

Abstract
Despite progress in identifying molecular drivers of cancer, it has been difficult to translate this knowledge into new therapies, because many of the causal proteins cannot be inhibited by conventional small molecule therapeutics. RNA interference (RNAi), which uses small RNAs to inhibit gene expression, provides a promising alternative to reach traditionally undruggable protein targets by shutting off their expression at the messenger RNA (mRNA) level. Challenges for realizing the potential of RNAi have included identifying the appropriate genes to target and achieving sufficient knockdown in tumors. We have developed high-potency Dicer-substrate short-interfering RNAs (DsiRNAs) targeting β-catenin and delivered these in vivo using lipid nanoparticles, resulting in significant reduction of β-catenin expression in liver cancer models. Reduction of β-catenin strongly reduced tumor burden, alone or in combination with sorafenib and as effectively as DsiRNAs that target mitotic genes such as PLK1 and KIF11. β-catenin knockdown also strongly reduced the expression of β-catenin–regulated genes, including MYC, providing a potential mechanism for tumor inhibition. These results validate β-catenin as a target for liver cancer therapy and demonstrate the promise of RNAi in general and DsiRNAs in particular for reaching traditionally undruggable cancer targets.
Introduction
The past decade has seen significant progress in translating the understanding of cell growth and cancer into targeted therapeutics. Multiple classes of proteins have been targeted, including protein kinases, GPCR-like proteins, and other targets such as hormone receptors.1,2,3,4,5 Despite the successes, many potential target proteins have remained “undruggable” because they lack features that would make them accessible to conventional therapeutics. For example, transcription factors function largely via protein–protein interactions that can be difficult to disrupt with small molecules.6 Many of these targets, such as MYC, HIF1α, and β-catenin, are implicated in a wide variety of cancers.7,8,9,10,11,12 As previously discussed,13 such transcription factors represent ideal targets for blocking signaling pathways involved in cancer.
RNA interference (RNAi) offers an approach to reach undruggable targets. RNAi is a normal mechanism of gene expression control involving regulation of messenger RNA (mRNA) translation and degradation via the binding of short strands of homologous RNA to target mRNA.14,15,16 Since its discovery little more than 10 years ago, RNAi has advanced rapidly to clinical applicability.17,18,19,20 Therapeutic agents designed as short strands of RNA complementary to target genes, termed siRNAs (small interfering RNAs), will cause degradation or “knockdown” of the target mRNA. Dicer-substrate siRNAs (DsiRNAs) represent a particularly useful design for siRNAs.21,22,23 DsiRNAs are longer than traditional “21mer” siRNAs and are recognized and cleaved by Dicer, an enzyme involved in RNAi processes such as microRNA formation (Supplementary Figure S1). Dicer is a component of the RNA-induced silencing complex (RISC) and is involved in RISC formation. DsiRNAs can be particularly potent at reducing target gene expression, possibly because Dicer facilitates transfer of the RNA duplex directly into RISC.15
β-catenin (human gene name: CTNNB1) is an ideal example of an undruggable protein suitable for targeting by RNAi. β-catenin acts as a transcription factor to mediate responses to Wnt growth factors.8,9 In the absence of Wnt, β-catenin is constitutively degraded. Wnt stimulation causes stabilization of β-catenin, translocation into the nucleus, and transcription of β-catenin's “downstream” genes (Supplementary Figure S2). Many of these downstream genes are relevant to cancer, including cell growth–related genes such as MYC. Consistent with this, β-catenin is overexpressed in multiple cancer types. β-catenin and the Wnt pathway are attractive targets for novel therapeutics, and multiple drug discovery programs have targeted the Wnt/β-catenin pathway.24 Unfortunately, these efforts have not led to an approved therapeutic, possibly because the relevant mutations and signaling components of the pathway are not easily druggable.
Overactivation of the Wnt pathway has been found to frequently occur in liver cancers, particularly in hepatocarcinomas (HCCs).25,26,27,28,29 These cancers carry poor prognoses and have few therapeutic options. HCC is the fifth most common cancer worldwide and the third leading cause of cancer death, with more than 600,000 new cases diagnosed annually and a 90+% mortality rate.30 The recent approval of sorafenib has provided only modest benefit; the Sorafenib HCC Assessment Randomized Protocol (SHARP) trial, for example, showed a survival benefit of 2.8 months.31,32,33 Thus, there is a strong need for new therapeutics for HCCs.
In this article, we describe the effective targeting of β-catenin with DsiRNAs in models of liver cancer. Through large-scale screening, we have identified a series of DsiRNAs that are high-potency inhibitors of β-catenin. These DsiRNAs were delivered in vivo using a lipid nanoparticle (LNP) system, leading to potent knockdown of β-catenin. Knockdown of β-catenin strongly reduced the burden of liver tumor and reduced the expression of MYC, which may represent a mechanism for tumor inhibition. Together, these results demonstrate the promise of DsiRNA therapeutics for traditionally undruggable cancer targets.
Results
DsiRNAs and the endogenous cellular RNAi pathway
DsiRNAs are short RNA-based duplex oligonucleotides that enter the endogenous RNAi pathway like natural microRNA precursors by being bound and cleaved by Dicer (Supplementary Figure S1). The “25/27mer” DsiRNA design in this study uses a 25-nucleotide sense strand (all RNA except two DNA nucleotides at the 3′ end) duplexed with a 27-nucleotide RNA antisense strand, yielding a blunt “right” end, and at the “left” end, two nucleotides overhanging at the 3′ end of the antisense strand. This asymmetric structure (overhang left end, blunt right end) promotes optimal RNAi activity by orienting processing of the DsiRNA by Dicer, resulting in cleavage and removal of a short stretch of the blunt end, transfer of the remaining RNA duplex into RISC, and preferential retention of the antisense strand in the mature RISC.34 In RISC, the retained antisense strand then directs recognition of the target mRNA, resulting in mRNA cleavage and reduced protein levels.
High-potency DsiRNAs to β-catenin identified by large-scale screening
To identify the most potent DsiRNAs for targeting β-catenin mRNA (“β-catenin DsiRNAs”), we performed large-scale screening for in vitro mRNA knockdown activity. The 1° screen included 488 DsiRNAs distributed across the mRNA, including both human-specific “Unique” DsiRNAs (i.e., with mismatches with mouse β-catenin) and “Common” DsiRNAs that match human and mouse (for a 21mer duplex remaining after cleavage of the DsiRNA by Dicer). DsiRNAs were tested via transfection into human HeLa cells at 1 nmol/l final concentration followed by reverse transcription–quantitative polymerase chain reaction (qPCR)–based detection of mRNA knockdown. Two separately located qPCR assays were used to confirm the results. Figure 1 shows β-catenin mRNA knockdown results. Strong RNAi activity, detected consistently by both assays, was seen for many DsiRNAs. For the 72 human-Unique DsiRNAs, 57 (79%) of the DsiRNAs showed >90% knockdown even at this low dose. This 1° screen successfully identified many potent β-catenin DsiRNAs, suitable for further optimization.
Figure 1.

Results of β-catenin Dicer-substrate short-interfering RNAs (DsiRNA) 1° screen. (a) Knockdown results for 368 Common DsiRNAs; (b) results for 72 human-Unique DsiRNAs. Red and blue symbols indicate results for two quantitative polymerase chain reaction assays. The 1° screen successfully identified active DsiRNAs, many of which knocked down β-catenin >90% at 1 nmol/l. mRNA, messenger RNA.
From the 1° screen, 96 potent DsiRNAs were retested to confirm activity, and from this 2° screen (data not shown), 32 top DsiRNAs were selected for optimization in the tertiary (3°) round of screening. For optimization, we used the 2′-O-methyl (2′-OMe) modification, a naturally occurring nucleotide modification that promotes oligonucleotide stability and reduces potential in vivo immunostimulatory activity.35,36,37 For each of the 32 DsiRNA sequences, six distinct versions were synthesized, each with a different pattern of 2′-OMe modification positions on the antisense strand, to identify the modification positions that allowed retention of high activity. On the basis of the results in the 1° through 3° screens, active DsiRNA sequences, with one or more antisense strand 2′-OMe modification patterns, were selected for quaternary (4°) screening, in which multiple sense strand 2′-OMe modification patterns were tested. Figure 2a shows examples of mRNA knockdown results obtained in 3° and 4° screening. DsiRNAs generally tolerated 2′-OMe modification well. High activity was retained for many sequences (e.g., sequence series 3659; the DsiRNA suffix, such as M0/M27, indicates the sense/antisense strand 2′-OMe pattern combination). Through this process, high-activity DsiRNAs were selected, each including sense and antisense strand 2′-OMe modification patterns identified as optimal for each DsiRNA.
Figure 2.
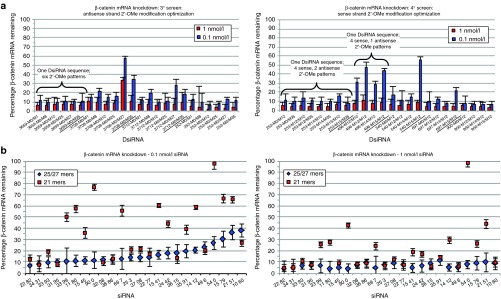
Optimization of β-catenin Dicer-substrate short-interfering RNAs (DsiRNAs). (a) Examples of 3° and 4° screening results. Each DsiRNA was tested both at 1 nmol/l (red bars) and at 0.1 nmol/l (blue bars) (mean + SEM). In the 3° screen, each DsiRNA sequence was tested with six different 2′-OMe patterns on the antisense strand. In the 4° screen, each DsiRNA sequence was then tested with one or two different antisense strand 2′-OMe patterns (identified from the 3° screen as optimal) combined with four different sense strand 2′-OMe patterns. Potent, 2′-OMe–modified DsiRNAs were identified. (b) Messenger RNA (mRNA) knockdown activity of β-catenin 25/27mer DsiRNAs, compared with activity of corresponding 21mer siRNAs (data for 21mers without 5′ phosphates is shown). Each duplex was tested at 0.1 nmol/l (left) and 1 nmol/l (right) (means ± SEM). Each 25/27mer DsiRNA (blue diamonds) was compared in activity with its corresponding 21mer (red squares); the siRNA pairs are ordered by rank activity of the DsiRNAs at 0.1 nmol/l. Many of the 25/27mer DsiRNAs showed higher knockdown activity than the corresponding 21mers.
Three DsiRNA sequences, β-cat-253, β-cat-900, and β-cat-3393, were selected for more detailed dose–response analyses (Supplementary Figure S3). Three versions of each DsiRNA sequence were tested, representing successive levels of 2′-OMe modification. The M0/M29 minimal modification pattern, which includes no 2′-OMe modifications on the sense strand and only three 2′-OMe nucleotides on the antisense strand, was used in 1° screening. The M0/M35 and M0/M12 versions include no 2′-OMe on the sense strand, but incorporate a higher number of antisense strand 2′-OMe modifications, and represent patterns found to allow high activity in 3° screening. The M14/M35 and M14/M12 versions were final leads identified from the 4° screen, with both sense and antisense strands 2′-OMe modified. All DsiRNAs retained high activity throughout 2′-OMe modification, yielding potent leads, including many with sub-picomolar IC50 values. The IC50 values were 0.45, 0.31, and 0.46 pmol/l for β-catenin-253-M0/M29, M0/M35, and M14/M35, respectively; 0.46, 0.89, and 0.10 pmol/l for β-catenin-900-M0/M29, M0/M12, and M14/M12 respectively; and 1.41, 0.30, and 1.70 pmol/l for β-catenin-3393-M0/M29, M0/M12, and M14/M12 respectively. Thus, as generally seen in the 3° and 4° screens, DsiRNAs tolerated significant 2′-OMe modification. DsiRNAs were also tested in Hepa1-6 mouse cells (Supplementary Figure S4). β-cat-900-M14/M12 and β-cat-3393-M14/M12 showed high knockdown activity; β-cat-253 is human specific and, as expected, did not show high activity in Hepa1-6.
As discussed earlier, DsiRNAs with a “25/27mer” structure are cleaved by Dicer; this yields shorter duplexes that are similar in size and structure to “21mer” siRNAs also used for RNAi. We were interested in comparing the activities of the DsiRNAs identified from screening with corresponding 21mers. Therefore, we selected 24 DsiRNAs distributed across the β-catenin mRNA, synthesized both the 25/27mer and 21mer versions and compared these for knockdown activity. To make an appropriate comparison, the 21mers were designed according to the predicted processing of 25/27mer DsiRNAs to 21mers by Dicer, such that both the DsiRNA and the 21mer would direct Ago2/RISC-mediated cutting of the target mRNA at the same base position. For a thorough comparison, the 21mers were synthesized both with and without 5′-phosphate groups on the antisense strand to identify any possible effect of this. Figure 2b shows the results for mRNA knockdown (21mers yielded the same results with and without 5′ phosphates, data not shown). For about half of the 24 sequences, the 25/27mer DsiRNA structure showed much better activity than the corresponding 21mer, and in several instances, the DsiRNA showed high activity, whereas the 21mer showed very low activity. Several 21mers also showed good activity; however, in all such cases, the corresponding 25/27mer DsiRNA also showed high activity. These results suggest that when trying to achieve target knockdown using RNAi, the DsiRNA design may allow more reliable success.
Testing of DsiRNAs for potential immunostimulatory activity
RNA oligonucleotides can potentially cause immunostimulation due to recognition by components of the innate immune system such as Toll-like receptors.35 This can lead to nonspecific or immunotoxic effects; however, this process can be reduced or eliminated by 2′-OMe modification.35 Before using the optimized β-catenin DsiRNAs for antitumor efficacy testing, they were screened in vivo for immunostimulatory activity using an assay based on the ability of an oligonucleotide to stimulate the production of antibodies to the PEGylated components of the LNP encapsulating the oligonucleotide.38 The DsiRNA β-cat-253-M14/M35 was a particular focus. Because this DsiRNA is human specific, it allows testing of the effect of β-catenin knockdown on the growth of human tumor xenografts in mice, without potential effects of β-catenin knockdown in mouse tissue. β-cat-253-M14/M35 was formulated in LNP F30.1. This LNP contains lipids that are known to facilitate immunostimulation,39 and was designed for stringent testing for immunostimulatory activity. DsiRNAs in LNP F30.1 were administered to mice in a single dose of 10 mg of DsiRNA per kg body weight (10 mg/kg), and anti-PEG-IgM titers were measured after 7 days. As shown in Figure 3, low anti-PEG antibody titers were seen in the control phosphate-buffered saline and empty particle groups, whereas high anti-PEG titers were observed for lightly modified DsiRNA (β-catenin-3393 M0/M11). This immunostimulatory effect was reduced by additional 2′-OMe modifications (sequences 3393-M14/M11 and M14/M12). β-catenin-253-M14/M35, and other β-catenin DsiRNAs tested in parallel, also showed very low anti-PEG titer, with levels comparable to phosphate-buffered saline or empty particle (dashed line). These results demonstrate that this heavily 2′-OMe–modified DsiRNA lacks significant immunostimulatory activity.
Figure 3.

Lack of immunostimulatory activity of 2′-OMe–modified Dicer-substrate short-interfering RNAs (DsiRNAs). Mice were dosed once intravenously, at 10 mg/kg, with β-catenin DsiRNAs in lipid nanoparticle (LNP) F30.1, a formulation designed to facilitate immunostimulation. Seven days postdose, serum was analyzed for an immune response, indicated by antibody to PEG. The heavily 2′-OMe–modified DsiRNA β-cat-253-M14/M35 did not show significant in vivo immunostimulation activity, compared with mice treated with phosphate-buffered saline (PBS) or with LNP F30.1 without DsiRNA (Empty Particle (EP), dashed line), in contrast with DsiRNA β-cat-3393-M0/M11 with lower 2′-OMe modification (**P < 0.01, relative to EP; mean ± SEM).
In vivo delivery and gene targeting efficacy of β-catenin DsiRNAs formulated in LNPs
To effectively and safely deliver DsiRNAs in vivo, we have developed a LNP delivery system termed “LNP2072”. These LNPs will be described in more detail elsewhere, but in brief, they are designed as a nanodelivery system with a lipid-nucleic acid aggregate, selected for double-stranded RNA binding activity, with a transfection lipid layer. These LNPs were designed for optimized in vivo delivery functions including accumulation in tumors, cell binding, and cytoplasmic release of oligonucleotides. In preliminary in vivo tolerability studies, shown in Supplementary Figure S5, no significant adverse effects of LNP administration have been observed using dosing levels and schedules like those used in the in vivo studies described below.
To measure effectiveness of LNP2072 delivery of DsiRNAs in vivo, β-cat-253-M14/M35 formulated in LNPs was administered to nude mice bearing established orthotopic tumors of Hep 3B cells (estimated tumor size: 300–500 mg) ~3 weeks after implanting cells, at 5 mg DsiRNA/kg intravenously twice a week × 2 weeks. In this initial experiment, all animals were cotreated with sorafenib (10 mg/kg orally (po), once a day (qd) × 14 days) in anticipation of efficacy testing conditions. Mice were killed 48 hours after the last LNP2072-DsiRNA dose, and tumor and liver tissue were isolated. In this model, Hep 3B tumors grow as distinct nodules, so normal liver and tumor can be isolated separately. DsiRNA content was quantitated by an oligonucleotide hybridization-based assay. LNPs accumulated in both liver and tumor, with an average DsiRNA concentration of 65 pg per mg tissue in liver and 84 pg DsiRNA per mg of tumor tissue (n = 4 mice each; no significant signal seen in control mice).
To evaluate the in vivo knockdown activity of the β-catenin DsiRNAs identified through screening, we measured the expression of β-catenin mRNA after LNP-DsiRNA administration. As shown in Figure 4a, significant β-catenin mRNA knockdown in mouse liver was seen with β-cat-900 and β-cat-3393, which match mouse and human β-catenin, but not with the human-specific DsiRNA β-cat-253. To further measure suppression of the Wnt-β-catenin pathway, we examined expression of Axin2, a known direct transcription target of β-catenin whose expression is induced by β-catenin.40 β-catenin DsiRNAs strongly reduced expression of Axin2, demonstrating inhibition of β-catenin function. Notably, the reduction of Axin2 mRNA was even greater than the reduction of β-catenin mRNA, indicating that the shutdown of β-catenin function may be even more complete than suggested by the residual amount of β-catenin mRNA. No reduction in β-catenin or Axin2 expression was seen in mice treated with a control DsiRNA to the gene hypoxanthine phosphoribosyltransferase 1 (HPRT1) (an active DsiRNA with an IC50 for in vitro HPRT1 mRNA knockdown of ~30 pmol/l, data not shown).
Figure 4.
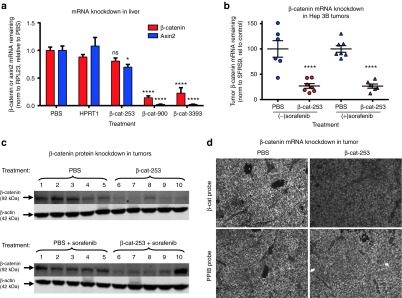
In vivo gene targeting efficacy of β-catenin Dicer-substrate short-interfering RNAs (DsiRNAs). (a) Inhibition of β-catenin and Axin2 messenger RNA (mRNA) expression in liver by β-catenin DsiRNAs. Mice were treated with β-cat-253-M14/M35, β-cat-900-M14/M12, or β-cat-3393-M14/M12, or the control DsiRNA HPRT1-716-M0/M26 to HPRT1 (5 mg/kg per dose, six doses over 2 weeks). Forty-eight hours after the last dose, mice were killed and liver mRNA levels were quantitated by quantitative polymerase chain reaction (qPCR). β-catenin was significantly knocked down by β-cat-900-M14/M12 and β-cat-3393-M14/M12 but not by human-specific β-cat-253-M14/M35. Corresponding with β-catenin knockdown, expression of Axin2, a β-catenin transcriptional target, was reduced (****P < 0.0001; *P < 0.05; means + SEM are shown). (b) Knockdown of β-catenin mRNA in Hep 3B orthotopic tumors, quantitated by qPCR. β-cat-253-M14/M35 was dosed at 5 mg/kg, in five doses over 15 days, starting ~2 weeks after Hep 3B cell implantation. Sorafenib was dosed at 10 mg/kg orally once a day × 14 days. Forty-eight hours after the last DsiRNA dose, mice were killed and tumors were isolated. β-catenin was knocked down by β-cat-253-M14/M35 (means ±SEM are shown; ****P < 0.0001). (c) Knockdown of β-catenin protein in tumors. Tumor samples from the experiment in Figure 5b were analyzed by immunblotting for β-catenin and for β-actin for normalization; 1 through 10 represent individual mice. (d) Knockdown of β-catenin mRNA in Hep 3B tumors, visualized by ViewRNA with hybridization probes specific for β-catenin (same experiment as Figure 4b,c; original magnification × 200; the phosphate-buffered saline (PBS) or β-catenin DsiRNA-treated mice from which these sections were analyzed also received sorafenib). ViewRNA analysis of the control housekeeping gene PPIB did not show reduced expression after β-catenin DsiRNA-LNP treatment. LNP, lipid nanoparticle.
To evaluate whether DsiRNA delivery yielded knockdown in tumor cells, we measured the expression of β-catenin mRNA in orthotopic Hep 3B tumors after LNP2072-DsiRNA administration (either without or with sorafenib cotreatment, conditions also used for tumor inhibition evaluation). β-cat-253-M14/M35 was dosed at 5 mg/kg, in 5 doses over 15 days, starting ~2 weeks after Hep 3B cell implantation. Sorafenib was dosed at 10 mg/kg po qd × 14 days. Forty-eight hours after the last DsiRNA dose, mice were killed and tumors were harvested. As shown in Figure 4b, β-catenin mRNA levels were dramatically reduced in Hep 3B tumors, in the absence or presence of sorafenib cotreatment. This mRNA knockdown led to reduced expression of β-catenin protein (Figure 4c; quantitation, Supplementary Figure S6). To evaluate the distribution of β-catenin mRNA knockdown, sections of tumor tissue were analyzed using the ViewRNA method, which uses gene-specific oligonucleotide probes coupled with fluorescence-linked probes to image gene expression levels. As shown in Figure 4d (quantitation, Supplementary Figure S7), β-catenin knockdown visualized with ViewRNA corresponded well with qPCR-detected knockdown and, moreover, demonstrated that knockdown occurred throughout the tumor section. ViewRNA analysis of the control housekeeping gene PPIB did not show reduced expression in tumor sections from LNP-DsiRNA–treated mice. In separate experiments, we also determined that target gene knockdown in response to DsiRNA-LNP administration is rapid, with knockdown in Hep 3B tumors seen at 6 hours after a single intravenous dose of DsiRNA-LNP (Supplementary Figure S8), indicating that the observed reduction in gene expression is a direct effect of the administered DsiRNA.
In vivo tumor growth inhibition by β-catenin DsiRNAs
Having determined that in vivo delivery of β-catenin DsiRNAs produces significant and specific gene knockdown, the effect on tumors was evaluated. In an initial experiment, DsiRNAs were administered in combination with sorafenib treatment, a standard therapy for HCC. As above, Hep 3B cells were implanted to form orthotopic liver tumors in nude mice. Approximately 2 weeks after implantation (estimated tumor size: 100–200 mg), dosing was initiated with LNP2072-DsiRNA. The 2-week delay in dosing allows evaluation of the treatment on tumors representative of advanced disease, rather than looking at an effect on tumor establishment. LNP2072-DsiRNA was administered at 5 mg/kg intravenously (six doses over 2 weeks), in combination with 10 mg/kg po sorafenib, qd × 14 days. Mice were killed 48 hours after the last DsiRNA dose, and tumors were dissected from the liver and weighed. Figure 5a shows the effect on tumor weights after treatment with a β-catenin DsiRNA or with DsiRNAs to the following three genes: KSP1 and PLK1, for which siRNA delivery has previously shown HCC tumor inhibition,41 and HIF1α, a transcription factor implicated in many cancers.11 DsiRNAs to KSP1, PLK1, or β-catenin all significantly reduced tumor weight. The HIF1α DsiRNA (HIF1α-1385-M0/M4), by contrast, did not reduce tumor weight. β-cat-253-M14/M35 demonstrated antitumor efficacy equivalent to that observed with the KSP1 and PLK1 DsiRNAs that were based on siRNAs previously selected for optimal activity.41 Because β-cat-253-M14/M35 is human specific, these results indicate that targeting β-catenin in the tumor cells alone and not in stromal cells or vasculature is sufficient for antitumor activity.
Figure 5.

In vivo tumor inhibition by β-catenin Dicer-substrate short-interfering RNAs (DsiRNAs). (a) Reduced orthotopic Hep 3B tumor weight in mice treated with β-cat-253-M14/M35 (5 mg/kg intravenously, six doses over 2 weeks) in combination with sorafenib (10 mg/kg orally (po), once a day (qd) × 14 days). Mice were killed 48 hours after the last DsiRNA dose, and tumors were dissected from the liver and weighed. Means ± SEM are shown; statistics (one-way ANOVA with Bonferroni posttest) were calculated relative to phosphate-buffered saline (PBS) + sorafenib. β-catenin DsiRNA administration reduced Hep 3B tumors (**P < 0.01), as did the DsiRNAs KSP-2455-M14/M3 and PLK1/H-p1497-M108/M85 to the genes KSP1 and PLK1. (b) Reduction of Hep 3B tumor weight by β-catenin DsiRNA treatment. DsiRNAs in lipid nanoparticle (LNP) 2072 were dosed at 5 mg/kg, in five doses over 15 days, starting ~2 weeks after implantation. Sorafenib was dosed at 10 mg/kg po qd × 14 days. Tumors were significantly reduced by β-cat-253-M14/M35 treatment, both alone and in combination with sorafenib (means ± SEM).
The efficacy of β-cat-253-M14/M35 was next tested with and without sorafenib co-treatment. The DsiRNA MCL1-1916-M14/M11 against the gene MCL110 was also tested. DsiRNAs were dosed at 5 mg/kg, in 5 doses over 15 days, starting ~2 weeks after implantation. Sorafenib was dosed at 10 mg/kg po qd x 14 days. As shown in Figure 5b, β-cat-253-M14/M35 significantly reduced tumor weight, with or without sorafenib. The MCL1 DsiRNA did not strongly reduce tumor weight. The MCL1 and HIF1α DsiRNAs, which were relatively ineffective for tumor inhibition, support β-catenin as a preferred target and also demonstrate the specificity of the β-catenin DsiRNA effect; both of these are highly active DsiRNAs, with IC50 values of 2.7 and 42 pmol/l, respectively for in vitro knockdown of their target genes (data not shown). In separate experiments using the Hep 3B model, a DsiRNA to HPRT1 and a nonspecific control DsiRNA, control-K, also did not show tumor inhibition, further demonstrating the specificity of the β-catenin DsiRNA efficacy (Supplementary Figures S9 and S10; also in these studies, the HIF1α DsiRNA again did not show tumor inhibition, whereas β-cat-253 and a published siRNA to PLK1 showed tumor inhibition).
As described in Materials and Methods, the tumor-bearing mice are routinely evaluated for serum levels of human alphafetoprotein (AFP) produced by the tumor cells. This is primarily done for presorting mice for comparable tumor burden before treatment; AFP levels provide an approximation of tumor size and were, therefore, not used as a primary measure of efficacy. However, mice treated with β-catenin DsiRNAs showed a reduction of AFP levels during the treatment period, consistent with the reduced final tumor weights (Supplementary Figure S11 provides an example from the study shown in Figure 5b).
As described above, using DsiRNAs formulated in LNP30.1, we found that β-cat-253 lacked immunostimulatory activity. To further confirm the specificity of the antitumor effects of β-cat-253, we performed studies to confirm that β-cat-253 is not immunostimulatory in LNP2072, by measuring cytokine expression in serum after β-cat-253/LNP2072 administration. As shown in Supplementary Figure S12, β-cat-253 caused no detectable cytokine induction, whereas cytokines were strongly induced by parallel treatment with a non-2′-OMe–modified immunostimulatory siRNA.
To further confirm the specific, on-target inhibition of Hep 3B tumor growth by β-catenin knockdown in vivo, two independent β-catenin DsiRNAs were compared for antitumor activity, at two doses. As shown in Figure 6a, both DsiRNAs effectively reduced Hep 3B tumor weight, even when administered at only 2 mg/kg. As shown in Figure 6b, both independent β-catenin DsiRNAs in this experiment knocked down β-catenin mRNA expression and reduced expression of Axin2. In addition to Axin2, another known direct transcriptional target of β-catenin is the oncogene MYC.42 In in vitro experiments with Hep 3B cells, we found that knockdown of β-catenin also reduced expression of MYC (Supplementary Figure S13). To determine whether this also occurred in vivo, MYC expression was quantitated in the tumor RNA. As shown in Figure 6b, MYC expression was significantly reduced by β-catenin DsiRNA treatment. This result further demonstrates successful suppression of the Wnt-β-catenin pathway in tumor cells in vivo and implicates MYC inhibition as a potential contributor to the tumor inhibition.
Figure 6.

Reduction of Hep 3B tumor burden and target gene expression by multiple β-catenin Dicer-substrate short-interfering RNAs (DsiRNAs). (a) Reduction of Hep 3B tumor weight. Intravenous administration of β-cat-253-M14/M35 or β-cat-900-M14/M12 DsiRNAs in lipid nanoparticle (LNP) 2072 (2 or 5 mg/kg three times a week × 2 weeks) began ~2 weeks after Hep 3B cells were orthotopically implanted. Forty-eight hours after the last dose, mice were killed, and tumors were weighed. Means ±SEM are shown; statistics were calculated using one-way ANOVA and Bonferroni posttest analysis. P < 0.0001 for all groups compared with phosphate-buffered saline (PBS). (b) Knockdown of β-catenin messenger RNA (mRNA) and reduced expression of downstream genes Axin2 and MYC in tumors by β-catenin DsiRNAs (means + SEM).
As discussed above, DsiRNAs are cleaved by Dicer to yield shorter RNA duplexes that are then incorporated into RISC. The 5′ end of the antisense siRNA strand in RISC, relative to the sequence in the target mRNA, determines the position of the RISC-mediated cleavage in the mRNA. To confirm in vivo RISC-mediated target mRNA cleavage, and in vivo DsiRNA cleavage by Dicer, we analyzed tumor RNA after β-cat-253 treatment in the experiment of Figure 6, for β-catenin mRNA cleavage (by 5′-RACE, PCR and sequencing). As described in Supplementary Figure S14, a β-catenin mRNA cleavage point was detected, which precisely corresponded to that predicted to occur after processing of the β-cat-253 DsiRNA antisense strand by Dicer into a 21mer, incorporation of this strand into RISC, and RISC-mediated target mRNA cleavage. Thus, LNP2072-mediated delivery of β-cat-253 into tumor cells is followed by Dicer processing of β-cat-253 and RISC-mediated mRNA cleavage.
Finally, to investigate whether β-catenin DsiRNAs could inhibit tumor growth in additional liver cancer models, we tested effects in mice bearing orthotopic xenograft tumors of the liver cancer cell line Hep G2. As shown in Figure 7, β-cat-253-M14/M35 greatly reduced Hep G2 tumor weight, as compared with control phosphate-buffered saline–treated mice or mice that received a control DsiRNA to the HPRT1 gene. This demonstrates that β-catenin is an appropriate molecular target in multiple models of liver cancer.
Figure 7.

Reduction of Hep G2 tumor burden by β-catenin Dicer-substrate short-interfering RNA (DsiRNA) treatment. Intravenous administration of β-cat-253-M14/M35 or the control DsiRNA HPRT1-716-M21/M36 to HPRT1 in LNP2072 (5 mg/kg three times a week × 2 weeks, seven doses total) began ~2 weeks after Hep G2 cells were implanted. Forty-eight hours after the last dose, animals were killed, and tumors were weighed. Means ± SEM are shown; statistics were calculated using one-way ANOVA and Bonferroni posttest analysis. β-cat-253-M14/M35, but not an HPRT1 DsiRNA, significantly reduced Hep G2 tumor weight.
Discussion
Two current challenges for realizing the potential of RNAi therapeutics in oncology are the identification of efficacious gene targets and the successful delivery of RNAi molecules into tumors to induce target gene knockdown. In this study, we have demonstrated that delivery of DsiRNAs using LNPs can effectively inhibit target gene expression, resulting in reduced tumor burden. Moreover, we have demonstrated that β-catenin is an appropriate target for therapy of liver cancers.
By a process of DsiRNA screening and optimization, we identified high-potency β-catenin DsiRNAs. In addition to β-catenin, we have followed this process for several other target genes (including multiple classically undruggable targets), with similar success. These results attest to the power and potential of RNAi therapeutics to reach previously undruggable targets and demonstrate the effectiveness of DsiRNAs, siRNAs designed to be substrates of Dicer. DsiRNAs showed exceptional potencies and in many cases induced strong mRNA knockdown even when the corresponding non-Dicer substrate 21mer siRNA functioned relatively weakly. In a recent study that also compared 21mer siRNAs and 25/27mer DsiRNAs, 2′-OMe modification of selected DsiRNAs resulted in reduced knockdown activity.43 This previous study used a single 2′-OMe modification pattern that was developed in 200736 rather than using the optimization process in this study. Using the process described here, we find that DsiRNAs can tolerate 2′-OMe modification quite well.
DsiRNAs mediated knockdown of β-catenin mRNA and protein and reduced expression of the β-catenin transcription targets Axin2 and MYC. Consistent with this knockdown, β-catenin DsiRNAs showed effective tumor inhibition. This efficacy, seen with sorafenib or alone, was similar to that observed with DsiRNAs to previously validated target genes such as PLK1 and KSP1. Tumor inhibition was seen using the human sequence-specific β-cat-253-M14/M35, which does not target endogenous mouse β-catenin (Figures 4a and Supplementary Figure S4), validating tumor cell β-catenin as a relevant target. In addition, efficacy was seen not only in the Hep 3B model, but also with Hep G2 liver cancer cells, both grown in the appropriate orthotopic liver location. Together, the results described here underscore the potential of RNAi therapeutics in general and in particular for oncology, demonstrate the effectiveness of DsiRNAs, and validate β-catenin as a target gene for liver cancers.
Materials and Methods
Large-scale DsiRNA screening. siRNA and DsiRNA strands were synthesized on solid support and purified at Integrated DNA Technologies (Coralville, IA). Electrospray ionization mass spectrometry was used to confirm sequences. RNA duplexes were concentration-normalized by ultraviolet absorbance at 260 nm. DsiRNA sequences were selected for screening based on human–mouse sequence conservation and previous DsiRNA screening results. The DsiRNAs in the primary screen had a “25/27mer” structure, with a 25-nucleotide sense strand composed of all RNAs except for two 3′-terminal DNA nucleotides, annealed to a 27-nucleotide all RNA antisense strand. This structure includes a two-nucleotide 3′ antisense strand overhang at the “left” end and a blunt “right” end. The DsiRNAs in the 1° screen were minimally 2′-OMe modified, at the three-terminal 3′ positions of the antisense strand as shown below (“Sense-M0/Antisense-M29” duplex pattern), where “N” indicates ribonucleotide, “n” indicates deoxyribonucleotide, and an underline indicates a 2′-OMe–modified nucleotide.
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M0
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M29
The antisense strand 2′-OMe patterns in the 3° screen were as follows:
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M1
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M8
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M12
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M25
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M27
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M35
The sense strand 2′-OMe patterns in the 4° screen were as follows:
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M0
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M12
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M14
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M15
The modification patterns used in 3° and 4° screens were based on previous DsiRNA screens we have performed. As a starting general guide, patterns are designed to include at least one to two unmodified nucleotides flanking the Dicer cut sites and the position of the Ago2 cut site, on both DsiRNA strands; modifying positions at these sites can inhibit activity of DsiRNAs (data not shown). For reasons not understood, tolerated modification positions outside these areas vary between sequences (and some DsiRNAs tolerate 2′-OMe modification even at the Dicer or Ago2 sites); however, the patterns shown above have been tolerated in many DsiRNAs. Therefore, a successful approach uses initial modifications that leave cut sites clear, followed by empirical evaluation of further 2′-OMe modification.
HeLa cells (American Type Culture Collection (ATCC), Manassas, VA) were transfected using Lipofectamine RNAiMAX (Life Technologies, Grand Island, NY). Before the transfections, siRNAs and DsiRNAs were incubated at room temperature for 20 minutes with RNAiMAX in OptiMEM (Life Technologies). Reverse transfections at final concentrations of 1 and 0.1 nmol/l were done in triplicate (0.1 nmol/l doses were tested in two sets of triplicate wells), with 20,000 cells per well in 96-well plates.
Following 24-hour transfections, total RNA was isolated (SV96 Total RNA Isolation System, Promega, Madison, WI) and cDNA was reverse transcribed (Superscript II, Life Technologies) using liquid-handling automation by implementing the QIAxtractor (Qiagen, Germantown, MD) and Janus (Perkin Elmer, Waltham, MA), respectively. RNA knockdown was quantified by real-time qPCR on an Applied Biosystems 7900 HT (Carlsbad, CA). In addition to qPCR for β-catenin, qPCR was performed for two housekeeping genes (HPRT1 and SRSF9) for normalization, and the data were analyzed using SDS software (Applied Biosystems). Mean β-catenin knockdown was calculated relative to negative control DsiRNA-transfected samples.
The 1° screen included 488 DsiRNAs (72 human-Uniques and 368 human-mouse-Commons, as well as 48 mouse-Uniques not used in this study). The DsiRNAs were ranked by knockdown activity seen in the 1°, and 96 top DsiRNAs were selected for 2° screening, with all the selected human-Uniques and Commons showing at least 93% β-catenin knockdown in HeLa cells in the 1° (at 1 nmol/l). Similarly, after the 2° screen, 32 top DsiRNAs were selected for 3° optimization, with all the selected human-Uniques and Commons showing at least 83% β-catenin knockdown at 0.1 nmol/l in HeLa in the 2°.
For IC50 determinations, Hep 3B cells (ATCC) were seeded at 50,000 cells/500 µl/well in 24-well plates in Eagle's minimal essential medium (EMEM) with 10% fetal bovine serum. One day after seeding, cells were transfected using RNAiMAX, with β-catenin DsiRNAs at doses from 2000 pmol/l fivefold down to 0.2 pmol/l. Controls were nonspecific DsiRNA (NC1-M11) and Mock (RNAiMAX without DsiRNA). Cells were washed with phosphate-buffered saline and lysed, 24 hours after transfection. RNA extraction and cDNA synthesis were carried out using the SV96 RNA isolation system and Transcriptor First strand cDNA synthesis kit (Roche, Indianapolis, IN), respectively. Knockdown was quantitated by real-time qPCR (iQ Multiplex Powermix, BioRad, Hercules, CA, #172–5849) using primer–probe sets for β-catenin and the housekeeping gene SFRS9 for normalization (multiplex qPCR with extension at 64 °C). β-catenin knockdown was calculated relative to Mock, and data were analyzed in Prism (GraphPad, La Jolla, CA) to generate IC50 values. For assays in Hepa1–6 cells (ATCC), qPCR assays were for mouse β-catenin and mouse HPRT1 (at 60 °C). For human MYC, the qPCR assays were for MYC and human HPRT1 (at 55 °C).
For the comparison of 25/27mer and 21mer siRNAs, 24 DsiRNAs were selected based on their representing a range of activities in the 2° screen (half the DsiRNAs, ranked 2–31, were “very active” = 90–95% knockdown at 0.1 nmol/l, whereas the other half, ranked 37–83 were “active” = 75–95% knockdown).
In vivo tumor models. Animals were treated in accordance with institutional ethical guidelines of animal care, handling, and termination; all studies were approved by Dicerna's Institutional Review Board. Male Fox-1 nude mice (Harlan Laboratories, Indianapolis, IN) were orthotopically implanted with 2 × 106 Hep 3B or 3 × 106 Hep G2 cells (ATCC) into the liver. Approximately 12 or 18 days after implantation (depending on study), before the DsiRNA treatment period, a serum sample was assayed for human AFP levels (R&D Systems, Minneapolis, MN) to enable presorting into treatment groups. Human AFP is specifically produced by the human tumor cells, and serum AFP levels, therefore, provide an approximation of tumor size. Animals were randomized into groups based on AFP levels, such that each group (including control and therapeutic treatment groups) had a similar average and range of AFP values, ensuring similar tumor burden between groups before treatment. DsiRNA was administered by tail vein injection at 5 mg/kg (unless otherwise specified) ~2 weeks after implantation. Forty-eight hours after the last dose, animals were killed and tumors dissected from the liver and weighed. Statistics were calculated using one-way ANOVA and Bonferroni posttest analysis (GraphPad Prism). For treatment with sorafenib, a stock solution of sorafenib tosylate (LC Laboratories, Woburn, MA), at four times the final concentration, was prepared in CremaphorEL (Sigma Aldrich, St. Louis, MO) and 95% ethanol (USP) 1:1 and stored at 4°. On the day of dosing, the stock was diluted 1:4 in sterile water.
DsiRNA sequences. DsiRNA strand sequences (RNA, except lower case DNA nucleotides) are provided in Table 1. Additional 2′-OMe patterns used (in non-β-catenin DsiRNAs) are as follows:
Table 1. Sequences of DsiRNA strands.

5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M16
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M21
5′ NNNNNNNNNNNNNNNNNNNNNNNnn 3′ S-M108
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M3
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M4
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M11
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M26
3′ NNNNNNNNNNNNNNNNNNNNNNNNNNN 5′ AS-M36
SUPPLEMENTARY MATERIAL Figure S1. DsiRNAs and the endogenous RNAi pathway. Figure S2. The canonical Wnt signaling pathway. Figure S3. β-catenin DsiRNA RNA knockdown IC50 curves in Hep 3B human cells in vitro. Figure S4. β-catenin DsiRNA RNA knockdown IC50 curves in Hepa1-6 mouse cells in vitro. Figure S5. Lack of in vivo toxicity of LNP2072. Figure S6. Quantitation of knockdown of β-catenin protein in Hep 3B tumors. Figure S7. Knockdown of β-catenin mRNA in Hep 3B tumors quantitated by ViewRNA vs. qPCR. Figure S8. Time course of in vivo target gene knockdown in response to DsiRNA-LNP treatment. Figure S9. Lack of Hep 3B tumor inhibition by DsiRNAs to HPRT1 and HIF1α. Figure S10. Lack of Hep 3B tumor inhibition by a DsiRNA to HPRT1 and control DsiRNA control-K. Figure S11. Reduced tumor-derived AFP levels in mice treated with β-cat-253 DsiRNA. Figure S12. Lack of cytokine induction in mice treated with β-cat-253 DsiRNA. Figure S13. Inhibition of expression of MYC by β-catenin DsiRNAs in Hep 3B cells. Figure S14. Detection of β-catenin mRNA cleavage product resulting from in vivo Dicer processing of β-cat-253-14/35 and RISC-mediated mRNA cleavage. Table S1. Primers and probes used in real-time qPCR.
Acknowledgments
We dedicate this paper to the memory of our friend and colleague Kathleen Wortham, whose enthusiasm and commitment to the search for new cancer therapeutics are reflected in this work. She will be greatly missed by all of her colleagues.
The authors thank Aftab Rashid, Kaisheng Jiao, and Shuhao Zhu for bioanalytical analyses; Rima Palkar, Rachel Brown, Shrirang Karve, and Kristy Wood for additional formulation contributions; Utsav Saxena, Lei Zhang, Chris Cowles, and Doug Fambrough for helpful discussions (all Dicerna); and Michael Christodoulou, Kyle McQuisten, and Scott Rose (all Integrated DNA Technologies) for additional screening and bioinformatics contributions. H.D., R.A., A.S., R.D., K.W., W.Z., B.H., A.S., R.F., W.C., Y.Z., H.Y., B.Y., and B.B. are employees of Dicerna Pharmaceuticals, which is developing DsiRNAs as therapeutics. M.B. is a scientific cofounder and shareholder of Dicerna Pharmaceuticals and a member of its Scientific Advisory Board.
Supplementary Material
References
- Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, et al. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev. 2001;21:499–512. doi: 10.1002/med.1022. [DOI] [PubMed] [Google Scholar]
- Minna JD, Dowell J. Erlotinib hydrochloride. Nat Rev Drug Discov. 2005. pp. S14–S15. [DOI] [PubMed]
- Bollag G, Freeman S, Lyons JF, Post LE. Raf pathway inhibitors in oncology. Curr Opin Investig Drugs. 2003;4:1436–1441. [PubMed] [Google Scholar]
- Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci USA. 2003;100:4616–4621. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31:2737–2746. doi: 10.1038/emboj.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:1–13. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012;20:483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Doudna JA. Structural insights into RNA interference. Curr Opin Struct Biol. 2010;20:90–97. doi: 10.1016/j.sbi.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JH, Armugam A, Jeyaseelan K. MicroRNAs: biogenesis, function and applications. Curr Opin Mol Ther. 2009;11:189–199. [PubMed] [Google Scholar]
- Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, et al. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rao DD, Senzer N, Nemunaitis J. RNA interference and cancer therapy. Pharm Res. 2011;28:2983–2995. doi: 10.1007/s11095-011-0604-5. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- Amarzguioui M, Lundberg P, Cantin E, Hagstrom J, Behlke MA, Rossi JJ. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat Protoc. 2006;1:508–517. doi: 10.1038/nprot.2006.72. [DOI] [PubMed] [Google Scholar]
- Zhou J, Song MS, Jacobi AM, Behlke MA, Wu X, Rossi JJ. Deep sequencing analyses of DsiRNAs reveal the influence of 3' terminal overhangs on dicing polarity, strand selectivity, and RNA editing of siRNAs. Mol Ther Nucleic Acids. 2012;1:e17. doi: 10.1038/mtna.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault S, Rickman DS, de Reyniès A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Laurent-Puig P. Genetic diversity of hepatocellular carcinomas and its potential impact on targeted therapies. Pharmacogenomics. 2007;8:997–1003. doi: 10.2217/14622416.8.8.997. [DOI] [PubMed] [Google Scholar]
- Teufel A, Staib F, Kanzler S, Weinmann A, Schulze-Bergkamen H, Galle PR. Genetics of hepatocellular carcinoma. World J Gastroenterol. 2007;13:2271–2282. doi: 10.3748/wjg.v13.i16.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci USA. 2007;104:14771–14776. doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- But DY, Lai CL, Yuen MF. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol. 2008;14:1652–1656. doi: 10.3748/wjg.14.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil BH, Venook AP. Hepatocellular carcinoma: the role of the North American GI Steering Committee Hepatobiliary Task Force and the advent of effective drug therapy. Oncologist. 2007;12:1425–1432. doi: 10.1634/theoncologist.12-12-1425. [DOI] [PubMed] [Google Scholar]
- Ma WW, Hidalgo M. Exploiting novel molecular targets in gastrointestinal cancers. World J Gastroenterol. 2007;13:5845–5856. doi: 10.3748/wjg.v13.i44.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Collingwood MA, Rose SD, Huang L, Hillier C, Amarzguioui M, Wiiger MT, et al. Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs. Oligonucleotides. 2008;18:187–200. doi: 10.1089/oli.2008.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- Tagami T, Nakamura K, Shimizu T, Yamazaki N, Ishida T, Kiwada H. CpG motifs in pDNA-sequences increase anti-PEG IgM production induced by PEG-coated pDNA-lipoplexes. J Control Release. 2010;142:160–166. doi: 10.1016/j.jconrel.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Ma Z, Li J, He F, Wilson A, Pitt B, Li S. Cationic lipids enhance siRNA-mediated interferon response in mice. Biochem Biophys Res Commun. 2005;330:755–759. doi: 10.1016/j.bbrc.2005.03.041. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A, et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest. 2009;119:661–673. doi: 10.1172/JCI37515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Foster DJ, Barros S, Duncan R, Shaikh S, Cantley W, Dell A, et al. Comprehensive evaluation of canonical versus Dicer-substrate siRNA in vitro and in vivo. RNA. 2012;18:557–568. doi: 10.1261/rna.031120.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.