

Abstract
Lentiviral vectors are widely used in basic research and clinical applications for gene transfer and long-term expression; however, safety issues have not yet been completely resolved. In this study, we characterized hepatocarcinomas that developed in mice 1 year after in utero administration of a feline-derived lentiviral vector. Mapped viral integration sites differed among tumors and did not coincide with the regions of chromosomal aberrations. Furthermore, gene expression profiling revealed that no known cancer-associated genes were deregulated in the vicinity of viral integrations. Nevertheless, five of the six tumors exhibited highly significant upregulation of E2F target genes, of which a majority are associated with oncogenesis, DNA damage response, and chromosomal instability. We further show in vivo and in vitro that E2F activation occurs early on following transduction of both fetal mice and cultured human hepatocytes. On the basis of the similarities in E2F target gene expression patterns among tumors and the lack of evidence implicating insertional mutagenesis, we propose that transduction of fetal mice with a feline lentiviral vector induces E2F-mediated major cellular processes that drive hepatocytes toward uncontrolled proliferation culminating in tumorigenesis.
Introduction
Human immunodeficiency virus (HIV)-1-based lentiviral vectors have been proven to be an efficient gene delivery system and are widely used in basic and applied research as a tool for overexpression of transgenes, gene silencing by shRNA, and generation of induced pluripotent stem cells as well as delivery of therapeutic genes in clinical trials.
Previous studies using γ-retroviral vectors for transduction of hematopoietic stem cells led to leukemias in animal models1 and in several patients enrolled in a gene therapy trial for the correction of severe combined immunodeficiency disease2 and caused myelodysplasia in patients treated for chronic granulomatous disease.3 The oncogenic capacity of retroviral vectors is attributed, in general, to insertional mutagenesis that is due to a combination of their bias for integration into promoter regions of genes involved in growth control and cancer, and their capacity to activate genes via their transcriptionally active long terminal repeat (LTR).1 To reduce the potential for activation of genes located downstream of the chromosomally integrated vector, γ-retroviral vectors devoid of the enhancer/promoter in the 3′ LTR have been constructed, rendering them self-inactivating (SIN). However, clonal expansion and oncogenesis have continued to be reported using these SIN constructs,4 possibly due to viral promoter activity in the 3′ LTR that has not been completely attenuated5 or by the presence of strong internal enhancers that trigger profound clonal imbalance.6
HIV-based lentiviral SIN vectors have been shown to be safe in the clinic as exhibited by therapeutic improvement for metachromatic leukodystrophy7 and Wiskott–Aldrich Syndrome.8 However, clonal dominance was observed following transplantation of transduced bone marrow cells in a patient with β-thalassemia.9 Furthermore, in animal studies, it was reported that tumors developed following the transduction of tumor-prone mouse models10,11 and lymphomas resulted in mice following the transplantation of genetically modified hematopoietic stem cells.12 In addition, hepatocellular carcinomas (HCCs) developed following the transduction of highly proliferating fetal livers with nonprimate lentiviral vectors.13,14
In this study, we used a SIN lentiviral vector derived from feline immunodeficiency virus (FIV), which is nonpathogenic in humans,15 for assessing the potential for correction of metabolic diseases at an early developmental stage using a liver-directed gene therapy approach. We transduced mice in utero with an FIV vector carrying the EGFP transgene driven by the human α-antitrypsin (hAAT) liver-specific promoter (FIV-hAAT-EGFP). Following transduction, efficient and long-term expression of EGFP was observed in the liver; however, three of the eight transduced mice each developed two HCCs. We found that although the location of deregulated genes and viral integrations sites in these tumors did not coincide, genes involved in the control of cell proliferation and genomic stability were mutually upregulated in five of the six tumors. Strikingly, a majority of these deregulated genes are also known E2F targets. Members of the E2F family of transcription factors are downstream effectors of the retinoblastoma, Rb/p16, tumor suppressor pathway and were shown to function in a wide range of biological processes, playing a major role in the control of cell cycle progression as well as DNA damage repair and apoptosis, depending on the cellular context.16,17,18 However, recent publications have shown that functions of E2F extend beyond the control of cell proliferation and may be implicated in the pathogenesis of multiple human cancers. A clear oncogenic role of E2F1, 2, and 3 has been described in human cancer, as frequent amplification of its target gene loci is observed in many cancers, including HCC.19 Furthermore, transgenic mice that overexpress E2F1 in the liver develop HCC.20
In light of the common upregulation of a significant number of E2F target genes in FIV-induced HCCs and the lack of cancer-associated gene upregulation in the vicinity of FIV integration sites, we suggest that the tumorigenesis observed following fetal transduction was a consequence of the activation of a global cellular mechanism in which E2Fs play a pivotal role in inducing uncontrolled cell proliferation.
Results
HCC development following FIV transduction of fetal mice
To test for transduction efficiency and oncogenic potential of FIV vectors following prenatal gene delivery, we transduced MF-1 fetal mice at 16 days of gestation, via the yolk sac vein, with the FIV-hAAT-EGFP vector, at an approximate multiplicity of infection (MOI) of 40. Viral administration to fetuses via the yolk sac vessel assures efficient delivery of large viral loads to the small liver.21 Liver biopsies taken at 5 months post-FIV transduction revealed that >50% of hepatocytes expressed the transgene (Figure 1a). We previously observed a significantly lower percentage of EGFP-positive hepatocytes following transduction of adult and neonatal Balb/c mice through tail vein and temporal vein administrations (1 and 17%, respectively),22,23 most likely due to the larger liver size (reducing the effective MOI) and the lower proliferation rate of postnatal hepatocytes. In this study, we used the outbred MF-1 mice that have a low frequency of spontaneous tumor development.14 Nevertheless, following in utero administration of the FIV vectors, we observed liver tumors in three of the eight transduced mice between the ages of 9 and 16 months (Figure 1b). One male and two females each developed two tumors that were pathologically identified as HCCs (Figure 1c).
Figure 1.

Transgene expression in hepatocytes and hepatocellular carcinoma development following transduction. Fetal mice were transduced with FIV-hAAT-EGFP at ~40 transducing units per cell. (a) Liver biopsies were analyzed for EGFP expression 5 months posttransduction by direct visualization (left panel; magnification 400×) and immunostaining (right panel; magnification 200×). (b) A representative photo image of two liver tumors that developed in one of the three mice ~1 year posttransduction. The scale on the ruler is 1 mm. (c) H&E staining of tissue sections taken from a liver tumor that developed in mouse M1 (left panel) and from the surrounding liver tissue (right panel). FIV, feline immunodeficiency virus; hAAT, human α-antitrypsin.
Determination of vector copy number and viral integrations sites within tumors
We first performed Southern blot analysis on DNA extracted from the tumors that revealed a unique band pattern for each tumor indicating clonal origin (Figure 2a). Then, we used quantitative polymerase chain reaction (qPCR) for determining the vector copy number (VCN) in the tumors and in their surrounding tissue as well as in livers of control tumor-free transduced mice. As seen in Figure 2b, tumors contained a significantly higher average of at least 42 viral copies per 100 cells as compared with 10 and 4 copies per 100 cells in their surrounding tissue and control livers, respectively (P < 0.03). One exception of tumor M1-T1, in which VCN was similar to that of the surrounding tissue in the same animal, was not included in the average. The VCN values obtained in our study are consistent with those determined in other studies involving lentiviral vector transduction.12,13 To determine the genomic locations of viral insertions, we amplified and sequenced genomic DNA-vector junctions and identified 28 FIV integration sites in the six tumors, between 1 and 8 integration sites per tumor (an average of 4.7 integrations per tumor) (Supplementary Table S1). This value is consistent with recent studies, in which 3.2–5.7 viral integrations were mapped per tumor.10,13,14 The low number of integrations could be explained by the growth of a tumor cell clone marked by a given number of integrations at the expense of other marked cells. Of note, in the FIV tumors, no overlap of mapped integration sites was observed between tumors and within a single tumor; integrations were at least 5 kb apart. Of the 28 FIV integration sites that we mapped, 16 (64%) occurred within known genes: 13 in introns, 2 in the 3′ UTR, and 1 integration was within an exon (Supplementary Table S1).
Figure 2.

Southern blot analysis and vector copy number in the lentiviral vector-induced liver tumors. (a) Southern blot analysis of genomic DNA extracted from liver tissue of an untransduced control mouse (lane 11), a tumor-free transduced mouse (lane 1), tissue surrounding tumors (lanes 2, 3, and 8), and vector-induced tumors (lanes 4–7 and lanes 9–10). (b) Vector copy number (VCN) per 100 cells was determined by quantitative polymerase chain reaction on the DNA samples. M, male; F, female; T, tumor; S, surrounding nontumorous tissue; TF, tumor-free transduced mouse; control, untransduced mouse.
It is known that overexpression of microRNAs, long-interfering noncoding RNAs, or epigenetic silencing of tumor suppressor genes is associated with HCC progression.24 We crossreferenced all mapped FIV viral integration sites with the location of microRNAs in the mouse genome and found no known microRNA sequences within 6 Mb of mapped FIV vector integration sites. The Affymetrix microarray used in our study lacked probes to microRNAs, long-interfering noncoding RNAs, or epigenetic markers. Therefore, we cannot exclude deregulated expression of some microRNAs and long-interfering noncoding RNAs or epigenetic changes resulting in tumor progression.
To explore the possibility that viral integration sites coincide with regions of chromosomal instability (CIN), such as common fragile sites (CFSs), we crossreferenced FIV viral integration sites with the syntenic location of 22 mapped human CFSs in the mouse genome (Supplementary Table S2) and found only one viral insertion in tumor F1-T2 that coincided with a syntenic CFS located in chromosome 4. Therefore, there was little evidence of preferential FIV integration into fragile sites in fetal hepatocytes. This is consistent with a previous study showing that HIV-based vectors do not have a preference for integrating into these unstable chromosomal regions.25
Chromosomal aberrations in tumors
Comparative genomic hybridization of FIV-induced tumors versus surrounding tissue revealed varying numbers and locations of chromosomal abnormalities among tumors, ranging from 1 to 50 aberrations per tumor (Supplementary Figure S1a–c; accession no GSE25900). Foremost, no individual chromosomal aberration was shared among all six tumors, but there were isolated incidences of aberrations shared by three tumors (F1-T2, M1-T1, and M1-T2) including a complete deletion of one copy of chromosome X, a small amplification in chromosome 9, and a small deletion in chromosome 17 (Supplementary Figure S1d). No significant correlation was found between the locations of chromosomal aberrations and syntenic CFSs, as only 5 of the 95 total mapped aberrations occurred within a CFS (Supplementary Table S2). Of note, although the majority of aberrations differed between the mice, numerous small common aberrations were detected within the two tumors that originated in the same mouse (seen in mice M and F2; Supplementary Figure S1a,c). This phenomenon may be due to tumor development from a precancerous lesion that carried chromosomal modifications as a result of FIV transduction, and subsequently gave rise to two separate tumors, which continued to accumulate additional independent aberrations at later stages of tumor development.26
No correlation was found between the number of vector copies per cell (Figure 2b) and the number of chromosomal abnormalities within a given tumor (Supplementary Figure S1). Furthermore, except for one viral integration that occurred within an amplified region of chromosome 18 (see F2-T1 in Supplementary Table S1 and Supplementary Figure S1a), no mapped viral integration sites coincided with a chromosomal aberration in any tumor.
No evidence for deregulation of genes located in the vicinity of mapped FIV integration sites
We performed genome-wide gene expression analysis on RNA isolated from the 6 FIV-induced liver tumors and matched nontumorous liver tissue, as well as from livers of tumor-free FIV-transduced animals and untransduced controls (accession no GSE19306). Gene expression data revealed separate clusters for male and female samples as well as for tumor and nontumor samples, excluding tumor F1-T2, whose gene expression pattern differed significantly from the five remaining tumors (Supplementary Figure S2). Foremost, analysis of expression of genes located within 100 kb of mapped FIV integration sites revealed no evidence of oncogene activation or tumor suppressor downregulation in all six tumors.
It has been reported in a clinical study for β-thalassemia9 and in preclinical studies27,28 that aberrant transcripts have arisen as a result of gene disruption by integration of HIV-based vectors that are known to have a strong preference (71%) for integrating into actively transcribed genes. We performed a high-throughput sequencing study on FIV-transduced murine liver cells (AML12) (Supplementary Table S3), which revealed >17,000 unique viral integration sites in which 37% occurred within transcription units similar to previously published estimates for random integrations into gene coding regions.29 This low preference for integration into transcription units suggests that aberrant transcription is less likely to occur with FIV vectors. In addition, it should be noted that only 271 of the unique 17,576 integration sites (1.5%) were located in or near a known oncogene according to the “allOnco” database (http://www.bushmanlab.org/links/genelists), a compilation of seven cancer gene databases including a murine oncogene list (RTGCD). This percentage is significantly lower than expected in the case of random integration.
E2F target genes associated with oncogenesis, DNA damage response, and CIN are activated in FIV-induced tumors
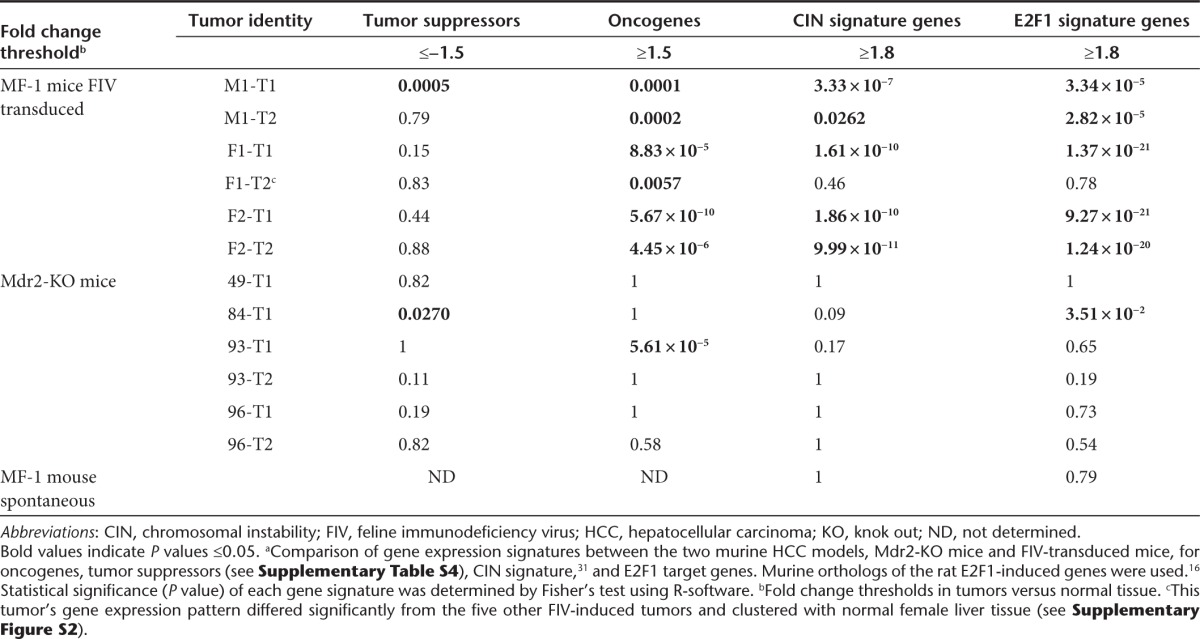
Our major finding from the analysis of the gene expression profiling of the FIV-induced tumors as compared with nontumorous tissue from the same mouse was the significant upregulation of E2F target genes in five of the six tumors, even though tumors differed in their vector integration sites and locations of chromosomal aberrations (Tables 1 and 2). Specifically, 46 E2F target genes were significantly upregulated (>1.8-fold) in four of the six tumors and 18 of these target genes were upregulated in a fifth tumor as well. Twelve additional E2F targets were upregulated in three FIV-induced HCCs.
Table 1. Genes upregulated in the FIV-induced tumors that are associated with major cellular processes.

Table 2. Gene signature enrichment in FIV-induced HCCs as compared to inflammation-induced tumorsa.
E2F1 plays a critical role in controlling both cell cycle progression and apoptotic cell death in response to DNA damage and oncogene activation (see review 17). The microarray data revealed that a majority of the upregulated E2F targets in the FIV-induced tumors are associated with oncogenesis, DNA damage response (DDR), and CIN (Table 1). Twelve known oncogenes were upregulated in at least three FIV-induced tumors; eight of them are also E2F targets.
DNA double-strand breaks (DSBs) and impairment of the DDR contribute to CIN that can lead to tumorigenesis.30 We found within the expression profiling data of the FIV-induced tumors, significant upregulation of known CIN signature genes31 in all five of the FIV-induced tumors as seen in Table 2. Of note, all upregulated CIN signature genes are also E2F target genes (see Table 1). In accordance with this finding, our data mining of several public HCC data sets revealed a strong correlation between CIN signature and E2F target gene upregulation (not shown). Furthermore, it has been demonstrated that HCCs, which developed in retinablastoma (Rb)-deficient mice, exhibited upregulation of CIN signature genes.32 Collectively, these data point to the pivotal role that E2F plays in FIV-induced liver tumors via its activation of oncogenes and its induction of genes associated with the DDR and CIN. Of note, a comparison of the gene expression patterns of the FIV vector-induced HCCs in our study with those of the lymphomas that developed in mice transplanted with HIV-transduced hematopoietic stem cells12 revealed a remarkable similarity in upregulated genes associated with the four aforementioned cellular processes (Table 1).
We chose seven of the highly upregulated E2F target genes in the FIV-induced tumors from the microarray data (H2afz, Mad2L1, Rrm2, Mcm2, Mcm5, Mcm6, and Mcm7) and validated their expression by quantitative real-time PCR on RNA extracted from the liver tumors. As seen in Supplementary Figure S3, tumors exhibited significantly higher expression levels of these genes as compared with their matched, nontumorous liver tissue, consistent with the expression array data.
E2F activation is an early event postlentiviral vector transduction
To examine the status of E2F at an early stage postfetal transduction, we harvested the livers of neonatal mice 10 days after birth and 2 weeks after in utero administration of FIV-hAAT-EGFP. Livers contained 25% ± 7.7 EGFP-positive cells (Figure 3a) and at least 0.60 ± 0.27 vector copies/100 cells (Figure 3b). Of note, already at this early time point posttransduction, we observed an average of 3.5-fold higher E2F1 nuclear protein levels in transduced mice as compared with untreated controls as measured by Western blot analysis (Figure 3c). Notably, the microarray data and qPCR validation of the seven aforementioned genes did not detect upregulation of E2F target genes in these neonatal transduced mice (data not shown). This is consistent with reports showing that E2F induction upon DNA damage does not necessarily lead to E2F target gene upregulation.33 Furthermore, the microarray data revealed that E2F target gene expression levels in young mice, whether transduced or untransduced, were higher than in adult mice, probably due to intensive hepatocyte proliferation at this age (Supplementary Figure S4).
Figure 3.

Analysis of livers from 10-day-old mice after in utero transduction. Fetal mice (at 16 days gestation) were transduced with FIV-hAAT-EGFP lentiviral vector at MOI ~40 and the livers were harvested 2 weeks posttransduction. (a) Tissue sections from livers of transduced mice were analyzed for EGFP expression by immunostaining. Left panel: 36% EGFP-positive cells; right panel: 15% EGFP-positive cells. (b) Vector copy number (VCN) per 100 cells was determined by quantitative polymerase chain reaction on genomic DNA extracted from transduced (T) and untransduced (control) siblings. (c) Nuclear protein levels of E2F1 were determined by immunoblotting analysis of livers of five transduced mice (T) and four untransduced siblings (C). Anti-lamin A/C was used as a loading control. Each column represents one mouse. FIV, feline immunodeficiency virus; hAAT, human α-antitrypsin.
To test the effect of lentiviral vector transduction on E2F levels in vitro, we used the HepaRG cells; a cell line derived from an HCV-associated liver tumor yet shares many features with normal adult hepatocytes, has a normal karyotype, and does not grow on soft agar.34 HepaRG cells were transduced with FIV-hAAT-EGFP at an MOI of 1.5 × 104, which led to 40% EGFP-positive cells. qPCR analysis performed 3 days posttransduction revealed upregulation of four out of five selected E2F target genes that were upregulated in the FIV-induced tumors (Figure 4a and Supplementary Figure S3). We next searched for phenotypic changes associated with cellular transformation in these transduced HepaRG cells. Notably, we found that FIV-transduced cells exhibited a significantly higher proliferation index than control cells, as well as a loss of contact inhibition (Figure 4b,c). These results suggest that FIV vector transduction of hepatocytes in vitro induces E2F targets, which cause phenotypic changes that may lead to cellular transformation. To test whether the observed cellular response was due to vector integration, per se, we attempted to use integrase-deficient (ID) FIV to transduce several hepatocyte-derived cell lines; however, only negligible transgene expression was detected as was previously observed using ID HIV-based vectors (Rafael Yanez-Munoz, personal communication). This may be due to heterochromatinization of the genome in episomal form.35 We also found that episomal VCNs in HepG2 cells transduced with ID FIV were at least fivefold lower than those of integrase-competent virus, probably due to lower viral titers. We did not use ID FIV in our in vivo model due to rapid dilution of vector copies that might have occurred in the massively proliferating fetal liver.
Figure 4.

Lentivector transduction of HepaRG cells induces phenotypic changes and E2F target gene upregulation. HepaRG liver cells were transduced with FIV-hAAT-EGFP lentivector. (a) Total RNA was extracted 24 and 72 hours posttransduction from transduced and untransduced control cells, respectively, and expression levels of E2F target genes were measured by quantitative polymerase chain reaction. Hypoxanthine-guanine phosphoribosyltransferase was used as an endogenous control. (b) Growth rates of transduced cells and untreated controls, as measured at the indicated days posttransduction, are represented as fold increase relative to the values detected 1 day postseeding. (c) Transduced cells and controls were harvested at subconfluency (50%) and 3 days after reaching confluency (100%). Cell cycle analysis was performed by fluorescence-activated cell sorting analysis after permeabilization and propidium iodide staining. Data represent mean ± SD (n = 8). *P > 0.05. FIV, feline immunodeficiency virus; hAAT, human α-antitrypsin.
Gene expression is altered in tumor-free livers of transduced mice
A comparison of microarray data between liver tissue of tumor-free FIV-transduced mice and that of control, untransduced mice revealed a set of genes that were deregulated in the transduced livers: we observed upregulation of the HCC-associated oncogene, Pdgfc, and the tumor suppressors Cdkn1a, Ddit4, and Ndrg1 (Supplementary Table S4). In addition, several HCC-associated tumor suppressors were downregulated, including Per3, Plk3, and Rnd3. Remarkably, many deregulated genes in the tumor-free, transduced livers are associated with p53 signaling, thus, bearing the following evidence of p53 activation: (i) upregulation of the p53 targets, Cdkn1a, Ddit4, Egr1, and Ndrg1, (ii) downregulation of Usp2 that results in p53 activation,36 and (iii) upregulation of Igfpb1, a known antagonist of p53-induced apoptosis in the liver.37 The deregulation of the aforementioned genes, ~1 year after transduction, suggests that the presence of the integrated FIV vector in the host genome induces a chronic cellular response, possibly aimed at creating homeostasis between proproliferative and proapoptotic signals.
Gene expression patterns differ between FIV-induced and inflammation-induced HCCs
To understand whether the aforementioned gene signatures observed in the FIV-induced tumors are also found in other murine models of HCC, we compared gene expression profiles of the six FIV-induced tumors with six inflammation-mediated tumors that developed in Mdr2-knockout (KO) mice.38 In the Mdr2-KO mice, lack of a multidrug resistance gene results in chronic inflammation, which leads to tumor development in all mice by the age of 12–16 months. On the basis of the literature, we compiled a list of 30 HCC-associated oncogenes and 79 tumor suppressors (Supplementary Table S5), in addition to a list of murine orthologs of the human CIN signature genes31 and rat E2F1-induced genes.39 We next tested each tumor for enrichment in genes from these four categories. As seen in Table 2, downregulation of tumor suppressors was not significant in either of the HCC models. However, the FIV-induced tumors exhibited a highly significant enrichment in genes belonging to the CIN, E2F, and oncogene families which was not observed in tumors of Mdr2-KO mice. Furthermore, when comparing the number of genes that were upregulated or downregulated in at least four tumors of each dataset, gene expression in the FIV-induced tumors exhibited significantly higher similarity to each other than the Mdr2-KO tumors (Supplementary Figure S5). This is remarkable, given that the Mdr2-KO tumor data were taken only from male mice of an inbred strain, whereas the FIV-induced tumor data were collected from tumors from both males and females that developed in the outbred MF-1 mouse strain. Furthermore, we did not observe either a strong CIN signature or massive induction of E2F target genes in a spontaneous liver tumor which developed in a nontransduced control MF-1 mouse (Table 2). Taken together, the data highly suggest a common mechanism underlying the FIV-induced tumorigenicity and indicate that not all murine HCCs are characterized by either a CIN signature or E2F activation.
Discussion
In this study, we characterized six HCCs that developed following fetal transduction of MF-1 mice with an FIV-based lentiviral vector. Upon sequencing of FIV integration sites, no overlap was observed between tumors and, within a single tumor, integrations were at least 5 kb apart. The distinct integration configuration in each tumor is in agreement with the unique band pattern that was obtained in the Southern blot analysis (Figure 2a). It is also consistent with our previous finding of different integration sites in each liver tumor following fetal transduction with another nonprimate lentiviral vector derived from equine infectious anemia virus.13 Furthermore, with one exception, mapped viral integration sites did not coincide with chromosomal aberrations in any tumor and no aberration was shared by all tumors. These data suggest that the site of FIV integration, per se, does not cause local chromosomal aberrations. Rather, we suggest that genomic instability, a hallmark feature of nearly all solid tumors, probably occurs with tumor progression.
Of note, no deregulation of known cancer-associated genes was detected in the vicinity of FIV integration sites. This is in contrast to reports of retroviral-induced tumors which were found to be the result of insertional mutagenesis1 in which upregulation of one specific oncogene, such as LMO2 or Mds1/Evi1, in the vicinity of a viral integration site, was observed in a majority of cohorts within each study and was the direct cause of tumor development.40,41,42 The lack of direct evidence implicating insertional mutagenesis in our data is in accordance with a previous publication which also reported no alterations in the expression of cancer-associated genes located near viral vector integration sites, in lymphomas that developed in mice transplanted with hematopoietic stem cells transduced with an HIV-based SIN vector.12
Our most notable finding was the upregulation of a large number of E2F target genes associated with DNA damage, CIN, and oncogenesis in FIV-induced tumors 1 year following transduction (Table 1). Oncogenes, including E2F1 itself, and a number of its targets, are known to induce a state of DNA replication stress and lead to the formation of DNA DSBs18 that recruit the cellular DDR which, in turn, can activate transcription and stabilize E2F1 protein via phosphorylation by the DDR-associated kinases, ATM/ATR.43 Markedly, 24 genes downstream of ATM/ATR, in the DNA repair pathway, were upregulated in at least three tumors. It should be noted that 21 of these 24 genes are also E2F targets (Table 1). In a study conducted by Ranzani et al. 10 using tumor-prone mice, HIV transduction resulted in HCCs that differed in vector integration sites from those targeted by FIV in our study. The Ranzani group identified four new oncogenes in the vicinity of viral integration sites; two of these genes (Fign and Braf) induced HCCs that exhibited upregulation of E2F transcriptional targets.
High levels of nuclear E2F1 protein were observed in the livers of FIV-transduced mice 2 weeks following transduction. The analysis of the newborn mouse livers indicates that E2F induction is an early event following FIV transduction and suggests that it plays a role early in the hepatocarcinogenic process.
We propose a model suggesting that murine fetal FIV transduction initiates oncogenesis by a global E2F-dependent cellular mechanism. It has recently become clear that the Rb/E2F pathway provides primary signals for both the control of cell proliferation and cell death depending on the cellular context.16,18 Many of the upregulated E2F target genes in the FIV-induced tumors include oncogenes that are known to cause the accumulation of DNA DSBs leading to CIN. We suggest that the observed E2F1 nuclear accumulation early on following fetal transduction may result in skewing of the E2F response toward uncontrolled proliferation rather than cell death, which results in HCC development in the adult animals.
Recently, it has been shown that HIV integration into human CD4 lymphocytes elicits a cellular double-stranded DDR, which influences cell fate and is dependent on viral integration, as this response is not obtained with ID HIV.44 It is tempting to speculate that FIV integration also initiates a DDR at viral-genome junctions which, in turn, has been shown to induce E2F.43
In contrast to the FIV-induced tumors in this study, not all HCCs exhibit a CIN or E2F signature, as only ~30% of human HCCs are highly proliferative and are characterized by overexpression of cell cycle genes that are E2F targets.45 Similarly, we observed that inflammation-induced HCCs that develop in the Mdr2-KO mice also do not exhibit a CIN or an E2F signature.
The initial events leading to tumor development following FIV transduction of fetal mice have not yet been elucidated, but one factor that may contribute to tumor formation is the high rate of fetal hepatocyte division, which was shown to render the fetal liver more prone to cancer development by chemical carcinogens.46 Furthermore, massively proliferating tissues contain a large number of DNA DSBs, most probably due to replication stress.47 Accordingly, we have recently demonstrated a link between HCC development and a high frequency of DNA DSBs present in proliferating hepatocytes of the inflamed regenerating liver of Mdr2-KO mice.48 Therefore, transduction of mice with high doses of an integrating FIV vector at the fetal stage may induce DNA replication stress and augment the incidence of DSBs leading to genomic instability.
In summary, our results strongly suggest that FIV vector-induced tumorigenesis following fetal transduction is a consequence of activation of a global cellular mechanism in which E2Fs play a pivotal role. We believe that characterization of these FIV vector-induced tumors, which share important features with certain human HCCs, may contribute to our understanding of the mechanism leading to liver tumor development and thus, present the opportunity for new therapeutic approaches.
Materials and Methods
Viral particle preparation. Viral particles carrying the FIV vector-expressing EGFP from the liver-specific human α1 antitrypsin promoter (FIV-hAAT-EGFP) were produced in HEK293T cells as previously described22 using the TransIT-LT1 transfection reagent (Mirus Bio LLC, Madison, WI). Viral particles were concentrated to a titer of ~1 × 109 transducing units/ml as previously described.22 Titer was determined by transduction of HepG2 human liver hepatoma cells and fluorescence-activated cell sorting analysis.
Immunohistochemistry. Tissue sections were stained with rabbit anti-GFP (Invitrogen, Grand Island, NY) followed by antirabbit HRP secondary antibody (Dako, Glostrup, Denmark) detected by DAB chromogen (Zymed, Grand Island, NY). Direct visualization was performed following DAPI staining (EMS).
Transduction of fetal mice. MF-1 fetal mice were transduced at 16 days gestation as previously described21 with 2 × 107 transducing units. All animal procedures were performed according to institution-approved protocols (Imperial College ethical review committees and the British Home Office).
Southern blot. Genomic DNA was extracted using the Wizard Genomic DNA purification kit (Promega, Madison, WI) and digested with HindIII that cleaves the FIV provirus once. Bands were separated on a 0.6% agarose gel, transferred to a Hybond N+ membrane (Hybond-N), and probed with an α-32P-CTP-labeled 700 bp EGFP probe.
Amplification of vector-genomic DNA junctions and sequencing of FIV integration sites. Viral integration sites in tumors were determined by amplification of vector-genomic DNA junctions by linear amplification mediated-PCR, ligation mediated-PCR, and inverse PCR on genomic DNA extracted from tumors or from FIV-transduced AML12 murine liver cells, 96 hours posttransduction using the Qiagen DNeasy kit (Qiagen, Valencia, CA).
Linear amplification mediated-PCR. Linear amplification was performed using two biotinylated primers from the FIV 5′ LTR: 5′-GTTCTCGGCCCGGATTCC and 5′-CCCGGATTCCGAGACCTC. A second DNA strand was synthesized using random primers and the resulting double-stranded DNA was digested with Tsp509I and ligated to a double-stranded linker (5′-GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTTAGG-3′ and 5′-AATTCCTAACTGCTGTGCCACTGAATTCAGATC-3′) followed by two rounds of PCR; first-round PCR primers: FIV LTR 5′-CTCGACAGGGTTCAATCTC-3′ and linker 5′- GACCCGGGAGATCTGAATTC-3′ and second-round PCR primers (nested): FIV LTR 5′-CTCAAAAGTCCTCAACAAAG-3′ and linker 5′ GATCTGAATTCAGTGGCACAG-3′. The second-round PCR products were cloned into a TOPO TA plasmid (Invitrogen). Positive clones were sequenced using the M13 universal primer.
Ligation mediated-PCR. Genomic DNA (1 µg) was digested with MseI and NarI and ligated to a double-stranded linker: 5′-TAGTCCCTTAAGCGGAG-3′ and 5′-GTAATACGACTCACTATA GGGCTCCGCTTAAGGGAC-3′, followed by two rounds of PCR; first-round PCR primers: FIV LTR 5′-CCTGTCGAGTATCTGTGTAATCTTTTTTACC-3′ and linker 5′-GTAATACGACTCACTATAGGGC-3′ second-round (nested) primers: FIV LTR 5′-GTGAGGTCTCGGAATCCGGGC-3′ and linker 5′-AAGGCTCCGCTTAAGGGA C-3′. The resulting PCR products were cloned and sequenced as described above.
Deep sequencing of FIV integration sites in AML12 cells was performed as previously described.49 First-round primers were as mentioned above. Second-round FIV LTR primer: 5′-GCCTCCCTCGCGCCATCAGNNNNGTGAGGTCTCGGAATCCGGGC-3′ NNNN represents a 4 nucleotide barcode that was incorporated into each replicate experiment and was one of the following: TAGC, CGTA, ATCG, or TCAG. Second-round linker primer: GCCTTGCCAGCCCGCTCAGAGGGCTCCGCTTAAGGGAC. The amplified vector-genome junctions were sequenced using the GS-FLX Genome Sequencer (Roche/454 Life Sciences, Branford, CT) pyrosequencing platform. Crude sequence reads were processed by CD-HIT (cd-hit.org) and mapped onto the mouse genome by an automated bioinformatics pipeline, ISA, developed by Martijn Brugman (Leiden University Medical Center, The Netherlands).
Inverse PCR. Inverse PCR was performed by digesting genomic DNA with CspI, self-ligation, and linearization with AlwNI. Two rounds of PCR followed; first-round PCR primers: FIV gag 5′-GAATGGACAGGGGCGAGATTGG-3′ and FIV LTR 5′-CCTCAAGGGAGAACTCAAAAGTC-3′ second-round (nested) PCR primers: FIV gag 5′-GTAATGTTGCTGTAGGAGTAGG-3′ and FIV LTR 5′-GACTCCTCGAAGTTTCACAAAGC-3′. The isolated DNA fragments were cloned and sequenced as above.
Quantitative PCR and real-time PCR. VCN was measured by qPCR assay performed on an AB 7900 HT fast real-time PCR system (Applied Biosystems, Foster City, CA) using the following primer and probe sequences of the FIV ψ packaging signal: forward primer: 5′-AGCAGAACTCCTGCTGACCTAAA; reverse primer: 5′-TCGAGTCTGCTTCACTAGAGATACTC; and probe sequence: FAM-ACTGTTAGCAGCGTCTGCTACTGCTTCCCT. A standard curve containing viral vector DNA, ranging from 101 to 107 copies, was mixed with a constant amount of control mouse liver DNA and run in duplicate. The amplification reactions of 25 μl contained: 12.5 μl of TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA), 100 nM of each primer and probe, and 100 ng genomic DNA. The linear regression coefficient of the standard curve was 0.99 and variation between the Ct of each duplicate was <0.5 Ct. Estimated DNA content of an adult rat hepatocyte was 7.7 pg.50 E2F target gene expression levels were measured by qPCR on cDNA prepared from total RNA extracted from cultured cells and mouse liver tissue using random primers, dNTPs, and M-Mlv RT (Promega) and were measured using Sybrgreen (Invitrogen) and the primers listed in Supplementary Table S6.
Microarrays. Microarrays (Affymetrix Mouse Genome Array 430A) were performed on total RNA isolated from the tumors or matched nontumorous liver tissues using the Trizol reagent (Invitrogen). Microarray data have been deposited into a public database and can be accessed using the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=htofneeuiwswkdm&acc=GSE19306.
Gene expression analysis was performed using “Partek” software and the GCRMA algorithm following median polish normalization. Statistical significance of the specific gene expression signatures has been evaluated using “R” statistical software and a strict Fisher's two-tailed test. Genes were considered deregulated when exhibiting a twofold change relative to control (matched, nontumorous) samples.
Comparative genomic hybridization. Genomic DNA was extracted using the Wizard Genomic DNA purification kit (Promega) and subjected to comparative genomic hybridization arrays (Miltenyi Biotech, Germany) on 4 × 44 K chips (Agilent, Santa Clara, CA). Comparative genomic hybridization data can be accessed from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=fzsljuusgoykavm&acc=GSE2590. The arrays were scanned in a dual-laser scanner (Agilent DNA Microarray Scanner) and the images were extracted and analyzed through Agilent Feature Extraction software and DNA Analytics software (v3.5), respectively.
In vitro transduction, proliferation, and cell cycle analysis. HepaRG, kindly donated by Jorg Heeren (University Medical Center Hamburg-Eppendorf), is a human bipotential liver cell line established from an HCV-associated liver tumor that shares many features with adult hepatocytes, has a normal karyotype, and does not grow on soft agar. The cells were grown in William's Medium (Gibco, Carlsbad, CA) supplemented with 10% fetal calf serum, hydrocortisone, and insulin. Cells were transduced with FIV-hAAT-EGFP viral particles at an MOI of 50 in the presence of 5 µg/ml polybrene. For the determination of proliferation rate, 2,500 transduced HepaRG cells were seeded per well in a 96-well plate and cell growth was measured three times a week using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI). Mean optical density values were compared with the mean optical density obtained 24 hours after seeding. Contact inhibition was determined by analyzing the percentage of cells in S-phase on subconfluent and confluent cell cultures. Cells were fixed and incubated in propidium iodide solution (working concentration 50 µg/ml) containing RNase A (500 U/ml) at 37°C for 30 minutes before evaluation on a flow cytometer.
SUPPLEMENTARY MATERIAL Figure S1. Distribution of chromosomal aberrations in the FIV-induced liver tumors. Figure S2. A graphical representation of the principal component analysis of genome scale gene expression profiling data obtained from FIV-transduced and control mice. Figure S3. Validation of genome-wide microarray expression data by qRT-PCR. Figure S4. High level of E2F target gene expression in livers of young mice. Figure S5. Comparison of deregulated genes between the two murine HCC models, namely, FIV-induced (current study) and inflammation-mediated Mdr2-KO mice34. Table S1. FIV integration sites within liver tumors. Table S2. Syntenic regions of human common fragile sites in the mouse genome. Table S3. Summary of FIV integration profiling following high throughput sequencing of transduced AML 12 cells. Table S4. Genes deregulated in livers of tumor-free, transduced mice, compared with untransduced controls. Table S5. HCC-associated oncogenes and tumor suppressors. Table S6. Primer sequences of E2F target genes used for qRT-PCR.
Acknowledgments
We thank Assaf Bester and Batsheva Kerem from the Hebrew University, Jerusalem, Israel for common fragile sites data and helpful discussion, Ali Nowrouzi from the German Cancer Research Center, Heidelberg, Germany, and Alessandra Recchia from the University of Modena and Reggio Emilia, Modena, Italy for help with the inverse and LM-PCR protocols and Aviezer Lifshitz for microRNA location analysis. This study was supported by ISF (E.G.), FP7 program LSHB-CT-2008-223317 (LIV-ES) (E.G.); the Kamea Scientific Foundation (D.G.); European Research Council grant "SomaBio" (S.M.K.B. and S.N.W.). Additional support was received from the Lillyan &Alfy Nathan (R.C.), Barbara Fox Miller, and Wolfson Foundations (E.G.). E.G. is part of the DFG SFB 841 program project. E.G. is supported by the I-CORE 41/11 ISF center of excellence center.
Supplementary Material
References
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn DB. Update on gene therapy for immunodeficiencies. Clin Immunol. 2010;135:247–254. doi: 10.1016/j.clim.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Buchholz CJ, Cichutek K. Is it going to be SIN?: a European Society of Gene Therapy commentary. Phasing-out the clinical use of non self-inactivating murine leukemia virus vectors: initiative on hold. J Gene Med. 2006;8:1274–1276. doi: 10.1002/jgm.966. [DOI] [PubMed] [Google Scholar]
- Xu W, Russ JL, Eiden MV. Evaluation of residual promoter activity in ?-retroviral self-inactivating (SIN) vectors. Mol Ther. 2012;20:84–90. doi: 10.1038/mt.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Schambach A, Brugman MH, Wicke DC, Knoess S, Li Z, et al. Leukemia induction after a single retroviral vector insertion in Evi1 or Prdm16. Leukemia. 2008;22:1519–1528. doi: 10.1038/leu.2008.118. [DOI] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzani M, Cesana D, Bartholomae CC, Sanvito F, Pala M, Benedicenti F, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013;10:155–161. doi: 10.1038/nmeth.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Cesana D. Genotoxicity assay for gene therapy vectors in tumor prone Cdkn2a-/- mice. Meth Enzymol. 2012;507:171–185. doi: 10.1016/B978-0-12-386509-0.00009-0. [DOI] [PubMed] [Google Scholar]
- Ginn SL, Liao SH, Dane AP, Hu M, Hyman J, Finnie JW, et al. Lymphomagenesis in SCID-X1 mice following lentivirus-mediated phenotype correction independent of insertional mutagenesis and gammac overexpression. Mol Ther. 2010;18:965–976. doi: 10.1038/mt.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themis M, Waddington SN, Schmidt M, von Kalle C, Wang Y, Al-Allaf F, et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol Ther. 2005;12:763–771. doi: 10.1016/j.ymthe.2005.07.358. [DOI] [PubMed] [Google Scholar]
- Nowrouzi A, Cheung WT, Li T, Zhang X, Arens A, Paruzynski A, et al. The fetal mouse is a sensitive genotoxicity model that exposes lentiviral-associated mutagenesis resulting in liver oncogenesis. Mol Ther. 2013;21:324–337. doi: 10.1038/mt.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeschla EM, Looney DJ. CXCR4 is required by a nonprimate lentivirus: heterologous expression of feline immunodeficiency virus in human, rodent, and feline cells. J Virol. 1998;72:6858–6866. doi: 10.1128/jvi.72.8.6858-6866.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR. Balancing the decision of cell proliferation and cell fate. Cell Cycle. 2009;8:532–535. doi: 10.4161/cc.8.4.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- Pickering MT, Kowalik TF. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene. 2006;25:746–755. doi: 10.1038/sj.onc.1209103. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19:5054–5062. doi: 10.1038/sj.onc.1203885. [DOI] [PubMed] [Google Scholar]
- Waddington SN, Mitrophanous KA, Ellard FM, Buckley SM, Nivsarkar M, Lawrence L, et al. Long-term transgene expression by administration of a lentivirus-based vector to the fetal circulation of immuno-competent mice. Gene Ther. 2003;10:1234–1240. doi: 10.1038/sj.gt.3301991. [DOI] [PubMed] [Google Scholar]
- Condiotti R, Curran MA, Nolan GP, Giladi H, Ketzinel-Gilad M, Gross E, et al. Prolonged liver-specific transgene expression by a non-primate lentiviral vector. Biochem Biophys Res Commun. 2004;320:998–1006. doi: 10.1016/j.bbrc.2004.06.044. [DOI] [PubMed] [Google Scholar]
- Grinshpun A, Condiotti R, Waddington SN, Peer M, Zeig E, Peretz S, et al. Neonatal gene therapy of glycogen storage disease type Ia using a feline immunodeficiency virus-based vector. Mol Ther. 2010;18:1592–1598. doi: 10.1038/mt.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX, et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679–1689. doi: 10.1002/hep.24563. [DOI] [PubMed] [Google Scholar]
- Bester AC, Schwartz M, Schmidt M, Garrigue A, Hacein-Bey-Abina S, Cavazzana-Calvo M, et al. Fragile sites are preferential targets for integrations of MLV vectors in gene therapy. Gene Ther. 2006;13:1057–1059. doi: 10.1038/sj.gt.3302752. [DOI] [PubMed] [Google Scholar]
- Letouzé E, Allory Y, Bollet MA, Radvanyi F, Guyon F. Analysis of the copy number profiles of several tumor samples from the same patient reveals the successive steps in tumorigenesis. Genome Biol. 2010;11:R76. doi: 10.1186/gb-2010-11-7-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana D, Sgualdino J, Rudilosso L, Merella S, Naldini L, Montini E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J Clin Invest. 2012;122:1667–1676. doi: 10.1172/JCI62189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiani A, Paleari Y, Sartori D, Mezzadra R, Miccio A, Cattoglio C, et al. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J Clin Invest. 2012;122:1653–1666. doi: 10.1172/JCI61852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Mayhew CN, Carter SL, Fox SR, Sexton CR, Reed CA, Srinivasan SV, et al. RB loss abrogates cell cycle control and genome integrity to promote liver tumorigenesis. Gastroenterology. 2007;133:976–984. doi: 10.1053/j.gastro.2007.06.025. [DOI] [PubMed] [Google Scholar]
- Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012;72:13–17. doi: 10.1158/0008-5472.CAN-11-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion MJ, Hantz O, Durantel D. The HepaRG cell line: biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol Biol. 2010;640:261–272. doi: 10.1007/978-1-60761-688-7_13. [DOI] [PubMed] [Google Scholar]
- Pelascini LP, Janssen JM, Gonçalves MA. Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum Gene Ther. 2013;24:78–96. doi: 10.1089/hum.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–986. doi: 10.1038/sj.emboj.7601567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes Dev. 2007;21:3095–3109. doi: 10.1101/gad.1567107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenellenbogen M, Mizrahi L, Pappo O, Klopstock N, Olam D, Jacob-Hirsch J, et al. Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol Cancer Res. 2007;5:1159–1170. doi: 10.1158/1541-7786.MCR-07-0172. [DOI] [PubMed] [Google Scholar]
- Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Baum C. Preventing and exploiting the oncogenic potential of integrating gene vectors. J Clin Invest. 2009;119:755–758. doi: 10.1172/JCI38831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Calmels B, Ferguson C, Laukkanen MO, Adler R, Faulhaber M, Kim HJ, et al. Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood. 2005;106:2530–2533. doi: 10.1182/blood-2005-03-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- Cooper A, García M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature. 2013;498:376–379. doi: 10.1038/nature12274. [DOI] [PubMed] [Google Scholar]
- Boyault S, Rickman DS, de Reyniès A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD, Rao KV, Mihailovich N. Neoplastic response of mouse tissues during perinatal age periods and its significance in chemical carcinogenesis. Natl Cancer Inst Monogr. 1979;51:239–250. [PubMed] [Google Scholar]
- Sorrentino B. Assessing the risk of T-cell malignancies in mouse models of SCID-X1. Mol Ther. 2010;18:868–870. doi: 10.1038/mt.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash H, R Gross E, Edrei Y, Ella E, Israel A, Cohen I, et al. Accelerated carcinogenesis following liver regeneration is associated with chronic inflammation-induced double-strand DNA breaks. Proc Natl Acad Sci USA. 2010;107:2207–2212. doi: 10.1073/pnas.0908867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuffi A, Barr SD. Identification of HIV integration sites in infected host genomic DNA. Methods. 2011;53:39–46. doi: 10.1016/j.ymeth.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Bibbiani C, Tongiani R, Viola-Magni MP. I. Quantitative determination of the amount of DNA per nucleus by interference microscopy. J Cell Biol. 1969;42:444–451. doi: 10.1083/jcb.42.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.