

Supplemental digital content is available in the text.
Key Words: Bacterial meningitis, ERK 1/2, Fas apoptotic inhibitory molecule 2 (Faim2), Fas/CD95, Hippocampal neurogenesis, Spatial learning and memory, Streptococcus pneumoniae
Abstract
Fas-apoptotic inhibitory molecule 2 (Faim2) is a neuron-specific membrane protein and a member of the evolutionary conserved lifeguard apoptosis regulatory gene family. Its neuroprotective effect in acute neurological diseases has been demonstrated in an in vivo model of focal cerebral ischemia. Here we show that Faim2 is physiologically expressed in the human brain with a changing pattern in cases of infectious meningoencephalitis.In Faim2-deficient mice, there was increased caspase-associated hippocampal apoptotic cell death and an increased extracellular signal-regulated kinase pattern during acute bacterial meningitis induced by subarachnoid infection with Streptococcus pneumoniae type 3 strain. However, after rescuing the animals by antibiotic treatment, Faim2 deficiency led to increased hippocampal neurogenesis at 7 weeks after infection. This was associated with improved performance of Faim2-deficient mice compared to wild-type littermates in the Morris water maze, a paradigm for hippocampal spatial learning and memory. Thus, Faim2 deficiency aggravated degenerative processes in the acute phase but induced regenerative processes in the repair phase of a mouse model of pneumococcal meningitis. Hence, time-dependent modulation of neuroplasticity by Faim2 may offer a new therapeutic approach for reducing hippocampal neuronal cell death and improving cognitive deficits after bacterial meningitis.
INTRODUCTION
Neuroplasticity within the adult mammalian brain comprises a variety of biological processes by which the CNS responds to aging and injury. At the single-cell level, apoptosis and neurogenesis are complementary processes involved in the structural neuroplasticity that follows neurodegeneration, ischemia, trauma and inflammation, and associated secondary injury (1). The molecular mechanisms regulating cell fate decisions are important because secondary injury, unlike most primary insults, usually takes place in a potential therapeutic time window. In rodents and humans, basal adult neurogenesis is observed in 2 anatomic regions: the subventricular zone of the lateral ventricles/olfactory bulb (referred to as “rostral migratory stream”) and the subgranular zone of the dentate gyrus (DG) of the hippocampal formation (2–4). It is generally accepted that pathological conditions can stimulate adult neurogenesis in these neurogenic regions. In contrast, the occurrence and significance of damage-induced neurogenesis in areas such as the neocortex, striatum, amygdala, and hypothalamus are a matter of debate (5).
Despite the development of new antibiotics and intensive care options, bacterial meningitis, which is most often (60%) caused by Streptococcus pneumonia, remains an infectious disease with high mortality, i.e. up to 30%, and frequently has long-term sequelae (6–8). Permanent neurological deficits after bacterial meningitis typically include hearing loss, epileptic seizures, cerebral palsy, and cognitive impairment, particularly affecting learning and memory. Because it is almost impossible to treat the primary insult prior to the start of antibiotic treatment, understanding secondary (mal-) adaptive molecular mechanisms is a prerequisite to decreasing the morbidity associated with bacterial meningitis.
FasL-Fas (CD95L-CD95), one of the best-known death receptor–mediated cell signaling systems, is involved a number of pathological conditions of the CNS (9, 10). In addition to cell death signaling, nonapoptotic functions also seem to depend on Fas/CD95 activation (10–13), supporting regenerative processes such as neurogenesis (14–16) and neuritogenesis (17–19). The molecular mechanisms responsible for the switch between apoptotic and nonapoptotic Fas/CD95 signaling are not completely understood.
Fas apoptotic inhibitory molecule 2 (Faim2) is a 35-kDa membrane protein that is predominantly expressed in neurons; it is a member of the Bax inhibitor-1 (BI-1) family, an evolutionary strongly conserved group of small transmembrane Bax inhibitor motifs (TMBIM)–containing proteins with cytoprotective and antiapoptotic properties (20–22). Faim2 directly interacts with Fas upstream of the Fas-associated death domain-containing protein (FADD) (23), possibly at lipid rafts microdomains (24). Faim2 is activated by the phosphatidyl inositol 3-kinase (PI 3-kinase)-Akt/protein kinase B (PKB) pathway (25). Murine Faim2 null mutants display Faim2-dependent effects on cerebellar development and, in adult mice, on cerebellar cell homeostasis (26). In hypoxic-ischemic conditions, Faim2 deficiency increases neuronal vulnerability in vitro and in vivo (27). Lentiviral Faim2 overexpression not only rescues this phenotype in Faim2 null mutants but also shows additional neuroprotection in Faim2 wild-type mice.
In this study, we characterized physiological and nonphysiological Faim2 expression in the human hippocampus (DG) and analyzed the influence of Faim2 on the course of murine meningitis caused by Streptococcus pneumoniae type 3 strain (SP3) in vivo, with special focus on cellular changes in the DG and its associated functions such as learning and memory.
MATERIALS AND METHODS
Autopsy Cases
Standard formalin-fixed, paraffin-embedded hippocampal tissue samples from 4 patients with neuropathologically confirmed bacterial meningoencephalitis and 4 control cases were studied (Table 1). The sections were analyzed with respect to inflammatory infiltration including granulocyte invasion of the adjacent meninges and hippocampal Faim2 expression by hematoxylin and eosin staining and anti-Faim2 immunohistochemistry. Clinical data of the patients were obtained by analyzing the digital or written clinical charts. No neuropathological abnormalities in the DG were found in the control cases; in particular, there was no evidence of inflammation, hypoxia, or neuronal damage. The study was approved by the Ethics Committee of the Medical Faculty of the RWTH University Aachen, Germany.
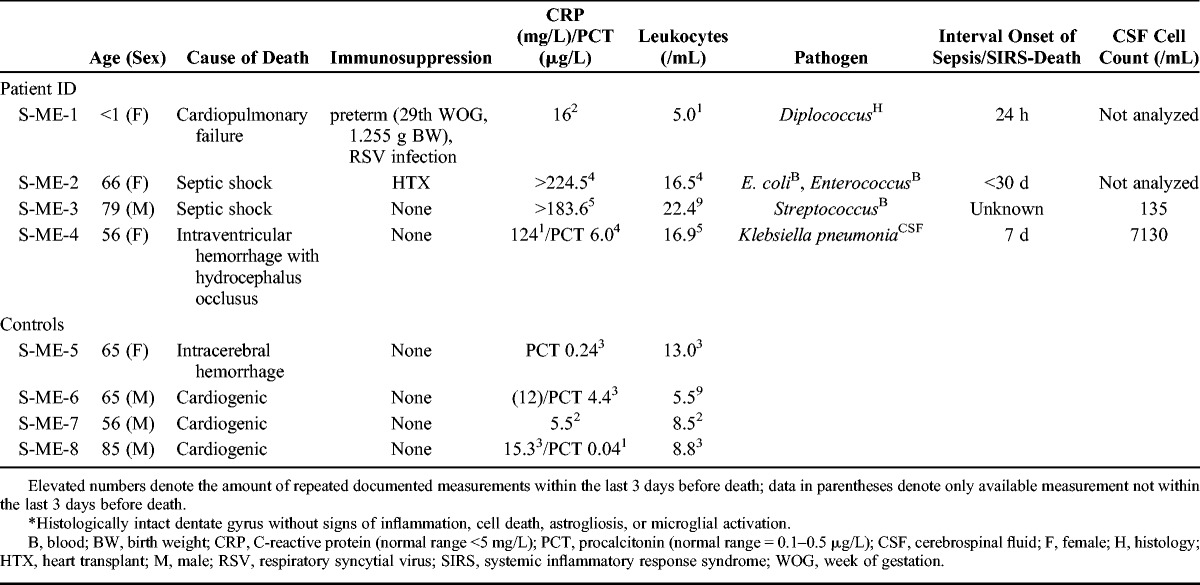
TABLE 1.
Clinical Data of Cases With Bacterial Meningoencephalitis and Controls*
Mouse Model and Experimental Design
For all experiments, murine 8- to 12-week-old male Faim2 null mutant (Faim2−/−) and wild-type littermates (Faim2+/+) were used. The generation, the morphological and spontaneous behavioral phenotype, as well as genotyping were previously described (27). Mice were habituated to a 12-hour shifted day/night cycle. The animal experiments were approved by the Animal Care Committee of the University Hospital Göttingen and by the District Government of Braunschweig, Lower Saxony as well as by the University Hospital Aachen and the District Government of Recklinghausen, North Rhine Westphalia, Germany.
Experimental procedures were adapted from earlier studies (28, 29). Mice were anesthetized with 100 mg/kg body weight ketamine and 10 mg/kg body weight xylazine intraperitoneally before subarachnoid injection of 10 μL of 0.9% NaCl containing 104 colony-forming units of SP3 or saline through the right frontolateral skull. After infection or sham injection of an equal volume of 0.9% saline, animals were divided into the following groups: one group of mice (“early meningitis”: n = 11 Faim2+/+, n = 10 Faim2−/−; and saline controls: n = 8 each group) remained untreated; these mice were killed 24 hours after infection or monitored for mortality. The second group (“late meningitis”: n = 10 Faim2+/+, n = 10 Faim2−/− [7 mice died during the course of meningitis]; saline control: n = 10 Faim2+/+, n = 13 control Faim2−/−) received subcutaneous treatment with ceftriaxone (100 mg/kg body weight; Roche, Grenzach-Wyhlen, Germany) twice daily starting 18 hours after infection until Day 5. These animals were also injected intraperitoneally with bromodeoxyuridine (BrdU; 50 mg/kg body weight; Sigma-Aldrich, St. Louis, MO) twice daily from Day 3 to Day 5 after infection. Mice killed 24 hours after infection were monitored for body weight, clinical, and motor performance before and at 12 and 24 hours after infection. In the long-term experiment, these parameters were checked at baseline, daily between Day 1 and Day 5 after infection, and once weekly from Week 2 to Week 7 after infection. At 8 weeks after infection, Morris water maze (MWM) and motor tasks (accelerod) tests were performed. Surviving animals were killed via the use of an overdose of xylazine 24 hours (early meningitis) or 9 weeks (late meningitis) after infection, and brains were removed for further analysis. The hippocampal formation was separated from the other brain structures. Cerebellum, spleen, and blood samples were processed to determine bacterial titers, as previously described (29).
Immunohistochemistry
Coronal slices of the formalin-fixed brains were embedded in paraffin. Four-micrometer-thick brain sections were prepared from the blocks that corresponded to coordinates from bregma −1.46 to −1.77 (30). After deparaffinization, sections were treated by microwaving for 5 minutes (3 times) in citric acid buffer (10 mmol/L, pH 6.0). Blocking with 10% fetal calf serum (Invitrogen, Life Technologies, Darmstadt, Germany) was followed by incubation with primary antibodies at the concentrations indicated at room temperature or at 4°C for indicated. The immunogen sequence of the polyclonal anti-human Faim2 antibody (rabbit, 1:500, no. HPA018790; Sigma-Aldrich, Taufkirchen, Germany) compromised aa 1–45 of the full-length protein. Dividing cells were visualized by rat anti-BrdU (1:200; Abcam, Cambridge, UK), and neuronal differentiation was determined by mouse anti-NeuN antibodies (1:100; Millipore, Schwalbach, Germany) incubated for 90 minutes at room temperature. For the detection of activated caspases, rabbit anti–cleaved caspase 3 (1:200; Cell Signaling Technology, Danvers, MA) and rabbit anti–cleaved caspase 8 (1:1000; Imgenex, San Diego, CA) antibodies were used. Phosphorylated Erk was detected by using rabbit anti-pERK1/2 antibody (1:100; Invitrogen Life Technologies). Sections were incubated at 4°C overnight. For visualization, the following secondary antibodies were used: goat anti-rabbit IgG (H and L) Alexa Fluor 488 (Invitrogen), goat anti-mouse IgG (H and L) Alexa Fluor 555 (Invitrogen), and rabbit anti-rat IgG (H and L) biotinylated (Vector Laboratories, Burlingame, CA). Control sections were incubated with isotype control antibody or without primary antibody. To assess the extent of meningeal inflammation and neuronal damage, paraffin sections were stained with hematoxylin and eosin following standard procedures.
For analysis of hippocampal apoptosis, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) was performed with the In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s protocol.
Real-time Reverse Transcription–Polymerase Chain Reaction
RNA was isolated from right hemispheric hippocampal tissue of Faim2−/− and Faim2+/+ mice using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reversely transcribed by M-MLV reverse transcriptase (Promega, Mannheim, Germany), as previously described (27). cDNA was amplified by real-time polymerase chain reaction (PCR) using SYBR Green (Thermo Fisher Scientific, Waltham, MA) and the following primers (Metabion, Martinsried, Germany): Faim2 (fwd 5′- AGAAGACATCATGACCCAGGG-3′, rev 5′-CTTTCTGGTCATCCCAGCTG-3′), Fas/CD95 (fwd 5′-CTGCGATGAAGAGCATGGTTT-3′, rev 5′-CCATAGGCGATTTCTG GGAC-3′) and murine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (fwd 5′-TGGC AAAGTGGAGATTGTTGCC-3′, rev 5′-AAGATGGTGATGGGCTTCCCG-3′) (annealing temperature for all 60°C) as housekeeping gene. Specificity of individual real-time PCR products was assessed by melting curve analysis. All sets of reactions were measured as duplicates, and each measurement included a nontemplate control. Stratagene Mx3000P sequence detection system (Agilent Technologies, Santa Clara, CA) was used to perform the molecular reactions. Quantitative analysis was carried out by applying the 2−ΔΔCt method (MxPro-Mx3000P, version 3.5.2; Agilent Technologies). All gene-specific mRNA expression values were normalized against GAPDH. PCR was performed with samples of 6 control and 8 meningitis mice.
Clinical Assessment
Mice were weighed and clinically scored at the indicated time points. The clinical score (range = 0–4) assessed global motor capabilities and vigilance: healthy status (score = 0), lethargic and walking (score = 1), somnolent and walking (score = 2), stuporous and unable to walk (score = 3), or dead (score = 4).
Motor Performance
The tightrope test (31) and accelerod were performed at the indicated time points. For the first test, mice were placed in the middle of a tightrope (length = 60 cm, thickness = 2 mm, height above ground = 50 cm) and the time to reach the platform at either end of the rope was measured. The needed time was converted into a score (29). In the accelerod test, mice were trained to run on a treadmill (47600-Maus-Rota-Rod, Ugo Basile, Comerio VA, Italy; acceleration: 10–40 rounds per min [rpm] either over 3 minutes), and the time on rod was recorded. At baseline, all animals met the criteria of successful test performance.
Visuospatial Learning and Memory
At 7 weeks after meningitis, hippocampal function was analyzed by applying adapted protocols of the MWM (diameter = 120 cm, height = 60 cm, mice-only use; TSE Systems, Bad Homburg, Germany) (32). The complete task consisted of cue learning (4 days), spatial acquisition (6 days learning, seventh-day reference memory [probe trial]), and spatial reversal (6 days of relearning, seventh-day new reference memory). Each animal performed a maximum of 4 trials per day (maximal duration = 120 seconds per trial, minimal intervals between trials = 45–60 minutes). Time for task completion, swimming speed, and swimming pattern were recorded (VideoMot2; TSE Systems). Nonperformers (“floaters”) were excluded from further testing and analyses.
Quantification of Immunoreactive Cells
The sections were examined by a blinded investigator using a 40× objective. Only immunoreactive cells within the granule cell layer and the subgranular zone of the DG were counted. An Analysis Software Imaging System (microscope BX51 [Olympus, Hamburg, Germany] and software AnylySIS 3.2 [Soft Imaging System, Münster, Germany]) was used to measure the area of the dentate granule cell layer. The density of immunolabeled cells was expressed as the number of marked cells per square millimeter of the area measured. The density of immunolabeled cells was evaluated in 3 coronal sections per mouse. Cells marked by TUNEL/in situ tailing were only counted if additional morphologic criteria of apoptosis such as cell shrinkage, fragmented, and condensed nuclei were observed at a 60× oil magnification. Six coronal sections per animal in 20-μm distances beginning at the coordinates defined above were quantified. Faim2-immunoreactivity was determined semiquantitatively by the ratio of labeled cells versus nonlabeled cells using the following scoring system: 0% of cells = 0, 1% to 10% of cells = 1, 11% to 50% of cells = 2, 51% to 100% of cells = 3. The inflammation score was assessed by evaluating the invasion of granulocytes into defined regions of the CNS: frontal interhemispheric region, hippocampal fissures, 3 superficial meningeal regions of the convexities and third ventricle. The degree of granulocyte density in 1 high-power field (diameter = 250 μm) per region was converted to scores as follows: no granulocytes = 0, 10 granulocytes = 1, 10 to 50 granulocytes = 2, and more than 50 granulocytes = 3; ranges of the scores (sum) were 0 to 21, as applied earlier (29), and 3 sections per animal were analyzed. Neuronal damage was assessed by typical morphological changes for necrosis such as condensation of chromatin structures, eosinophilia, nuclear, and cell swelling and sometimes surrounding inflammatory reaction and was analyzed in the following regions: hippocampus, DG, basal ganglia, and cortex. The number of damaged neurons was quantified by conversion into scores as follows: 0 = no damaged cells, 1 = 10%, 2 = 11% to 30%, 3 = more than 30% of neuronal cells with necrotic or apoptotic morphology; range of the score (sum) was 0 to 12 (32), and 3 coronal sections per animal were analyzed. Confocal images were obtained on a LSM700 microscope (Carl Zeiss, Jena, Germany) using the 405- and 488-nm laser lines for excitation of DAPI and Alexa-488. Three coronal sections per animal were analyzed.
Statistical Analysis
Survival time was expressed in hours and evaluated by Kaplan-Meier plots that were statistically compared by logrank test. If not stated otherwise, data were expressed as mean ± SEM. Two groups were compared by unpaired t-test, whereas for comparison of more than 2 groups (e.g. genotype and treatment), 2-way analysis of variance and Bonferroni post hoc test were used. p < 0.05 was considered statistically significant. Statistical analyses were carried out using GraphPad Prism (Version 5.00, La Jolla, CA).
RESULTS
Physiological and Disease-Related Expression of Faim2 in the Human DG
In most control autopsies, there was Faim2-specific staining of cytoplasm and axons in more than 50% of neurons in the fascia dentata (Fig. 1A; Table 2). In contrast, in most cases with bacterial meningitis, less than 50% of neurons were Faim2-positive. Furthermore, there was additional nuclear staining for Faim2 in the meningitis cases (Fig. 1B). Despite the low sample size and heterogeneity with respect to age, sex, underlying disease, and bacterial pathogen (Table 1), there was a trend toward downregulated Faim2 expression in the DG in patients with bacterial meningitis. This is the first evidence of Faim2 expression in the human CNS and of probable differential expression in a pathological condition.
FIGURE 1.
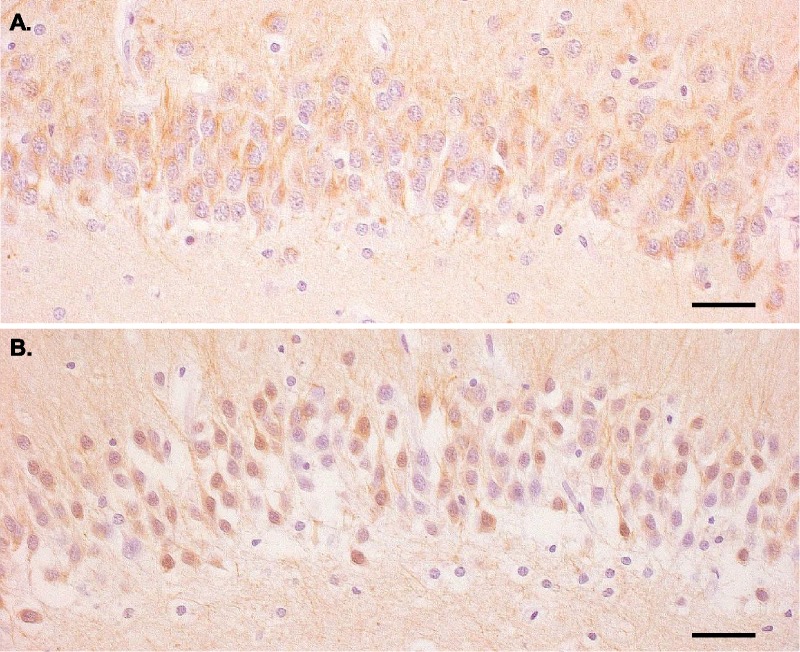
Fas apoptotic inhibitory molecule 2 (Faim2) expression in the human hippocampus in patients without and with bacterial meningitis. (A) Faim2 immunoreactivity is observed in the cytoplasm of many dentate gyrus granule cells in a control case (S-ME-6) without neuropathological abnormalities (hematoxylin counterstain; scale bar = 45 μm). (B) In contrast, fewer hippocampal granule cells are Faim2-immunoreactive in a meningoencephalitis case (S-ME-4); in addition, there is nuclear immunoreactivity of labeled neurons (hematoxylin counterstain; scale bar = 45 μm).
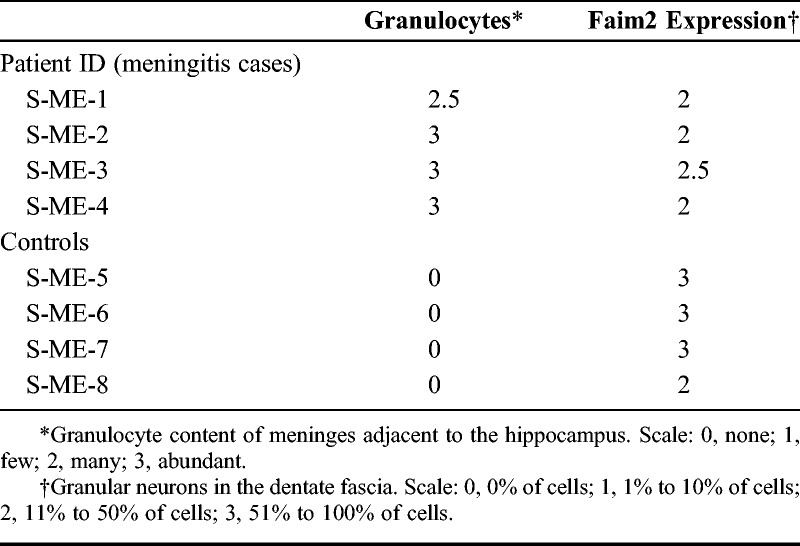
TABLE 2.
Neuropathological Scoring of Granulocyte Infiltration and Faim2 Expression in Patients’ Hippocampi
Genotype-Independent Clinical Course of Early Meningitis
We used Faim2-deficient mice (10) to evaluate the effects of Faim deficiency in experimental meningitis. Bacterial titers in blood, spleen, and cerebellum and histological evidence of meningeal granulocyte infiltration did not differ significantly between Faim2 null mutants and wild-type littermates 24 hours after subarachnoid infection with SP3. As expected, titers were highest in the cerebellum. Necrosis was detectable in all infected mice during meningitis and was most pronounced in the hippocampal formation and to a much lesser extent in the cortex (Supplementary Fig. 1A–D, http://links.lww.com/NEN/A537). Neuronal damage assessed by the necrosis score did not differ significantly between the phenotypes (6 ± 0.09 in Faim2+/+ mice vs 5.2 ± 0.73 in Faim2−/− mice, p = 0.5091). Similarly, the clinical course of early meningitis did not vary with the Faim2 genotype. All animals experienced weight loss (7%–8%), impaired motor performance (tightrope test, accelerod), and deterioration of the clinical phenotype within 24 hours (Fig. 2). In a separate experiment, subarachnoid infection with SP3 without antibiotic treatment proved to be lethal for all mice independent of genotype (Fig. 2).
FIGURE 2.
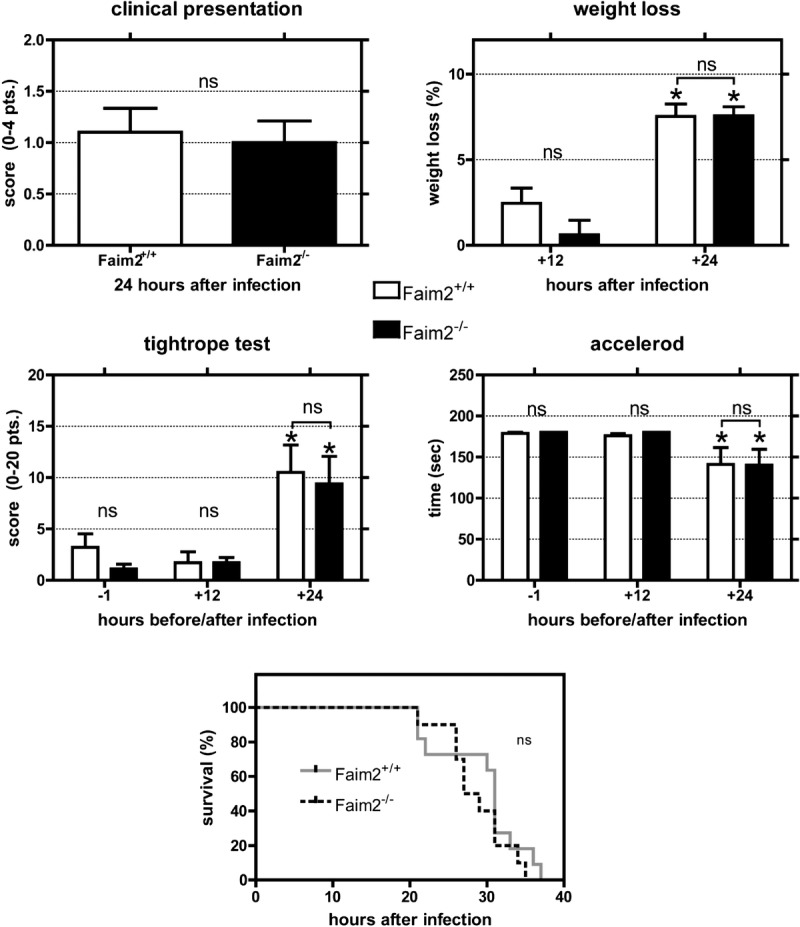
Clinical course, motor performance, and survival in mice with early meningitis. Comparisons of clinical presentation, extent of weight loss, and motor performance assessed by tightrope test and Rotarod treadmill during bacterial meningitis between Faim2+/+ and Faim2−/− mice. The decrease in motor performance (tightrope test p < 0.01, accelerod p < 0.05) and the extent of weight loss (p < 0.001) 24 hours after infection were significant and are typical features of the acute phase of meningitis but did not differ significantly between the genotypes. Spontaneous survival time after meningitis in Faim2+/+ and Faim2−/− mice without antibiotic treatment was recorded in hours and evaluated by Kaplan-Meier plot illustrating no significant difference in survival between the 2 genotypes during early meningitis (p = 0.313). ns, not significant.
Reciprocal Regulation of Fas/CD95 and Faim2 Gene Expression in Early Meningitis
At 20 hours after infection, hippocampal mRNA analysis of Faim2+/+ mice revealed a significant 5.5-fold increase in Fas/CD95 expression and a significant decrease by 0.5 in Faim2 expression versus saline-treated controls (p < 0.01 and p < 0.001, n = 8 meningitis, n = 6 control, unpaired t-test). This inverse regulation during the acute phase of a disease resembled the expression pattern observed in cerebral ischemia (27).
Genotype-Dependent Degree of Neuronal Apoptosis in the DG in Early Meningitis
Quantification of TUNEL staining in the DG at 24 hours after infection revealed a genotype-dependent significant increase in apoptosis in the Faim2−/− mice versus wild-type controls (Fig. 3A). Baseline cell death density was lower and not dependent on the genotype. Double staining by immunofluorescence confirmed that cell death mechanisms were associated with activated initiator and effector caspases (i.e. colocalization of TUNEL with cleaved caspase 8 and cleaved caspase 3) and specificity for neurons (i.e. colocalization of NeuN with cleaved caspase 8 and cleaved caspase 3) (Fig. 3D–G).
FIGURE 3.
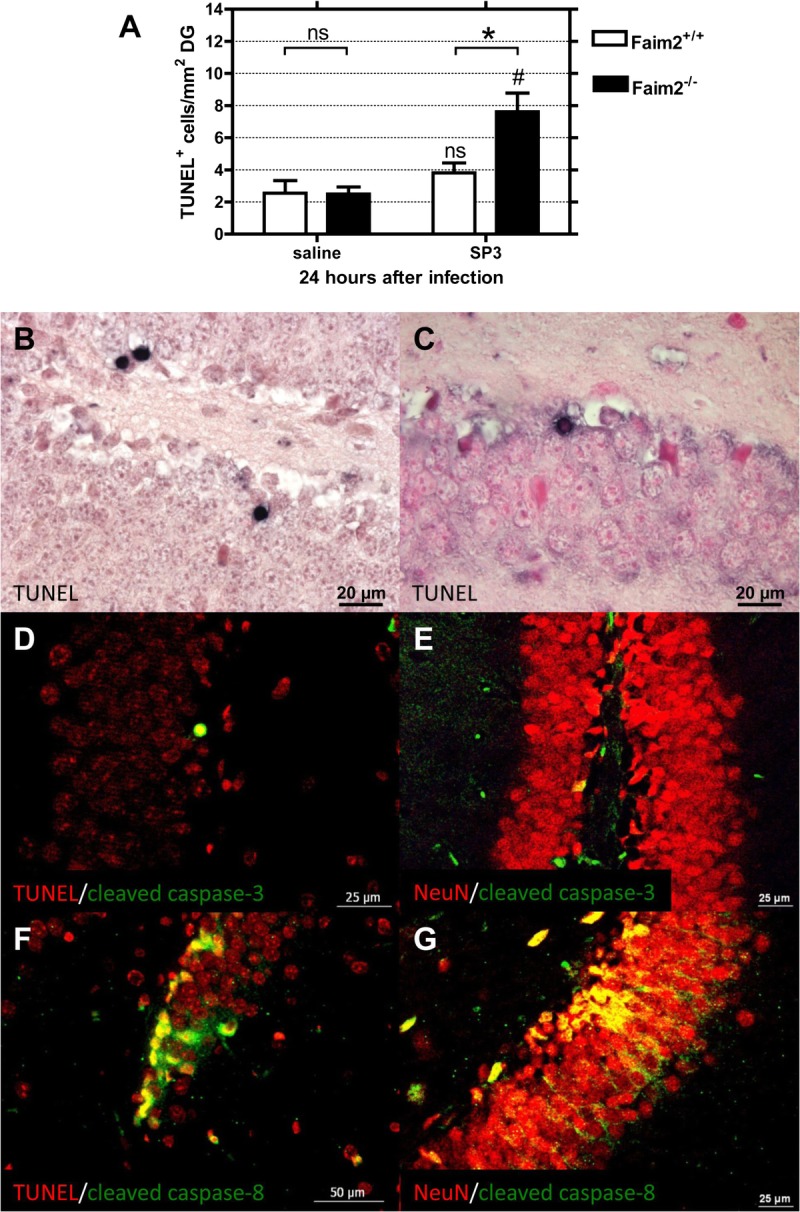
Fas apoptotic inhibitory molecule 2 (Faim2)–dependent hippocampal neuronal cell death in early meningitis due to Streptococcus pneumoniae type 3 strain (SP3). (A) Comparison of the density of apoptotic neurons in the dentate gyrus (DG) between Faim2+/+ and Faim2−/− mice reveals greater numbers of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells in Faim2-null mutants 24 hours after infection (mean ± SEM) (*Faim2+/+ vs Faim2−/−, p < 0.05; #Faim2−/− SP3 vs saline, p < 0.01). (B, C) Examples of TUNEL-positive cells in Faim2−/− (B) and Faim2+/+ (C) mice identifying apoptotic neurons in the DG of infected animals. (D–G) Double immunofluorescence staining with colocalization of TUNEL (red, D) or NeuN (red, E) and cleaved caspase 3 (green, merge in yellow) and TUNEL (red, F) or NeuN (red, G) with cleaved caspase 8 (green, merge in yellow) (Faim2−/− mice with SP3 infection) indicating that cell death mechanisms were associated with caspases 3 and 8 and were specific for neurons.
Clinical Course and Motor Performance in Late Meningitis
A 7-week follow-up after subarachnoid SP3 infection and rescue by antibiotic treatment from Day 1 to Day 6 displayed characteristic postmeningitis abnormalities of clinical status and behavior in all mice. The tested parameters changed mostly during the first 5 days after infection and then gradually returned to baseline during the following weeks. Results of the tightrope test and clinical scoring and weight did not differ between Faim2−/− and Faim2+/+ genotypes (data not shown).
Genotype-Dependent Recovery of Hippocampal Function in Late Meningitis
The MWM is a frequently applied test of spatial learning (acquisition and reversal) and reference memory (probe trial) tasks; the results of this test strongly correlate with processes of hippocampal plasticity (33). The general test accessibility of all experimental animals was demonstrated by successful cue training (Fig. 4A). SP3-infected mice displayed increased latencies and slower learning curves compared to sham-infected animals in the first 1 to 3 days of cue training independent of Faim2 genotype. At Day 4, statistically significant differences within treatment groups or genotypes were no longer observed (data not shown). Overall, in this control condition, all mice learned how to swim to a cued goal (platform) and performed equally well at final testing.
FIGURE 4.
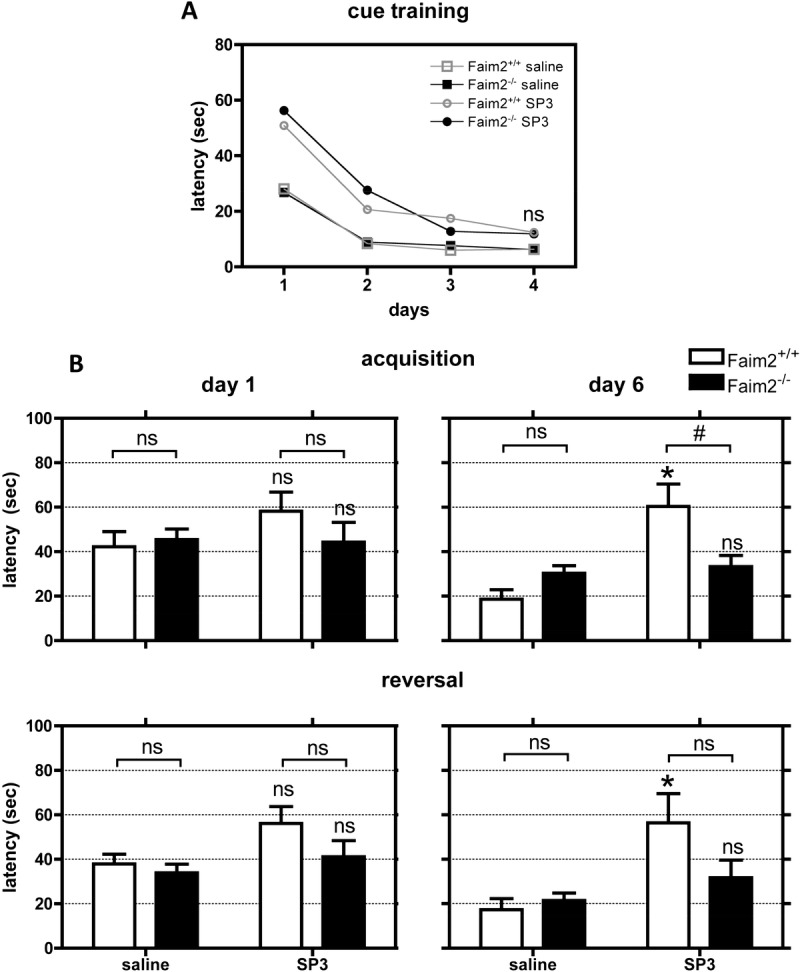
Genotype-dependent differences in visuospatial learning in late meningitis due to Streptococcus pneumoniae type 3 strain (SP3). At 7 weeks after meningitis, impairment in hippocampal function was evaluated by Morris water maze: The complete task consisted of cue learning (4 days), spatial acquisition (6 days learning, seventh-day reference memory [probe trial]) and spatial reversal (changed position of platform, 6 days relearning, seventh-day new reference memory, not all data of experiments shown). In general, all mice with bacterial meningitis performed worse than sham-infected controls. (A) The general test accessibility of all experimental animals was demonstrated by successful cue training, where at the end on Day 4, no significant differences in latency to find the platform were detected. (B) SP3-infected Faim2+/+ mice performed significantly worse at the end of acquisition and reversal trials (Day 6) in comparison to SP3-infected Faim2−/− animals. Performance of SP3-infected Faim2+/+ mice was significantly worse in comparison to sham-infected Faim2+/+ littermates (mean ± SE; Faim2+/+ [saline] n = 10, Faim2−/− [saline] n = 13, Faim2+/+ [SP3] n = 6, Faim2−/− [SP3] n = 7; acquisition: *, p = 0.001 Faim2+/+ [saline] vs Faim2+/+ [SP3]; #, p < 0.01; reversal: *, p < 0.001 Faim2+/+ [saline] vs Faim2+/+ [SP3], 2-way analysis of variance followed by Bonferroni post hoc analysis).
Mice that had SP3 infection and survived as a consequence of antibiotic treatment performed worse in all hippocampus-specific tasks of the MWM (acquisition, reversal, probe trial) at 7 to 9 weeks after infection compared to sham-infected littermates (acquisition and reversal, Day 6: p < 0.01 sham- vs SP3-infected mice [data not shown]). The only exception to this general observation was the performance of sham-infected Faim2 null mutants in the probe trials (similar to SP3-infected mice); they failed to show correct quadrant preferences in both probe trials. This may suggest a latent spontaneous Faim2−/− phenotype.
Detailed analyses of the acquisition and reversal trials revealed that the significant differences in these spatial learning tasks at the end of training (Day 6) were mainly due to the reduced abilities of SP3-infected Faim2 wild-types to learn (acquisition: interaction [p = 0.001], treatment [p = 0.0002], genotype [p = 0.1596], p < 0.01 Faim2+/+ [SP3] vs Faim2−/− [SP3], p < 0.001 Faim2+/+ [saline] vs Faim2+/+ [SP3]; reversal: interaction [p = 0.0396], treatment [p = 0.0009], genotype [p = 0.1353], p < 0.001 Faim2+/+ [saline] vs Faim2+/+ [SP3]) (Fig. 4B). By contrast, SP3-infected Faim2 null mutants showed similar learning curves during training and results at the end of training as the sham-infected mice (acquisition and reversal, Day 6: ns Faim2−/− [saline] vs Faim2−/− [SP3]).
Genotype-Dependent Differences in Exploratory Behavior and Locomotor Ability in Late Meningitis
The evaluation of exploratory behavior was assessed by open field test and balance and coordination by Rotarod treadmill. The analysis revealed no differences in distance and rearing between infected Faim2 wild-type mice and noninfected controls, but exploratory behavior was more pronounced in infected Faim2−/− mice with a significantly longer distance in comparison to uninfected Faim2−/− (p < 0.01), as well as in comparison to infected Faim2+/+ mice (p < 0.05). The tendency was also observed for the number of rearings, but this did not reach statistical significance (Fig. 5). The performance within the treadmill was also significantly better in infected Faim2−/− mice in comparison to wild-type controls (p < 0.05; mean ± SE, Faim2+/+ [saline] n = 10, Faim2−/− [saline] n = 13, Faim2+/+ [SP3] n = 6, Faim2−/− [SP3] n = 7; 2-way analysis of variance followed by Bonferroni post hoc analysis) (Fig. 5).
FIGURE 5.
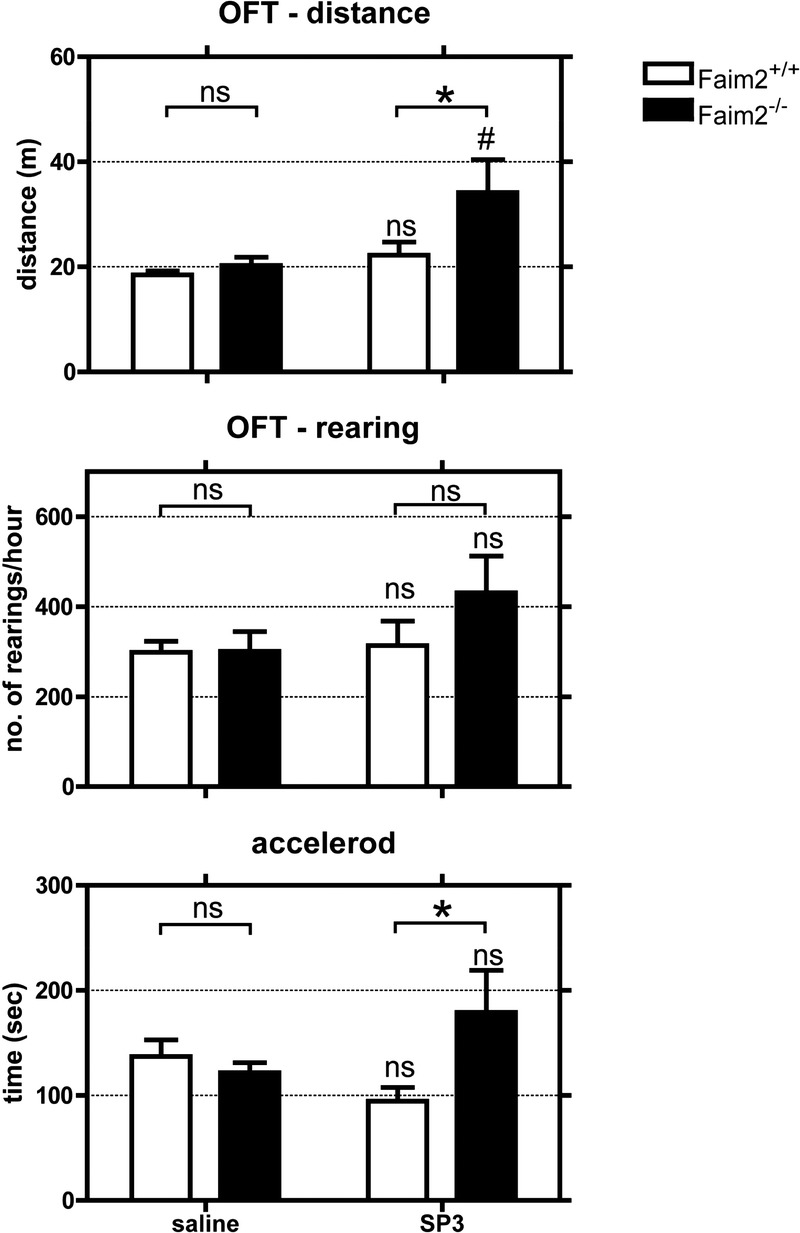
Genotype-dependent differences in exploratory behavior and locomotor ability in late meningitis due to Streptococcus pneumoniae type 3 strain (SP3). There were no differences in the performance in the open field test (OFT) and accelerod between infected Faim2 wild-type mice and noninfected controls. Faim2−/− mice performed significantly better (OFT: distance; accelerod) than infected littermates and in part better than noninfected controls (OFT: distance: *, p < 0.05; #, p < 0.01 Faim2−/− [saline] vs Faim2−/− [SP3]; accelerod: *, p < 0.05 Faim2+/+ [SP3] vs Faim2−/− [SP3]; 2-way analysis of variance followed by Bonferroni post hoc analysis).
Genotype-Dependent Increase in Hippocampal Neurogenesis and Evidence for Alternative Fas/CD95 Signaling After Meningitis
Application of BrdU on Days 3 to 5 after subarachnoid SP3 infection labeled dividing cells in a time window where neurogenesis was increased in previous studies (29, 34). Quantification of BrdU/NeuN double staining in the DG at 7 weeks after infection revealed a significant increase in newly differentiated NeuN-immunoreactive neurons in Faim2−/− versus Faim2+/+ littermates (Fig. 6AB). In infected mice but not healthy controls, we observed cells positive for phosphorylated ERK (Fig. 6C). Comparing the staining for phosphorylated ERK in the DG at 0, 10, and 20 hours after infection displayed a time-dependent activation pattern (Fig. 6D). This finding indicates activation of alternative Fas/CD95 signaling in the DG in bacterial meningitis.
FIGURE 6.
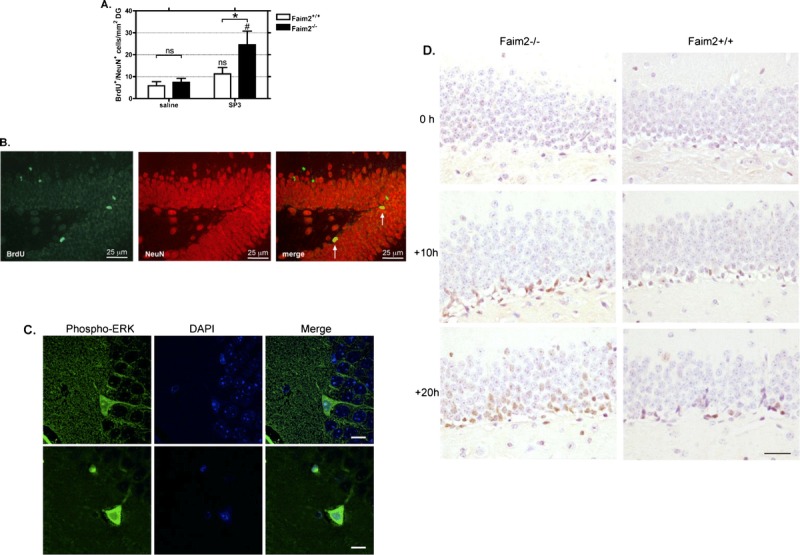
Fas apoptotic inhibitory molecule 2 (Faim2)–dependent hippocampal neurogenesis after Streptococcus pneumoniae type 3 (SP3) meningitis. (A) Quantification of the density of BrdU-immunopositive/NeuN-immunopositive cells in the dentate gyrus reveals a significant increase of newly formed differentiated granule neurons in Faim2−/− mutants versus littermates at 13 weeks after infection indicating increased neurogenesis in knockout mice after meningitis (mean ± SEM; *, p < 0.05 Faim2+/+ [SP3] vs Faim2−/− [SP3]; #, p < 0.01 Faim2−/− [saline] vs Faim2−/− [SP3]). (B) Detection of BrdU (green) and NeuN (red) by double-label fluorescence immunohistochemistry. Merge of both markers reveals newly formed neurons 13 weeks after bacterial meningitis in a Faim2−/− mouse. (C) Confocal images of sections from infected Faim2+/+ mice stained with phospho-ERK1/2 (green) and DAPI (blue) (scale bar = 10 μm). (D) Staining samples of phospho-ERK1/2 in the DG at 0, 10, and 20 hours after infection in Faim2−/− and Faim2+/+ mice showing a time-dependent increase and a tendency for a more pronounced ERK immunoreactivity in Faim2−/− mice versus Faim2+/+ mice.
DISCUSSION
Our results show that Faim2 influences caspase-associated hippocampal neuronal cell death in early bacterial meningitis as well as reactive hippocampal neurogenesis in the later course of meningitis. Despite increased early hippocampal apoptosis, Faim2 null mutant mice showed marked neurogenesis that enabled functional recovery of spatial learning back to baseline levels after bacterial meningitis.
Faim2 neither influenced the extent of bacterial burden, the clinical course, nor the mortality rate in early meningitis. Levels of bacterial titers and the degree of meningeal inflammation did not differ between Faim2-deficient and wild-type mice in this study or in comparison to historical cohorts (28). Consistent with the concept of a lack of Faim2 to influence the host resistance to SP3 infection, it is known that genetically disrupted Fas/CD95 signaling (Faslpr and Fasgld mice) does not alter the inflammatory response in bacterial meningitis, as measured by granulocyte infiltration/extravasation, granulocyte apoptosis, and blood-brain barrier permeability (35, 36). The same applies for the clinical course of bacterial meningitis in these mice. In view of this uniform inflammatory response, the histological and functional differences that were observed between Faim2 null mutants and wild-type littermates are due to Faim2-dependent differences in the way the brain responds to the neuronal injury.
As in other acute phases of neurological diseases (10), the expression of Fas/CD95 dramatically increased during early bacterial meningitis (36, 37), while the expression of Faim2 significantly decreased, as previously observed in focal brain ischemia (27). This inverse dynamic of Fas/CD95 and Faim2 expression and the fact that genetic disruption of Faim2 increased disease-related hippocampal cell death and neurogenesis support the idea of stage-dependent Fas/CD95 signaling influenced by Faim2, i.e. in healthy individuals, high Faim2 expression aims at preventing Fas/CD95 triggered cell death, whereas downregulation in early disease stages facilitates initiation of regenerative mechanisms, possibly via alternative Fas/CD95 signaling.
Faim2 expression can be regulated by the PI 3-kinase-Akt/PKB signaling cascade (25), a pathway known for its neuroprotective effects triggered by a number of growth factors and neurotrophins including insulin-like growth factor, nerve growth factor, brain-derived nerve growth factor, and erythropoietin (38–41). Involvement of brain-derived nerve growth factor in neurogenesis after bacterial meningitis and protection against disease-associated forms of cell death has been previously demonstrated (29, 42). Whether the PI 3-kinase-Akt/PKB signaling cascade-inhibiting proneurotrophins (43) negatively influence Faim2 expression is not known, but Faim2 as a target of neurotrophin signaling remains intriguing.
Activation of various caspases during early phases of the disease is common to experimental pneumococcal meningitis models (44). Upregulation of Fas/CD95, activation of caspase 8 and 3, as well as apoptotic cell death represent hallmarks of classic Fas/CD95 cell death signaling. Quantitative differences can be explained by time course (temporary activation), convergence of receptor-mediated cell signals, and nonapoptotic alternative signaling of activated caspases (45–47). TUNEL-positive cell death was mainly observed in cells of the subgranular layer of the DG, a pattern that had been described for Streptococcus pneumoniae–related hippocampal cell death before and presumably affects immature neurons (42). The fact that only SP3-infected Faim2−/−mice showed a significant increase in hippocampal cell death compared to sham-infected controls and SP3-infected wild-type littermates supports the hypothesis that a major amount of disease-associated apoptotic cell death is prevented via Faim2-dependent mechanisms.
Growing evidence suggests that Fas/CD95 signaling supports regenerative processes in the CNS such as neurogenesis (14, 15) and neuritogenesis (17, 19). In a model of cortical trauma, Fas/CD95 signaling had a positive impact on clinical outcome (48). In line with these results, the present study showed that Faim2 deficiency increased hippocampal neurogenesis and spatial learning performance in the regenerative phase after bacterial meningitis. An association between the number of newly formed neurons and the performance on hippocampal-dependent memory tasks such as the MWM has been shown in several models (49–53). Involvement of Faim2 in cerebellar plasticity, especially affecting Purkinje cells and cerebellar granule neurons, is indicated in a number of experiments (24–26). The improved performance of Faim2-deficient mice compared to wild-type littermates in late meningitis in the accelerod is in line with this hypothesis. Because spatial distribution of locomotion in the open field test (i.e. ratio of center/border and of horizontal/vertical activity) was comparable among all groups (data not shown), it seems unlikely that the difference in exploratory behavior in the open field test was significantly confounded by fear or emotion (54). It was recently shown that stimulation of adult hippocampal neurogenesis combined with voluntary exercise results in increased exploratory behavior (55) and that plasticity within the DG contributes to the origin of exploration (56). Therefore, it seems plausible that Faim2 influenced exploratory behavior in the course of bacterial meningitis via modulation of stimulated neurogenesis in the DG. However, constitutive absence of Faim2 is not always helpful in terms of hippocampal functioning because uninfected Faim2-deficient mice were not able to perform the probe trial of the MWM, i.e. failed to show place preference after successful spatial learning.
The changes in Faim2 expression in mice were mimicked in the human cases where a tendency for lower immunoreactivity was observed in meningitis cases in comparison to controls. We hypothesize that the downregulation in pathological conditions facilitates the initiation of regenerative mechanisms via alternative signaling. Because of the small size of the human cohorts, we cannot draw further conclusions, but the evidence of human Faim2 expression and of meningitis-related expression changes in the brain further strengthens the idea that Faim2 might be a therapeutic target to facilitate endogenous neuroplasticity in the course of human CNS disease.
The involvement of Faim2 in different disease pathologies (i.e. antiapoptotic effects in ischemic and inflammatory conditions) and the strong evolutionary conservation of Faim2 with a growing number of BI-1 family members (22) argue in favor of Faim2 being part of a general neuroprotective mechanism. So far, the only known directly interacting partner of Faim2 is the eponymous death receptor Fas/CD95 (23, 24). Therefore, the biological relevance of Faim2 is closely linked to the modulation of apoptotic and nonapoptotic Fas/CD95 signaling (10), but this does not exclude that possibility that disease-related effects of Faim2 are due to interference with not-yet-identified mediators.
Lack of Faim2 apparently potentiated caspase-associated hippocampal apoptotic cell death as well as ERK activation during the acute phase of bacterial meningitis. However, during the repair phase of the disease, Faim2 deficiency led to increased hippocampal neurogenesis. This was associated with improved hippocampal spatial learning and memory performance of Faim2-deficient mice 7 weeks after infection.
In summary, Faim2 deficiency influenced both degenerative and regenerative processes in a mouse model of pneumococcal meningitis. Hence, time-dependent modulation of neuroplasticity by Faim2 may offer a new therapeutic approach for reducing hippocampal neuronal cell death and improving cognitive deficits after bacterial meningitis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sabine Hamm and Stephanie Bunkowski for excellent technical assistance and Stefan Gründer for access to the confocal microscope.
Footnotes
Simone C. Tauber and Kristian Harms contributed equally.
The authors report no funding or conflicts of interest regarding the contents of the article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.jneuropath.com).
REFERENCES
- 1. Brown A, Weaver LC. The dark side of neuroplasticity. Exp Neurol 2011; 235: 133– 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deng W, Aimone JB, Gage FH. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 2010; 11: 339– 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gage FH. Mammalian neural stem cells. Science 2000; 287: 1433– 38 [DOI] [PubMed] [Google Scholar]
- 4. Ming GL, Song H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011; 70: 687– 702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gould E. How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 2007; 8: 481– 88 [DOI] [PubMed] [Google Scholar]
- 6. Swartz MN. Bacterial meningitis–a view of the past 90 years. N Engl J Med 2004; 351: 1826– 28 [DOI] [PubMed] [Google Scholar]
- 7. Thigpen MC, Whitney CG, Messonnier NE, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364: 2016– 25 [DOI] [PubMed] [Google Scholar]
- 8. Weisfelt M, Hoogman M, van de Beek D, et al. Dexamethasone and long-term outcome in adults with bacterial meningitis. Ann Neurol 2006; 60: 456– 68 [DOI] [PubMed] [Google Scholar]
- 9. Choi C, Benveniste EN. Fas ligand/Fas system in the brain: Regulator of immune and apoptotic responses. Brain Res Brain Res Rev 2004; 44: 65– 81 [DOI] [PubMed] [Google Scholar]
- 10. Reich A, Spering C, Schulz JB. Death receptor Fas (CD95) signaling in the central nervous system: Tuning neuroplasticity? Trends Neurosci 2008; 31: 478– 86 [DOI] [PubMed] [Google Scholar]
- 11. Lambert C, Landau AM, Desbarats J. Fas-beyond death: A regenerative role for Fas in the nervous system. Apoptosis 2003; 8: 551– 62 [DOI] [PubMed] [Google Scholar]
- 12. Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev 2003; 14: 53– 66 [DOI] [PubMed] [Google Scholar]
- 13. Peter ME, Legembre P, Barnhart BC. Does CD95 have tumor promoting activities? Biochim Biophys Acta 2005; 1755: 25– 36 [DOI] [PubMed] [Google Scholar]
- 14. Ceccatelli S, Tamm C, Sleeper E, et al. Neural stem cells and cell death. Toxicol Lett 2004; 149: 59– 66 [DOI] [PubMed] [Google Scholar]
- 15. Corsini NS, Sancho-Martinez I, Laudenklos S, et al. The death receptor CD95 activates adult neural stem cells for working memory formation and brain repair. Cell Stem Cell 2009; 5: 178– 90 [DOI] [PubMed] [Google Scholar]
- 16. Ricci-Vitiani L, Pedini F, Mollinari C, et al. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J Exp Med 2004; 200: 1257– 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Desbarats J, Birge RB, Mimouni-Rongy M, et al. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol 2003; 5: 118– 25 [DOI] [PubMed] [Google Scholar]
- 18. Ruan W, Lee CT, Desbarats J. A novel juxtamembrane domain in tumor necrosis factor receptor superfamily molecules activates Rac1 and controls neurite growth. Mol Biol Cell 2008; 19: 3192– 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuliani C, Kleber S, Klussmann S, et al. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1). Cell Death Differ 2006; 13: 31– 40 [DOI] [PubMed] [Google Scholar]
- 20. Reimers K, Choi CY, Bucan V, et al. The Bax inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med 2008; 8: 148– 56 [DOI] [PubMed] [Google Scholar]
- 21. Reimers K, Choi CY, Mau-Thek E, et al. Sequence analysis shows that Lifeguard belongs to a new evolutionarily conserved cytoprotective family. Int J Mol Med 2006; 18: 729– 34 [PubMed] [Google Scholar]
- 22. Hu L, Smith TF, Goldberger G. LFG: A candidate apoptosis regulatory gene family. Apoptosis 2009; 14: 1255– 65 [DOI] [PubMed] [Google Scholar]
- 23. Somia NV, Schmitt MJ, Vetter DE, et al. LFG: An anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci U S A 1999; 96: 12667– 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fernandez M, Segura MF, Sole C, et al. Lifeguard/neuronal membrane protein 35 regulates Fas ligand–mediated apoptosis in neurons via microdomain recruitment. J Neurochem 2007; 103: 190– 203 [DOI] [PubMed] [Google Scholar]
- 25. Beier CP, Wischhusen J, Gleichmann M, et al. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B–dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci 2005; 25: 6765– 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hurtado de Mendoza T, Perez-Garcia CG, Kroll TT, et al. Antiapoptotic protein Lifeguard is required for survival and maintenance of Purkinje and granular cells. Proc Natl Acad Sci U S A 2011; 108: 17189– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reich A, Spering C, Gertz K, et al. Fas/CD95 regulatory protein Faim2 is neuroprotective after transient brain ischemia. J Neurosci 2011; 31: 225– 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerber J, Raivich G, Wellmer A, et al. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol 2001; 101: 499– 508 [DOI] [PubMed] [Google Scholar]
- 29. Tauber SC, Stadelmann C, Spreer A, et al. Increased expression of BDNF and proliferation of dentate granule cells after bacterial meningitis. J Neuropathol Exp Neurol 2005; 64: 806– 15 [DOI] [PubMed] [Google Scholar]
- 30. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2nd ed San Diego, CA: Academic Press, 2001 [Google Scholar]
- 31. Wellmer A, Noeske C, Gerber J, et al. Spatial memory and learning deficits after experimental pneumococcal meningitis in mice. Neurosci Lett 2000; 296: 137– 40 [DOI] [PubMed] [Google Scholar]
- 32. Wellmer A, Gerber J, Ragheb J, et al. Effect of deficiency of tumor necrosis factor alpha or both of its receptors on Streptococcus pneumoniae central nervous system infection and peritonitis. Infect Immun 2001; 69: 6881– 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vorhees CV, Williams MT. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 2006; 1: 848– 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerber J, Bottcher T, Bering J, et al. Increased neurogenesis after experimental Streptococcus pneumoniae meningitis. J Neurosci Res 2003; 73: 441– 46 [DOI] [PubMed] [Google Scholar]
- 35. Fecho K, Cohen PL. Fas ligand (gld)- and Fas (lpr)-deficient mice do not show alterations in the extravasation or apoptosis of inflammatory neutrophils. J Leukoc Biol 1998; 64: 373– 83 [DOI] [PubMed] [Google Scholar]
- 36. Paul R, Angele B, Sporer B, et al. Inflammatory response during bacterial meningitis is unchanged in Fas- and Fas ligand–deficient mice. J Neuroimmunol 2004; 152: 78– 82 [DOI] [PubMed] [Google Scholar]
- 37. Fassbender K, Eschenfelder C, Hennerici M. Fas (APO-1/CD95) in inflammatory CNS diseases: Intrathecal release in bacterial meningitis. J Neuroimmunol 1999; 93: 122– 25 [DOI] [PubMed] [Google Scholar]
- 38. Kilic E, Kilic U, Soliz J, et al. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J 2005; 19: 2026– 28 [DOI] [PubMed] [Google Scholar]
- 39. Segal RA. Selectivity in neurotrophin signaling: Theme and variations. Annu Rev Neurosci 2003; 26: 299– 330 [DOI] [PubMed] [Google Scholar]
- 40. Siren AL, Fratelli M, Brines M, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A 2001; 98: 4044– 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng WH, Kar S, Quirion R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J Neurochem 2002; 80: 1049– 61 [DOI] [PubMed] [Google Scholar]
- 42. Bifrare YD, Kummer J, Joss P, et al. Brain-derived neurotrophic factor protects against multiple forms of brain injury in bacterial meningitis. J Infect Dis 2005; 191: 40– 45 [DOI] [PubMed] [Google Scholar]
- 43. Ibanez CF. Jekyll-Hyde neurotrophins: The story of proNGF. Trends Neurosci 2002; 25: 284– 86 [DOI] [PubMed] [Google Scholar]
- 44. von Mering M, Wellmer A, Michel U, et al. Transcriptional regulation of caspases in experimental pneumococcal meningitis. Brain Pathol 2001; 11: 282– 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. D’Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ 2010; 17: 1104– 14 [DOI] [PubMed] [Google Scholar]
- 46. Kuranaga E. Beyond apoptosis: Caspase regulatory mechanisms and functions in vivo. Genes Cells 2012; 17: 83– 97 [DOI] [PubMed] [Google Scholar]
- 47. Troy CM, Akpan N, Jean YY. Regulation of caspases in the nervous system implications for functions in health and disease. Prog Mol Biol Transl Sci 2011; 99: 265– 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beier CP, Kolbl M, Beier D, et al. CD95/Fas mediates cognitive improvement after traumatic brain injury. Cell Res 2007; 17: 732– 34 [DOI] [PubMed] [Google Scholar]
- 49. Drapeau E, Montaron MF, Aguerre S, et al. Learning-induced survival of new neurons depends on the cognitive status of aged rats. J Neurosci 2007; 27: 6037– 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dobrossy MD, Drapeau E, Aurousseau C, et al. Differential effects of learning on neurogenesis: Learning increases or decreases the number of newly born cells depending on their birth date. Mol Psychiatry 2003; 8: 974– 82 [DOI] [PubMed] [Google Scholar]
- 51. Leuner B, Mendolia-Loffredo S, Kozorovitskiy Y, et al. Learning enhances the survival of new neurons beyond the time when the hippocampus is required for memory. J Neurosci 2004; 24: 7477– 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marin-Burgin A, Schinder AF. Requirement of adult-born neurons for hippocampus-dependent learning. Behav Brain Res 2012; 227: 391– 99 [DOI] [PubMed] [Google Scholar]
- 53. Shors TJ, Miesegaes G, Beylin A, et al. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001; 410: 372– 76 [DOI] [PubMed] [Google Scholar]
- 54. Karl T, Pabst R, von Horsten S. Behavioral phenotyping of mice in pharmacological and toxicological research. Exp Toxicol Pathol 2003; 55: 69– 83 [DOI] [PubMed] [Google Scholar]
- 55. Sahay A, Scobie KN, Hill AS, et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011; 472: 466– 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saab BJ, Georgiou J, Nath A, et al. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 2009; 63: 643– 56 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.