance ratios corresponded to values corresponding to genome-containing AAV particles (~1.5) and AAV empty capsids (~0.7).30,31 DNA isolation from tissues was performed using the DNA Easy kit (Qiagen, Venlo, Netherlands) according to the manufacturer's protocol. Hundred nanogram of DNA from each sample was used for quantitative real-time polymerase chain reaction. Sybr green real-time polymerase chain reaction was performed to quantify the gag or hF.IX gene to determine viral genome copy number. The primer sequence used for the gag transgene was: 5′-TCCCTCTGCCAAAAATTATG-3′ (forward) and 5′-TTCCTTTATTAGCCAGAAGTCAG-3′ (reverse). The primer sequence used for the hF.IX transgene was: 5′-ACCAGCAGTGCCATTTCCA-3′ (forward) and 5′-GAATTGACCTGGTTTGGCATCT-3′ (reverse). Samples were run on an iCycler (Bio-Rad, Hercules, CA) in triplicate. The Gag and hF.IX plasmid DNA at 1 µg/ml, 100 ng/ml, 10 ng/ml, 1 ng/ml, 100 pg/ml, 10 pg/ml, 1 pg/ml, 100 fg/ml, and 10 fg/ml in 10 ng/μl salmon sperm DNA were used on each real-time polymerase chain reaction plate as copy number standard controls. Using this data, we determined the number of viral genome copies per microgram of total DNA for each vector.
Immunization of mice. Ad-AAV8cap vectors were diluted in sterile phosphate-buffered saline (PBS) to a total volume of 100 µl and intramuscularly injected into the hind legs of mice. AAV8 vectors were diluted in 200 µl of sterile PBS and injected i.v. into the tail vein of mice.
ELISA for human FIX in mouse plasma. Nunc 96-well plates were coated with monoclonal anti-human FIX antibody diluted 1: 600 in 0.1 mol/l carbonate-coating buffer (pH: 9.6). Plates were washed with 0.05% Tween-20/PBS, blocked with 1% BSA, 0.05% Tween20 in PBS, incubated with diluted mouse plasma samples, and detected by 1: 500 dilution of HRP-conjugated goat anti-human FIX antibody (Enzyme Research Laboratories, South Bend, IN). Levels of human FIX were determined by OD450 and quantified against the linear standard curve generated with serially diluted purified human plasma FIX.
In vivo proliferation assay. Lymphocytes were isolated from spleens of donor mice (Thy1.2 BALB/c or OT-1 mice), and red blood cells were lysed with 1× red blood cell lysing buffer (eBioscience, San Diego, CA). Cells were then labeled with 7 µmol/l CFSE using the CellTrace CFSE Cell Proliferation Kit (Invitrogen) and transferred to recipient mice (Thy1.1 BALB/c or C57BL/6 mice) in 200 µl PBS by tail vein injection. At 10 days after transfer, splenocytes or liver lymphocytes of recipient mice were harvested and stained with antibodies to Thy1.2, CD8, Vα2, and/or the AAV8 capsid-tetramer (tet). They were then analyzed by flow cytometry. Gates were set to identify antigen-specific CD8+ T cells of donor origin. These cells were then analyzed for decreases in CFSE expression, with a decrease being indicative for cell proliferation. The experimental approach is shown for testing of T cells to endogenous epitopes in Figure 6, which also shows gating for one example. For testing of SIINFEKL-specific immune responses, the approach was similar; the main difference was that donor mice were unvaccinated OT-1 mice. The proliferation index (PI) and % divided (%D) were calculated as described in http://www.flowjo.com/v9/html/proliferation.html.
Statistics. Significance was determined by Student's t tests comparing the results obtained with individual mice from one group to those obtained with individual mice from the other group. Significance was set at P ≤ 0.05.
Acknowledgments
This work was funded by P01 HL078810 from NIH/NHLBI. W.T.-L. and H.L. conducted the experiments. H.C.J.E. spearheaded experimental design and interpretation of data. W.T.-L., H.L., and H.C.J.E. wrote sections of the manuscript. S.F. and F.W. conducted vector quality control studies. K.H. contributed by discussing the data and critically reviewing and revising the manuscript. H.L. and H.C.J.E. have no conflict of interest regarding the data presented in this manuscript. We acknowledge the help of Joseph Rabinowitz (Temple University, Philadelphia, PA) for his assistance and guidance on silver stain and genome copy analyses.
References
- Arruda VR, Schuettrumpf J, Herzog RW, Nichols TC, Robinson N, Lotfi Y, et al. Safety and efficacy of factor IX gene transfer to skeletal muscle in murine and canine hemophilia B models by adeno-associated viral vector serotype 1. Blood. 2004;103:85–92. doi: 10.1182/blood-2003-05-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder RO, Miao CH, Patijn GA, Spratt SK, Danos O, Nagy D, et al. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat Genet. 1997;16:270–276. doi: 10.1038/ng0797-270. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, McIntosh J, Ng CY, Zhou J, Spence Y, et al. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood. 2007;109:1414–1421. doi: 10.1182/blood-2006-03-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe LH, Wang L, Somanathan S, Zhi Y, Figueredo J, Calcedo R, et al. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat Med. 2006;12:967–971. doi: 10.1038/nm1445. [DOI] [PubMed] [Google Scholar]
- Li H, Murphy SL, Giles-Davis W, Edmonson S, Xiang Z, Li Y, et al. Pre-existing AAV capsid-specific CD8+ T cells are unable to eliminate AAV-transduced hepatocytes. Mol Ther. 2007;15:792–800. doi: 10.1038/sj.mt.6300090. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78:3110–3122. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tuyishime S, Wu TL, Giles-Davis W, Zhou D, Xiao W, et al. Adeno-associated virus vectors serotype 2 induce prolonged proliferation of capsid-specific CD8+ T cells in mice. Mol Ther. 2011;19:536–546. doi: 10.1038/mt.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenaia O, Mingozzi F, Hauck B, Zhou S, High KA, Wright F. A capsid composition variant containing four VP proteins caused by a novel start codon at position 219 of the nucleotide sequence of AAV8 Cap. Abstract 403, ASCGT: Seattle, WA. Mol Ther. 2011;19:403. [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- Lux K, Goerlitz N, Schlemminger S, Perabo L, Goldnau D, Endell J, et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J Virol. 2005;79:11776–11787. doi: 10.1128/JVI.79.18.11776-11787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen D, Cantwell ER, O'Brien T, Johnson PA, Mahon BP. Adeno-associated virus serotype 2 induces cell-mediated immune responses directed against multiple epitopes of the capsid protein VP1. J Gen Virol. 2009;90 Pt 11:2622–2633. doi: 10.1099/vir.0.014175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–117. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, et al. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood. 2011;117:6459–6468. doi: 10.1182/blood-2010-10-314518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, et al. Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang H, Bell P, McCarter RJ, He J, Calcedo R, et al. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther. 2010;18:118–125. doi: 10.1038/mt.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pañeda A, Vanrell L, Mauleon I, Crettaz JS, Berraondo P, Timmermans EJ, et al. Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum Gene Ther. 2009;20:908–917. doi: 10.1089/hum.2009.031. [DOI] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- Baek KH. Cytokine-regulated protein degradation by the ubiquitination system. Curr Protein Pept Sci. 2006;7:171–177. doi: 10.2174/138920306776359740. [DOI] [PubMed] [Google Scholar]
- Akache B, Grimm D, Shen X, Fuess S, Yant SR, Glazer DS, et al. A two-hybrid screen identifies cathepsins B and L as uncoating factors for adeno-associated virus 2 and 8. Mol Ther. 2007;15:330–339. doi: 10.1038/sj.mt.6300053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douar AM, Poulard K, Stockholm D, Danos O. Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation. J Virol. 2001;75:1824–1833. doi: 10.1128/JVI.75.4.1824-1833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- Wright JF, Zelenaia O. Vector characterization methods for quality control testing of recombinant adeno-associated viruses. Methods Mol Biol. 2011;737:247–278. doi: 10.1007/978-1-61779-095-9_11. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, et al. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- Sommer JM, Smith PH, Parthasarathy S, Isaacs J, Vijay S, Kieran J, et al. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 2003;7:122–128. doi: 10.1016/s1525-0016(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Wright JF, Le T, Prado J, Bahr-Davidson J, Smith PH, Zhen Z, et al. Identification of factors that contribute to recombinant AAV2 particle aggregation and methods to prevent its occurrence during vector purification and formulation. Mol Ther. 2005;12:171–178. doi: 10.1016/j.ymthe.2005.02.021. [DOI] [PubMed] [Google Scholar]