

Abstract
Within the oncolytic virus field, the extent of virus replication that is essential for immune stimulation to control tumor growth remains unresolved. Using infected cell protein 0 (ICP0)-defective oncolytic Herpes simplex virus type 1 (HSV-1) and HSV-2 viruses (dICP0 and dNLS) that show differences in their in vitro replication and cytotoxicity, we investigated the inherent features of oncolytic HSV viruses that are required for potent antitumor activity. In vitro, the HSV-2 vectors showed rapid cytotoxicity despite lower viral burst sizes compared to HSV-1 vectors. In vivo, although both of the dICP0 vectors initially replicated to a similar level, HSV-1 dICP0 was rapidly cleared from the tumors. In spite of this rapid clearance, HSV-1 dICP0 treatment conferred significant survival benefit. HSV-1 dICP0–treated tumors showed significantly higher levels of danger-associated molecular patterns that correlated with higher numbers of antigen-presenting cells within the tumor and increased antigen-specific CD8+ T-cell levels in the peripheral blood. This study suggests that, at least in the context of oncolytic HSV, the initial stages of immunogenic virus replication leading to activation of antitumor immunity are more important than persistence of a replicating virus within the tumor. This knowledge provides important insight for the design of therapeutically successful oncolytic viruses.
Introduction
Most conventional cancer therapeutics achieve limited efficacy leading to the search for novel ways to treat cancer. Oncolytic viruses selectively propagate in and kill cancer cells while displaying minimal adverse effects in healthy cells.1,2 The collection of gain or loss of function mutations of a given cancer type determine the nature of the selective growth advantage over normal cells for an oncolytic virus.1 Herpes simplex virus type 1 (HSV-1) was the first virus used to show that gene deletion can render a virus oncolytic.3 The HSV-1 immediate early gene infected cell protein 0 (ICP0) encodes a protein responsible for overcoming the host interferon (IFN) response at multiple points.4,5 HSV-1 lacking functional ICP0 showed oncolytic activity in cancer cells with a decreased capacity to respond to IFN, leaving healthy cells intact.6 KM100 is one of the ICP0 null oncolytic viruses that has been used in different murine tumor models ranging from a highly immunogenic polyoma middle T model to nontolerized and tolerized transgenic HER-2/neu models, with variable survival benefits in tumor-bearing mice.6,7
Since its early inception, the field of oncolytic virology has rapidly advanced and clinical trials are underway to utilize oncolytic viruses in human cancer patients. There continues to be new viruses reported from multiple sources that will yield new opportunities for selectively attacking cancer cells. At some point, however, simple expansion of the repertoire of oncolytic vectors must be accompanied by detailed comparison.8 Comparison studies of oncolytic viruses are especially desired to understand what inherent features of oncolytic viruses dictate their therapeutic success.
Using different classes of RNA and DNA oncolytic viruses and a glioma xenograft model, it was shown that viruses that replicate and spread fast in vitro show better response in vivo.9 Similar work was carried out using two HSV-1–based oncolytic viruses, G207 and NV1020, that have varying in vitro replication capacities. In a xenograft cancer model, in vitro replication and cytotoxicity correlated with in vivo antitumor activity.10 Importantly, these studies utilized nude mice that lack the cell-mediated adaptive immune system. Comparison of ICP0 null oncolytic HSV-1 and vesicular stomatitis virus in immunocompetent mice showed that in vitro replication does not always correlate with in vivo oncolytic capacity.7 However, these two viruses differ in many aspects of viral biology. Comparing different viruses in one model is difficult owing to differences in tropism, virus dose, and route of administration. As a result, we chose to compare oncolytic viruses that are based on similar ICP0 mutations within HSV-1 and HSV-2, two alphaherpesviruses with nearly an identical set of ~75 colinear genes.
This study was carried out to identify inherent features of oncolytic HSV viruses that are associated with better antitumor immunity and survival in immune competent tumor-bearing mice. We report that in vitro cytotoxicity and in vivo virus persistence do not correlate with efficacy. Instead, HSV-mediated therapeutic benefit was associated with the induction of immunogenic apoptotic cell death, characterized by increased intratumoral expression of heat shock protein-70 (HSP-70) and elevated serum high mobility group box 1 (HMGB1) levels in the peripheral blood, increased infiltration of antigen-presenting cells and a subsequent stimulation of antigen-specific CD8+ T-cell responses.
Results
HSV-1 and HSV-2 ICP0 mutants show different CPE, viral burst size, and effects on cellular metabolism in vitro
We have previously shown that HSV-1 ICP0 mutants exert antitumor activity in various murine cancer models.6,7 Given the safety profile of the HSV-2 ICP0 mutant viruses dNLS and dICP0 in immunocompetent mice,11 we engineered HSV-1 and HSV-2 dNLS and dICP0 recombinants expressing both green fluorescent protein (GFP) and firefly luciferase to enable direct in vitro and in vivo comparisons of replication, cellular toxicity, and oncolytic activity. We sequenced the ICP0 region, using DNA extracted from purified virus stocks, to confirm the presence of the attenuating mutations (data not shown). Additionally, functional assays were used to verify the IFN sensitivity of these ICP0 mutant viruses11 (data not shown).
In HSV-1, the dICP0 mutation is more attenuating than the dNLS mutation with respect to cytopathic effect (CPE) in the murine HER-2/neu breast tumor line, TUBO, despite elevated production of GFP (Figure 1a,b). At 24 hours postinfection, HSV-2 dICP0 showed higher levels of CPE than HSV-1 dICP0, particularly at higher multiplicity of infection (MOI) (Figure 1a,b), while 48 hours postinfection, both viruses showed similar levels of CPE at MOIs >1 (data not shown). Consistent with the CPE data, cellular metabolism was consistently lower following HSV-2 infection in an MOI-dependent fashion (Figure 1c,d). While the viral burst sizes of wild-type HSV-1 (157 ± 23.4), dNLS (149 ± 36.5), and dICP0 (136.3 ± 44.8) were similar, that of wild-type HSV-2 (14.0 ± 1.5) was 10-fold lower (Figure 1e), despite a higher degree of CPE (Figure 1a,b). The HSV-2 dNLS (3.6 ± 0.5) and dICP0 (1.3 ± 0.1) viruses showed the lowest burst sizes (Figure 1e). These results indicate that HSV-1 and HSV-2 ICP0–defective oncolytic viruses have differences in in vitro cytotoxicity that do not strictly correlate with virus replication, as evidenced by GFP expression, or production of infectious virus.
Figure 1.
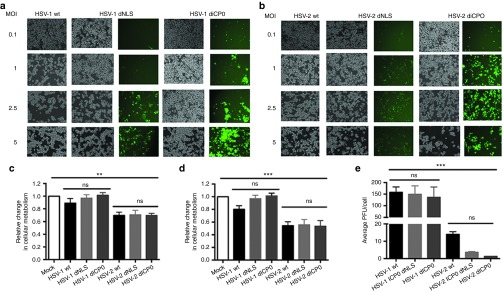
Herpes simplex virus (HSV)-1 and HSV-2 ICP0 mutants show differences in cytopathic effect, cellular metabolism and viral burst size in vitro. Bright field and green fluorescent protein fluorescence images of TUBO cells infected with (a) HSV-1 or (b) HSV-2 ICP0 mutants for 24 hours. Cellular metabolism of TUBO cells 24 hours following infection with HSV-1 or HSV-2 ICP0 mutants at (c) MOI 1 and (d) MOI 5. Burst sizes of HSV-1 and HSV-2 ICP0 mutants in TUBO cells 48 hours following infection with an (e) MOI of 0.5. The average and standard deviations are from three independent experiments each run with three biological replicates. The statistical significance of the mean differences between groups was analyzed using ANOVA. ***P < 0.0005, **P < 0.005.
HSV-2 oncolytic mutants induce early apoptotic cell death and release of HMGB1 in vitro
Given that HSV-1 and HSV-2 dNLS and dICP0 show differences in CPE, cellular metabolism and viral burst size, we next wanted to examine the kinetics of apoptotic cell death induced by these oncolytic viruses. Caspase-3, a 35 kDa protein, is cleaved by initiator caspases during apoptosis, forming activated proteins of 17 and 12 kDa. TUBO cells were treated with staurosporin as a positive control for caspase 3 cleavage (Figure 2a). Protein samples were harvested from HSV-infected TUBO cells and the level of cleaved caspase 3 was measured. All HSV-1 and wild-type HSV-2 viruses were able to block apoptosis throughout the experimental time course (Figure 2b), while HSV-2 dICP0- and dNLS-infected cells showed levels of cleaved caspase 3 above mock samples starting 24 hours postinfection. Among the four ICP0 mutant virus infections, HSV-2 dICP0–infected cells had a strikingly higher level of apoptosis that peaked 30 hours postinfection. We consistently detected low levels of cleaved caspase 3 in mock samples at later times as these monolayers reached confluency (data not shown).
Figure 2.

HSV-2 dICP0–infected TUBO cells show an early apoptotic cell death. Western blot analysis of cleaved caspase 3 in TUBO cells treated with (a) 62.5 nm staurosporin for 16 hours or (b) following mock treatment or infection with HSV wild-type (MOI 0.5) or ICP0 mutant (MOI 2.5) viruses. The blots were used to calculate the ratio of cleaved caspase 3 to actin by densitometry using ImageJ software. Shown is a representative of three independent experiments. (c) HMGB1 levels in the supernatant after infection with different MOIs of HSV-1 and HSV-2 mutants for 24-hour period. The plots are values generated from an ELISA using pooled samples from triplicate independent experiments.
Additionally, we assayed cellular supernatants for HMGB1 release following infection of TUBO cells with HSV-1 and HSV-2 viruses. Although we failed to detect release of HMGB1 over a 24-hour infection period with any HSV-1 virus, we detected HMGB1 in supernatants following infection with all three HSV-2 viruses, in an MOI-dependent fashion (Figure 2c).
In vivo efficacy of HSV ICP0 mutants does not correlate with in vitro replication or induction of apoptosis
Having the in vitro observation that the ICP0 mutants of HSV-1 and HSV-2 show different CPE, viral burst size, apoptotic cell death, and HMGB1 release, we wanted to test the in vivo therapeutic efficacy of these mutant viruses to determine what characteristic(s) is most important. The four mutant viruses were used to treat subcutaneous TUBO tumors in BALB/c mice as depicted in Figure 3. The Kaplan–Meier estimates of survivals and relative fold changes of tumor volumes are shown in Figure 4. Tumor-bearing mice treated with phosphate-buffered saline (PBS) had rapidly growing tumors (Figure 4a) reaching end point with a median survival of 12 days, with no mice suriving to the end of the study. HSV-2 dICP0–treated mice showed the most similar pattern of survival to the PBS-treated mice with a median survival of 19.5 days and 10% survival at the end of the study period. The other HSV ICP0 mutants (HSV-1 dICP0, HSV-1 dNLS, and HSV-2 dNLS) conferred slower tumor growth and higher survival benefit compared to HSV-2 dICP0. The HSV-1 dNLS–treated mice showed overall survival of 44%. HSV-2 dNLS and HSV-1 dICP0 treatments resulted in the highest survival rates, with 50% survivors at the end of the study. However, only HSV-1 dICP0 treatment showed a statstically significant survival benefit over PBS treatment (Figure 4a). Mice demonstrating complete tumor regression were rechallenged with 5 × 105 TUBO cells on the contralateral flank and tumor formation was monitored. All mice were refractory to tumor rechallenge (data not shown).
Figure 3.
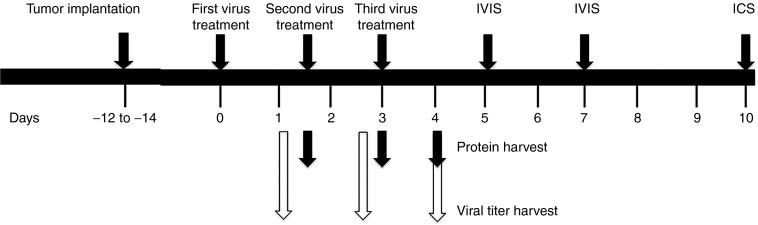
Schematic diagram of experimental procedures performed. Illustrated are the relative time frames of tumor implantation, oncolytic virus treatment, harvesting of tumors for protein and virus quantitation, IVIS imaging, and blood collection for intracellular cytokine staining.
Figure 4.

Oncolytic HSV ICP0 mutants show different levels of therapeutic efficacy in vivo. A total of 5 × 105 TUBO cells were implanted into BALB/c mice by subcutaneous injection into the left flank. When tumors reached treatable size, 1 × 107 total pfu of each virus was administered intratumorally three times every 36 hours. (a) Kaplan–Meier survival analysis following treatment. (b) Fold change in tumor volume relative to the tumor volume at the start of treatment. **P < 0.005.
Taken together, HSV-1 dICP0 caused the least CPE and failed to affect cellular metabolism, despite a high burst size. In contrast, HSV-2 dICP0 caused significant CPE and decreased cellular metabolism, despite the lowest burst size. Moreover, HSV-1 dICP0 and HSV-2 dICP0 induced the lowest and highest level of apoptosis, respectively. Surprisingly, in vivo, HSV-1 dICP0 is the only virus treatment that provided significantly increased survival, while HSV-2 dICP0 treatment was similar to PBS treatment. Since the largest discrepancies were observed between HSV-1 and HSV-2 dICP0 viruses, subsequent studies focused on these two oncolytic viruses.
HSV-1 dICP0 is cleared from tumors at a faster rate than HSV-2 dICP0
Given that HSV-1 and HSV-2 dICP0 oncolytic viruses showed a large difference in viral burst size in vitro, we wanted to study if the extent of virus replication in vivo contributed to antitumor activity. All of the ICP0 mutant viruses used in this study express firefly luciferase. Thus, we used the in vivo imaging spectrum to monitor the luminescence signal detected following expression of luciferase within the tumors of treated mice. Comparison of background adjusted total photons shows that while luciferase production was similar over the first 5 days of virus treatment, the luminescence signal from HSV-1 dICP0–treated mice was undetectable by day 7 (Figure 5a). To validate the results of in vivo imaging, which measures luciferase production, but not productive virus replication per se, we resected tumors to quantify the amount of virus 24 hours after each intratumoral virus injections (Figure 3). Both HSV-1 and HSV-2 dICP0–treated tumors had comparable total plaque-forming unit (pfu)/mg of tumor 24 hours after the start of treatment (Figure 5b). However, HSV-1 dICP0 titers/mg of tumor declined at a faster rate than HSV-2 dICP0. Indeed, while HSV-1 dICP0 was cleared from the tumor by 96 hours (44.2 ± 9.7 pfu/mg of tumor), significant levels of HSV-2 dICP0 persisted in the tumor bed (2064.9 ± 918.3 pfu/mg of tumor). Thus, despite similar initial viral titers 24 hours after treatment, HSV-1 dICP0 is more rapidly cleared from tumors than HSV-2 dICP0.
Figure 5.

Locally administered HSV-1 dICP0 is rapidly cleared from subcutaneous tumors. Replication of HSV dICP0 mutants in subcutaneous tumors of TUBO cells in BALB/c mice. (a) Log of total flux calculated following IVIS imaging of tumor-bearing mice treated with firefly luciferase expressing oncolytic HSV dICP0 viruses. Luciferase activity was measured following injection of mice with D-luciferin (n = 5 for each treatment). (b) Viral titers from tumors that were resected and homogenized at the indicated times posttreatment.
HSV-1 dICP0–treated tumors have increased levels of immunomodulatory molecules suggestive of an immunogenic type of apoptosis
There is strong evidence that the ability of oncolytic virotherapy to stimulate the host antitumor immune response contributes positively to the therapeutic efficacy.12 Since HSV-1 dICP0 replicates very well in vitro and gets cleared at a faster rate than HSV-2 dICP0, we hypothesized that HSV-1 dICP0 might induce a type of cancer cell death that is more immunogenic than HSV-2 dICP0. Tumors from treated mice were harvested at 1.5, 3, and 4 days posttreatment (three mice per time point) to quantify protein levels of cleaved caspase 3 and the immunomodulatory protein HSP-70. At all time points, treated tumors demonstrated higher levels of apoptosis than PBS-treated tumors, with higher levels routinely observed in mice treated with HSV-1 dICP0 (Figure 6a–c). While the expression of HSP-70 remained low at early times posttreatment, by day 4 posttreatment, HSV-1 dICP0–treated tumors consistently showed higher expression of HSP-70 compared to HSV-2 dICP0–treated tumors (Figure 6a–c).
Figure 6.

HSV-1 dICP0 treatment leads to a higher expression of immunomodulatory molecules compared to HSV-2 dICP0. In vivo assessment of immunogenic cell death in tumor-bearing BALB/c mice treated with HSV dICP0 oncolytic viruses. Tumors (three per group) were harvested at (a) 36, (b) 72, and (c) 96 hours after the first oncolytic virus treatment, and the levels of cleaved caspase 3 and HSP-70 were measured by western blot analyses. (d) Serum levels of HMGB1 were measured by ELISA 24 hours after the first treatment with HSV dICP0 oncolytic viruses.
Overall, the cleaved caspase 3 data suggest that both HSV-1 and HSV-2 dICP0–treated tumors were able to induce apoptosis, but HSV-1 dICP0–treated tumors had higher expression of HSP-70, suggesting immunogenic apoptotic cell death. To examine if HSV-1 dICP0 virus-infected tumors have generated a systemic immunogenic response, we quantified the level of serum HMGB1 at 24 hours posttreatment, when the viruses showed comparable titers. HSV-1 dICP0–treated mice showed significantly higher serum HMGB1 levels compared to either HSV-2 dICP0– or PBS–treated tumor-bearing mice (Figure 6d). At later time points, we saw fluctuations in HMGB1 levels (data not shown). All together, these results suggest that HSV-1 dICP0 more efficiently stimulates an immunogenic type of cell death characterized by increased expression of immunomodulatory molecules with in the tumor and the peripheral blood.
HSV-1 dICP0–treated tumors show increased infiltration of antigen-presenting cells and HER-2–specific CD8+ T-cell response
Immunogenic cell death induced by anticancer chemotherapeutics has been shown to modulate the intratumoral dynamics of antigen-presenting cells for effective antigen presentation and activation of antitumor immunity.13,14 We resected tumors treated with PBS or dICP0 viruses and isolated tumor-infiltrating lymphocytes at a time where there are differential growth patterns of tumors within the treatment groups. We saw a trend of increased infiltration of total CD45+ CD11b+ tumor-infiltrating lymphocytes in HSV-1 dICP0–treated tumors and, to a lesser extent, HSV-2 dICP0–treated tumors (Figure 7a). The enhanced variability in the response to HSV-1 dICP0 correlates with the variability in outcome (50% survival versus 10% for HSV-2 dICP0; Figure 4a). Moreover, the HSV-1 dICP0–treated tumors had the highest number of conventional dendritic cells (CD45+ CD11b+ CD11c+) and inflammatory dendritic cells (CD45+ CD11b+ Ly6Chi) compared to the other treatments (Figure 7 b,c). However, both HSV-1 dICP0– and HSV-2 dICP0–treated tumors had similar levels of neutrophil influx (CD45+ CD11b+ Ly6Chi) (Figure 7d).
Figure 7.

HSV-1 dICP0–treated tumors have higher infiltration of antigen-presenting cells and neutrophils. Tumors were treated with three doses of HSV dICP0. Ten days after treatment, the tumors were resected and CD45+ immune cells isolated using EasySep magnetic bead positive selection. Cells were directly stained with the indicated surface markers. The average and standard deviation of cell numbers were generated from five mice per each treatment group. ns, not significant, *P < 0.05.
Next, we compared the HER-2–specific CD8+ T-cell response against this tumor antigen in the peripheral blood of mice treated with the HSV dICP0 mutants (Figure 8a). Blood was collected day 10 postinfection, during the peak of the immune response.7 As shown in Figure 8a, a HER-2–specific CD8+ T-cell response was barely detected in the blood of HSV-2 dICP0–treated mice, similar to the PBS-treated mice. In contrast, a significant tumor antigen-specific CD8+ T-cell response was observed in HSV-1 dICP0–treated tumor-bearing mice, with 7.8 times more antigen-specific CD8+ T cells than HSV-2 dICP0–treated mice.
Figure 8.

HSV-1 dICP0–treated tumor-bearing mice have higher antigen-specific cytotoxic CD8+ T cells. (a) Percentage of HER-2–specific CD8+ T cells in the peripheral blood of treated tumor-bearing BALB/c mice. Blood samples were processed, and cells were restimulated with the HER-2–immunodominant epitope peptide and stained for the cell surface marker CD8 and intracellular IFN-γ to identify HER-2–specific CD8+ T cells (n = 5 for each treatment). The χ2 test was used to test the statistical significance of the differences in proportion of antigen-specific CD8+ T cells. (b) Viability assessed by measurement of cellular metabolism in TUBO cells left untreated or cocultured with CD8+ T splenocytes harvested from tumor-bearing mice treated with PBS or HSV dICP0. The values are average of three wells that used pooled splenocytes from five mice. ***P < 0.0005.
To verify that these antigen-specific CD8+ T cells are cytotoxic to TUBO cells, splenocytes from treated tumor-bearing mice were harvested to isolate CD8+ T cells for in vitro cytotoxicity assays. Compared to mock-treated cells, cells incubated with splenocytes harvested from treated mice showed a reduction in cellular metabolism, with splenocytes from HSV-1 dICP0–treated mice having the largest effect (Figure 8b).
We also investigated the level of CD4+ foxp3+ Tregs within the tumors of treated mice. Although there was a trend for lower levels of Tregs in HSV-1 dICP0–treated tumors, there was no statistically significant difference in the level of Tregs between the treatment groups (Supplementary Figure S1).
Discussion
By comparing four different, but highly related, ICP0 mutant viruses that vary in their in vitro replication and cytotoxicity, we addressed whether any in vitro property correlated with antitumor activity in vivo. HSV dNLS mutants do not display remarkable differences in their antitumor activity in vivo, and only showed distinct differences in their burst size in vitro. In terms of in vivo antitumor activity, the significant difference was observed between HSV-1 and HSV-2 dICP0. These two viruses were thus the focus of further characterization. Of interest, HSV-1 dICP0 showed a log higher burst size in vitro, while in vivo, both viruses initially showed similar virus titers, with HSV-1 dICP0 being rapidly cleared from tumors. This observation confirms our previous findings that in vitro replication does not always translate in vivo.7 Moreover, we observed that prolonged persistence of virus within the tumor is not a requirement of effective antitumor activity.
It is becoming increasingly clear that the immune system has evolved to recognize and eliminate dying and dead cells and translates the history of premortem stress into an immune response.13,14,15,16 Immunogenic cell death plays a significant role in the initiation of potent antitumor immune responses in murine models13,16 and human patients.17 Oncolytic viruses such as CD40L expressing Adenovirus18 and oncolytic Measles virus19 induce features of immunogenic cancer cell death such as calreticulin surface exposure and ATP and HMGB1 release into the extracellular environment during infection of tumor cells in vitro, along with activation of antitumor immune response that translates into survival benefit in preclinical models. However, unlike the characterization of immunogenic cancer cell death during chemotherapy in vivo, these oncolytic virus studies did not show direct evidence of immunogenic cell death in murine models in tumor-bearing mice.
While both HSV dICP0 viruses induced apoptosis in vivo, HSV-1 dICP0 additionally increased the expression of the immunomodulatory molecule HSP-70. HSPs have been characterized as danger-associated molecular patterns, inducers of sterile inflammation and innate immunity.20 Higher expression of HSP-70 activates dendritic cells and enhances T-cell–mediated immune killing of cancer cells.21,22,23,24,25 Similar to HSP-70, there was a significantly higher serum level of HMGB1 at early times after HSV-1 dICP0 therapy. HMGB1 is a host protein that functions as an inflammatory signal that alerts the host to stress, cell death, and microbial invasion.26,27 HMGB1 is released passively from dying tumor cells or actively secreted in response to inflammatory stimuli by immune cells such as macrophages, natural killer cells, neutrophils, and mature dendritic cells, functioning as a cytokine-like molecule.16,28 Extracellular release of HMGB1 during acute cell death of tumor cells leads to activation of the innate immune system that leads to establishment of long-standing antitumor immunity. Although HSV-1 dICP0 does not induce significant cell death and HMGB1 release in TUBO cells in vitro, given that in vitro observations do not translate directly in vivo, it is difficult to ascertain if HMGB1 is being released from infected tumor cells. The early spike in serum HMGB1 in vivo may emanate from immune cells that sense the initial burst of HSV-1 dICP0 replication in treated tumors. Similarly, the highest levels of cleaved caspase 3 and HSP-70 were observed following clearance of HSV-1 dICP0 from the tumor.
We have also verified that this immunogenicity is not due to the disparity in the particle:pfu ratio between HSV-1 and HSV-2 dICP0 (Supplementary Table 1). Indeed, HSV-2 dICP0 has ~10-fold higher particle:pfu ratio than HSV-1 dICP0, which is opposite to what one might predict if immunogenicity is linked to virus particle amount. One likely explanation is that HSV-1 dICP0 treatment triggers immune cells to amplify tumor cell death. In fact, this study showed a trend of increased infiltration of antigen-presenting cells and neutrophils in HSV-1 dICP0–treated tumors. Further work is needed to characterize the mechanism(s) of induction of immunogenic cell death, particularly in response to HSV-1 dICP0 infection, to determine direct and indirect effects of virus treatment.
Similar to virus replication, we found that in vitro cytotoxicity does not translate into in vivo immunogenicity, as HSV-1 dICP0 induced the least amount of CPE in vitro, yet displayed the highest level of immunogenic cell death in vivo. A main conclusion from this study is that the rapid induction of immunogenic tumor cell death is the best indicator of antitumor immune induction and survival benefit. Specifically, only treatment with HSV-1 dICP0 showed a significantly higher percentage of HER-2–specific CD8+ T cells that are cytotoxic to TUBO cells. Early studies have used nude mice29 to show that HSV-1 oncolytic virotherapy requires the immune system to exert its therapeutic effect. Subsequent work showed that CD8+ T-cell depletion abrogates HSV-mediated therapeutic efficacy.7,30,31,32,33 Although we did not measure anti-HSV immune responses within the tumor, it is likely that increased immunogenic cell death and subsequent adaptive immune response induction led to the rapid clearing of HSV-1 dICP0.
In summary, this study investigated what inherent features of oncolytic HSV viruses are required for a potent antitumor activity. Using HSV ICP0 mutant oncolytic viruses that show differences in their in vitro replication and cytotoxicity, we discovered that the in vitro replication capacity of the virus does not translate into in vivo virus persistence. Moreover, the initial stages of immunogenic virus replication leading to activation of the antitumor immunity are more important than prolonged replication of the virus within the tumor. This study provides a useful insight for the design of potent oncolytic viruses, along with the selection of appropriate combination therapies to enhance immunogenicity.34
Materials and Methods
Cells culture. Human osteosarcoma cells (U2OS; ATCC, Manassas, VA) were maintained in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% fetal bovine serum (FBS). TUBO is a cloned cell line generated from a spontaneous mammary gland tumor from a BALB-neuT mouse and highly expresses HER-2 protein on the cell membrane.35 TUBO cells were maintained in DMEM with 10% FBS. Vero cells (ATCC) were maintained in DMEM supplemented with 10% FBS. All media contained 2 mmol/l l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island, NY). All cell lines were grown at 37 °C under humidified conditions.
Antibodies. The following mAbs were used in flow cytometry assays: anti-CD16/CD32 (clone 2.4G2) to block FcRs, anti-CD3 (clone 17A3), anti-CD4 (clone RM4-5), anti-CD8 (clone 53-6.7), anti-CD11b (clone M1/70), anti-CD11c (clone HL3), Ly6C (clone AL-21), Ly6G (clone 1A8) for detecting cell surface markers, and anti–IFN-γ (clone XMG1.2) and foxp3 (clone FJK-16s) (all from BD Biosciences, San Diego, CA) for intracellular staining.
Viruses. To quantify virus replication using live imaging, luciferase expressing HSV-1 and HSV-2 ICP0 mutants were made. Recombinant viruses were generated by homologous recombination using infectious DNA of luciferase-expressing wild-type HSV-1 KOS/Dluc/oriL36 or HSV-2 MS-luc.11
HSV dNLS viruses encode a GFP-tagged protein that lacks the ICP0 NLS region and a portion of the C-terminal oligomerization domain.11 The HSV-2 dICP0 virus has been previously reported as HSV-2 d81011 and contains a deletion of 810 aa with in the ICP0 protein, resulting in GFP being fused to the C-terminal 15 aa of ICP0. HSV-1 dICP0 differs slightly in that the entire ICP0 coding region has been removed.
All HSV-1 ICP0 mutants were propagated and titered on U2OS cells in the presence of 3 mmol/l hexamethylene bisacetamide (Sigma, St Louis, MO). Wild-type HSV-1 strain KOS was propagated and titered on Vero cells. All HSV-2 viruses were propagated and tittered on U20S cells maintained at 34 °C.11 All viruses were purified and concentrated via sucrose cushion ultracentrifugation.
Cytopathic effect assays. TUBO cells were plated in 12-well plates at a density of 1 × 105 cells per well. The next day, the cells were either mock treated or infected with various HSV ICP0 mutants at MOIs 1, 2.5, and 5. After a 1-hour inoculation, medium containing 5% FBS was added back. Twenty-four hours later, cells were imaged under bright field using a Leica DM-IRE2 inverted fluorescent microscope.
Cell metabolism assays. TUBO cells were plated in 96-well plates at a density of 1 × 104cells/well. The next day, cells were infected with the different HSV ICP0 mutants at MOI of 5. After a 1 hour inoculation, maintenance medium was added back, and cells were incubated for 24 hours before measurement of cellular metabolism using the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (Sigma). Absorbance measured at 570 nm (background absorbance 690) was used to generate cellular metabolism relative to uninfected controls.
Virus burst size. TUBO cells were infected with 0.5 MOI of the different viruses. After 1 hour infection, cells were washed three times with PBS and 5% FBS DMEM was applied. Forty-eight hours after infection, cells and supernatants were harvested and frozen. Samples were freeze-thawed three times and cell debris was removed by centrifugation. Burst size was calculated by dividing the titers (pfu/ml) with the cell count to generate values in pfu per cell.
Western blotting. Whole-cell extracts were prepared by scraping the cells and pelleting with 14,000g for 15 minutes. Cell pellets were resuspended in radioimmunoprecipitation assay buffer with protease inhibitors, and lysis was allowed to proceed for 20 minutes at 4 °C. Whole-cell lysates were clarified by centrifugation at 13,000g for 10 minutes at 4 °C. Protein quantification was carried out using the Bradford assay kit (Bio-Rad Laboratories, Hercules, CA). Protein samples were loaded onto sodium dodecyl sulfate–polyacrylamide gels, resolved, and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA) at 100 V for 1 hour. All blots were blocked in 5% skim milk in Tris-buffered saline (TBS) for 1 hour, followed by an overnight incubation at 4 °C in rabbit anti-mouse cleaved caspase 3 (Cell Signaling Technology, Beverly, MA), mouse anti-human HSP-70 (Enzo Lifesciences, NY) or goat anti-human β-actin (Santa Cruz Technology, Santa Cruz, CA) diluted in blocking buffer. The next day, the blots were incubated for 1 hour with 5% skim milk in TBS–Tween20 (0.1%; TBST) containing the appropriate horseradish peroxidase–conjugated secondary antibodies for 1 hour and visualized by chemiluminescence.
Mouse experiments. Mice were maintained at the McMaster University Central Animal Facility with all procedures performed in full compliance with the Canadian Council on Animal Care and approved by the Animal Research Ethics Board of McMaster University. Six- to 7-week-old BALB/c mice (Charles River Laboratories, Wilmington, MA) were used to implant 5 × 105 TUBO cells subcutaneously on the left flank. To minimize experimental variability, low passage TUBO cells were used for subcutaneous injections. Twelve to 14 days after injection, the tumors reached treatable average tumor volume. Since tumor sizes are heterogeneous, mice are randomized so that each treatment group has tumors of variable volume at the start of treatment. The tumors were treated by local administration of three 50 µl doses of either PBS or 1 × 107 total pfu HSV ICP0 mutant virus.7 Tumors were measured every 3 days, and fold changes in tumor volumes were calculated relative to the tumor volume at the start of treatment.7 Mice having tumors of 10 mm in length and width (volume of 525 mm3) were classified as end point. Mice were imaged using in vitro imaging spectrum 5 and 7 days after the first virus injection. In vitro imaging spectrum imaging was carried out after intraperitoneal injection of D-luciferin (PerkinElmer, Waltham, MA) in anesthesized mice and images were taken every 5 minutes for 45 minutes. Nonsaturating signal images were used to compare the different viruses using Living Image 4.2 (PerkinElmer).
Intracellular cytokine staining. Peripheral blood samples were collected from treated mice and processed using ACK lysis buffer as described previously.7 Lymphocyte peptide (re) stimulation was carried out by incubation for 4 hours (in the presence of Brefeldin A; BD GolgiPlug) with the LELTYVPANASLSFL peptide, corresponding to the BALB/c immunodominant epitope of rat HER-2 (HER-2 aa 63-77; 1 μg/ml), and stained for the surface marker CD8 and intracellular IFN-γ as described previously.7 Data were acquired using a FACSCanto flow cytometer (BD Pharmingen, San Jose, CA) and analyzed with FlowJo (Tree Star, Ashland, OR).
In vitro cytotoxicity assay. Spleens were harvested from anesthetized and euthanized tumor-bearing mice treated with PBS or HSV dICP0 virus. Splenocytes harvested from groups of five mice were pooled and used for isolation of CD8+ T cells using the CD8− selection kit (EasySep; StemCell Technologies, Vancouver, BC). Splenocytes were cocultured with TUBO cell monolayers at an effector:target ratio of 1,000:1. The cocultures were incubated for 16 hours before viability was assessed using the MTT assay.
Isolation of tumor-infiltrating immune cells. Tumors infiltrating immune cells were isolated as previously described.37 Briefly, tumors were digested in a mixture of 0.5 mg/ml collagenase type I (Gibco), 0.2 mg/ml DNase (Roche, Indianapolis, IN), and 0.02 mg/ml hyalorunidase (Sigma) prepared in Hank's buffered saline (Sigma; 10 ml/250 mg of tumor). The digested material was passed successively through 70 and 40 μm nylon cell strainers, and lymphocytes were purified using mouse CD45.2-positive selection by magnetic separation (EasySep; StemCell Technologies). The purified cells were used for direct surface staining using different surface markers. Samples were stained using the near-infrared fluorescent reactive dye to gate viable cells.
Tumor harvesting and isolation of protein. Mice were anesthesized and euthanized before resection of their tumors. Tumors were cut into small pieces and homogenized in the presence of radioimmunoprecipitation assay lysis buffer. Homogenized tumors were incubated on ice for 30 minutes. Whole-tumor lysates were clarified by two sequential centrifugations at 13,000g for 10 minutes at 4 °C. Twenty micrograms of total protein was used for western blot analysis.
Titering of tumor-associated virus. HSV dICP0–treated tumors were resected after sacrificing anesthesized tumor-bearing mice. The tumors were weighed and homogenized in 2 ml DMEM. Cell debris was pelleted by centrifugation at 3,000 rpm for 10 minutes. Titers were done on U20S cells and used to calculate the total pfu per milligram of tumor.
HMGB1 ELISA. Peripheral blood was drawn from mice and allowed to clot at room temperature for 25 minutes. Cells were pelleted twice by centrifugation at 2,000 rpm for 10 minutes, and the supernatant was collected. Serum samples were aliquoted and immediately frozen at −80 °C. Blood samples were processed with care to avoid hemolysis that gives false-positive results for HMGB1. Twenty-four hours after infection with the various HSV ICP0 mutants, medium was collected, and supernatant cleared of cell debris by centrifugation at 2,000 rpm for 10 minutes. Serum samples and supernatants from infected cells were used to detect the level of HMGB1 using an ELISA kit developed by Shino Test (IBL international, GMBH, Hamburg, Germany). The ELISA was done following the manufacturer's protocol outlined for normal sensitivity format of the assay.
Data analysis. For each statistical analysis used, normality of the distributions and variance assumptions were tested before running the statistical tests. Log-rank (Mantel-Cox) test and log-rank test for trend was used for determining the statistical significance of the difference in Kaplan–Meier survival between the treatments. ANOVA was used to analyze the significance of the differences in average virus titer, cellular metabolism in vitro, and mean tumor-infiltrating lymphocyte number per gram of tumor. χ2 test was used to determine the statistical significance of the differences in CD8+ T-cell response. The null hypothesis was rejected for P values <0.05. All data analyses were carried out using GraphPad Prism (La Jolla, CA, USA).
SUPPLEMENTARY MATERIAL Figure S1. HSV dICP0 treated tumors have no significant differences in CD4+foxp3+ Tregs infiltrating the tumors. Table S1. Virus particle to pfu ratios.
Acknowledgments
We thank Yonghong Wan (McMaster University) for reagents and Susan Collins for technical assistance. These studies were supported by operating grants to KLM from the Canadian Cancer Society (formerly the Canadian Breast Cancer Research Alliance) and the Cancer Research Society. The authors declare no potential conflicts of interest.
Supplementary Material
References
- Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- Mossman KL, Smiley JR. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J Virol. 2002;76:1995–1998. doi: 10.1128/JVI.76.4.1995-1998.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, Smiley JR. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J Virol. 2000;74:2052–2056. doi: 10.1128/jvi.74.4.2052-2056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel JL, Safroneeva E, Mossman KL. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther. 2005;12:1101–1110. doi: 10.1016/j.ymthe.2005.07.533. [DOI] [PubMed] [Google Scholar]
- Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther. 2011;19:335–344. doi: 10.1038/mt.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandha H, Melcher A, Harrington K, Vile R. Oncolytic viruses: time to compare, contrast, and combine? 5th international meeting on replicating oncolytic virus therapeutics. Banff, Alberta, Canada, 18-22 March 2009. Mol Ther. 2009;17:934–935. doi: 10.1038/mt.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmann G, Tattersall P, van den Pol AN. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79:6005–6022. doi: 10.1128/JVI.79.10.6005-6022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JJ, Delman KA, Burt BM, Mariotti A, Malhotra S, Zager J.et al. 2002Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Ther 9935–945. [DOI] [PubMed] [Google Scholar]
- Halford WP, Püschel R, Rakowski B. Herpes simplex virus 2 ICP0 mutant viruses are avirulent and immunogenic: implications for a genital herpes vaccine. PLoS ONE. 2010;5:e12251. doi: 10.1371/journal.pone.0012251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010;70:9062–9072. doi: 10.1158/0008-5472.CAN-10-1825. [DOI] [PubMed] [Google Scholar]
- Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012;72:2327–2338. doi: 10.1158/0008-5472.CAN-11-2975. [DOI] [PubMed] [Google Scholar]
- Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013;20:7–15. doi: 10.1038/gt.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidera K, Gaitanou M, Stellas D, Matsas R, Patsavoudi E. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J Biol Chem. 2008;283:2031–2041. doi: 10.1074/jbc.M701803200. [DOI] [PubMed] [Google Scholar]
- Qin Z, DeFee M, Isaacs JS, Parsons C. Extracellular Hsp90 serves as a co-factor for MAPK activation and latent viral gene expression during de novo infection by KSHV. Virology. 2010;403:92–102. doi: 10.1016/j.virol.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multhoff G, Hightower LE. Distinguishing integral and receptor-bound heat shock protein 70 (Hsp70) on the cell surface by Hsp70-specific antibodies. Cell Stress Chaperones. 2011;16:251–255. doi: 10.1007/s12192-010-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, Sanchez-Perez L, Diaz RM, Kottke T, Thompson J, Lai M, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol. 2004;22:1125–1132. doi: 10.1038/nbt1007. [DOI] [PubMed] [Google Scholar]
- Sanchez-Perez L, Kottke T, Daniels GA, Diaz RM, Thompson J, Pulido J, et al. Killing of normal melanocytes, combined with heat shock protein 70 and CD40L expression, cures large established melanomas. J Immunol. 2006;177:4168–4177. doi: 10.4049/jimmunol.177.6.4168. [DOI] [PubMed] [Google Scholar]
- Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Guo ZS, Liu Z, Bartlett DL, Tang D, Lotze MT. Life after death: targeting high mobility group box 1 in emergent cancer therapies. Am J Cancer Res. 2013;3:1–20. [PMC free article] [PubMed] [Google Scholar]
- Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385–393. doi: 10.1089/10430349950018832. [DOI] [PubMed] [Google Scholar]
- Wong RJ, Patel SG, Kim S, DeMatteo RP, Malhotra S, Bennett JJ, et al. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther. 2001;12:253–265. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- Zager JS, Delman KA, Malhotra S, Ebright MI, Bennett JJ, Kates T, et al. Combination vascular delivery of herpes simplex oncolytic viruses and amplicon mediated cytokine gene transfer is effective therapy for experimental liver cancer. Mol Med. 2001;7:561–568. [PMC free article] [PubMed] [Google Scholar]
- Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7:741–747. doi: 10.1016/S1525-0016(03)00120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Fu X, Rousseau R, Chen SY, Zhang X. Destruction of nonimmunogenic mammary tumor cells by a fusogenic oncolytic herpes simplex virus induces potent antitumor immunity. Mol Ther. 2004;9:658–665. doi: 10.1016/j.ymthe.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Workenhe ST, Pol JG, Lichty BD, Cummings DT, Mossman KL. Cancer Immunol Res. doi:10.1158/2326-6066.CIR-13-0059-T (epub ahead of print); 2013. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. [DOI] [PubMed] [Google Scholar]
- Rovero S, Amici A, Di Carlo E, Bei R, Nanni P, Quaglino E, et al. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–5142. doi: 10.4049/jimmunol.165.9.5133. [DOI] [PubMed] [Google Scholar]
- Summers BC, Leib DA. Herpes simplex virus type 1 origins of DNA replication play no role in the regulation of flanking promoters. J Virol. 2002;76:7020–7029. doi: 10.1128/JVI.76.14.7020-7029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinshtein N, Ventresca M, Margl R, Bernard D, Yang TC, Millar JB, et al. High-dose chemotherapy augments the efficacy of recombinant adenovirus vaccines and improves the therapeutic outcome. Cancer Gene Ther. 2009;16:338–350. doi: 10.1038/cgt.2008.89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.