

Abstract
According to Thomas Kuhn, the success of ‘normal science,’ the science we all practice on a daily basis, depends on the adherence to, and practice of, a paradigm accepted by the scientific community. When great scientific upheavals occur, they involve the rejection of the current paradigm in favor of a new paradigm that better integrates the facts available and better predicts the behavior of a particular scientific system. In the field of Alzheimer’s disease, a recent example of such a paradigm shift has been the apparent rejection of the ‘amyloid cascade hypothesis,’ promulgated by Hardy and Higgins in 1992 to explain the etiology of Alzheimer’s disease, in favor of what has been referred to as the ‘oligomer cascade hypothesis’. This paradigm shift has been breathtaking in its rapidity, its pervasiveness in the Alzheimer’s disease field, and its adoption in an increasing number of other fields, including those of Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and the prionoses. However, these facts do not mean, a priori, that the experiments extant, and any re-interpretation of them, should be accepted by rote as support for the new paradigm. In the discussion that follows, I consider the foundational studies leading to the oligomer cascade hypothesis and evaluate the current state of the paradigm. I argue here that, more often than not, insufficient rigor has been applied in studies upon which this new paradigm has been based. Confusion, rather than clarity, has resulted. If the field is to make progress forward using as its paradigmatic basis amyloid β-protein oligomerization, then an epistemological re-evaluation of the amyloid β-protein oligomer system is required.
Review
The failure of the amyloid cascade hypothesis and the emergence of the oligomer cascade hypothesis
The history of amyloid has been suggested to have begun as early as the 17th century [1]. However, the recognition of Alzheimer’s disease (AD) may be said to have begun with Alois Alzheimer’s presentation of the case of Auguste Deter to the South-West German Society of Alienists. Alzheimer reported histologic evidence for the presence of what we now know are amyloid plaques and neurofibrillary tangles [2]. Approximately 80 years later, protein sequencing [3,4] and gene cloning [5-8] led to the identification of the amyloid β-protein precursor (APP), an achievement that enabled modern molecular biological studies of the APP/amyloid β-protein (Aβ) system. The most important of these first studies revealed that APP gene expression and Aβ production were normal physiologic processes [9], meaning that it was not the existence of Aβ per se that caused AD but rather that an epigenetic etiology was involved in the majority of cases of sporadic (that is, non-familial) AD.
An obvious hypothesis was that amyloid plaques, the pathognomonic histological feature of AD, caused the disease. In the mid-20th century, the pioneering electron microscopic work of Cohen and Kidd revealed that amyloid was composed of fibrils [10,11]. Aβ fibrils thus became the raison d’être of the AD field. Intense efforts to understand fibril formation and the biological activities of fibrils thus ensued. Early work showed that Aβ was not toxic unless it formed fibrils [12,13]. The linkage between fibrils and disease thus was established, both clinically and in vitro, a linkage that came to be known as the ‘amyloid cascade hypothesis’ [14].
If amyloid fibrils were the proximate neurotoxins in AD, then a reasonable therapeutic approach would be to target fibril formation. Knowledge-based approaches for fibril-centric therapies require the elucidation of fibril structure and the mechanism of fibril formation. Tremendous progress has been made by structural biologists in these areas. Electron microscopy, fiber X-ray diffraction, X-ray crystallography, NMR spectroscopy, and other spectroscopic techniques have provided detailed information on the morphology and structure (secondary, tertiary, and quaternary) of fibrils (see [15] and references therein). These experiments have been complemented by in vitro studies of the kinetics and thermodynamics of the fibril formation process [16-18]. Most techniques in common use by researchers are ‘population average’ in nature. This means that data derived from the technique (for example, by circular dichroism spectroscopy, NMR, thioflavin T, and infrared spectroscopy) are the average of the contributions of each of the structures probed by the technique. To reveal structural features, dynamics, and cellular interactions of individual structures, single-molecule spectroscopic methods have been used, including atomic force spectroscopy [19], fluorescence [20,21], and fluorescence resonance energy transfer [22]. However, by definition, single-molecule methods cannot show how oligomerization and higher-order assembly occur. For this reason, in silico (computational) studies of Aβ conformational dynamics, oligomerization, and fibril formation have been particularly valuable because they show the step-dependent structures of each Aβ monomer and all of its individual atoms as a simulation proceeds (for reviews, see [23-25]).
The most important conceptual breakthrough in studies of Aβ assembly was the recognition that Aβ fibril formation was not a simple nucleated polymerization reaction, akin to actin polymerization [26], in which monomers self-associate to form a small oligomeric nucleus from which subsequent monomer addition results in polymer growth. Instead, Aβ assembly has been found to be a remarkably complex process comprising aggregation events that are ‘on-pathway’ and ‘off-pathway’ for fibril formation (Figure 1) (for a recent review, see [27]). The implication of these discoveries was that assemblies other than amyloid fibrils might also play a role in AD pathogenesis. This postulation was strengthened by the relatively poor correlation between the amounts and regional distribution of amyloid deposits, and the clinical status of AD patients [28]. In fact, some have suggested that amyloid deposition is protective [29].
Figure 1.

Amyloid β-protein assembly. Amyloid β-protein appears to be an intrinsically disordered protein and thus exists in the monomer state as an equilibrium mixture of many conformers. ‘On-pathway’ fibril assembly requires the formation of a partially folded monomer that self-associates to form a nucleus for fibril elongation, a paranucleus, which in this case contains six monomers. Nucleation of monomer folding is a process distinct from fibril nucleation. Fibril nucleation is unfavorable kinetically (), which explains the lag phase of fibrillogenesis experiments, a period during which no fibril formation is apparent. Paranuclei self-associate readily () to form protofibrils, which are relatively narrow (approximately 5 nm), short (<150 nm), flexible structures. These protofibrils comprise a significant but finite number X of paranuclei. Maturation of protofibrils, through a process that is poorly understood but is favorable (), yields classical amyloid-type fibrils (approximately 10 nm diameter, indeterminate (but often >1 μm) length). Other assembly pathways produce a variety of oligomers, including annular, pore-like structures; globular dodecameric (and higher-order) structures; and amylospheroids. Adapted from [27].
If fibrils are not the key neurotoxins in AD, what is? Seminal studies by the Mucke group showed that transgenic animals displayed neurologic deficits prior to plaque formation [30,31]. Subsequent studies confirmed and extended these results by demonstrating that Aβ oligomer (Aβ*56 [32]) concentration, not fibril load, correlated better with functional deficits in a mouse model of AD [33]. In humans, an ‘Arctic’ form of AD, caused by an APP mutation producing a Glu22 → Gly amino acid substitution in Aβ, appears to be a disease caused by protofibrils, the immediate precursors of fibrils [34-36].
Clinical trials of potential AD drugs have been disappointing. An early active immunization trial involving aggregated Aβ42 plus an adjuvant was terminated prior to completion due to the occurrence of life-threatening aseptic meningoencephalitis and leukoencephalopathy [37,38]. Some patients in the trial did show significant decreases in amyloid load, but no significant cognitive improvement. Two subsequent phase III immunotherapy trials with humanized Aβ-specific antibodies both failed to achieve their primary endpoints – a change in cognitive and functional performance compared to a placebo [39,40]. In fact, to date, all Aβ-directed clinical trials have shown either no efficacy or subtle effects only in sub-groups of the clinical trial populations.
It is important to distinguish explicitly the failure of hypothesis testing from the failure of a hypothesis. Clinical trial results thus far do not support the amyloid cascade hypothesis, but the results also are not a refutation of the hypothesis. Trial design, including the selection of study populations and the therapeutic agents themselves, may be flawed, thus providing some hope that improved therapeutic approaches may have merit [41]. Our discussion here focuses on oligomers, but the reader should note that the role fibrils play in AD remains unclear. Are they protective [29]? Are they pathologic [42]? These answers remain moot.
The oligomer question
In considering oligomersa and their role in AD, it is useful to establish the position of our current research efforts within the larger context of biomedical and clinical research. The goals of this research are to prevent, treat, or cure disease. To do so, one must first understand disease etiology. In the case of microbial diseases, Robert Koch suggested four criteria for identifying a causative agent [43,44]. Koch’s postulates, or more modern variants thereof, are foundational elements of pathobiology. The application of these postulates in microbiology and virology is relatively straightforward. This is not the case for AD, for which the agent has not been: (1) isolated from an AD patient; (2) produced in pure form in vitro; (3) used to infect a susceptible host (in this case, for ethical reasons, non-human primates);b and (4) re-isolated from that new host. Nevertheless, if the etiology of AD is to be understood, then it is obligatory that AD researchers establish an appropriate set of criteria for determining what causes AD.
Let us begin with the strategic goal of identifying a causative agent. The first question is whether there is a causative agent. The data extant support the conclusion that there is not, if we define ‘causative agent’ as a single biological entity, for example, a single protein of defined structure. AD is a multi-factorial disorder [45,46] involving a complex pathway(s) comprising both genetic (for example, APP, presenilin gene, apolipoprotein E gene, and γ-secretase activating protein gene) and epigenetic (inflammatory insults, and oxidation reactions) components. However, we can stipulate that a majority of researchers believe that Aβ is involved, particularly Aβ oligomers [27,47]. Whether this majority opinion, in the future, will be supported by sufficient evidence remains at issue.
The amyloid β-protein oligomer
I now arrive at the kernel of the oligomer cascade hypothesis, namely the Aβ oligomer. What is it? Answer – no formal definition exists (although a nebulous generic definition certainly does (see below)). How is the field to apply the scientific method to studies of AD causation and cure if a key variable in the field, the Aβ oligomer, remains undefined? The answer is to define it, which brings us to our second conundrum – is there an ‘it,’ that is, a single Aβ oligomer structure? Again, the answer is ‘no’.
Aβ oligomerization is a protein-folding process that involves two interrelated sub-processes: monomer folding and monomer self-association. Each sub-process, and thus the entire oligomerization process itself, has no single pathway, just as protein folding, in general, has no single pathway [48-50]. The de facto working hypothesis that Aβ folding produces a single structural entity is wrong. The related hypothesis, that there exists a single Aβ assembly that causes AD, also is likely wrong. I now examine why, and the implications emerging from the answer.
Scientists are taught, to a significant degree, to think linearly; for example, when they are taught about glycolysis, chemical synthesis, or atomic decay. However, protein folding is non-linear. It is a stochastic process that has as its thermodynamic foundation the free energy of formation of each specific protein conformer (structure). The overall difference in the free energies of the initially disordered nascent protein and its natively folded form can be highly favorable; that is, the native fold is a very stable, low-energy state. However, kinetic considerations (for example, the ability of the folding process to overcome high-energy barriers in the folding pathway) often become the key determinants in the outcome of the folding process. For this reason, kinetic ‘traps’ have the potential to block the formation of native protein folds, regardless of the overall thermodynamic favorability of the folding process. In these cases, non-native folds may be the end state of the folding process.
The energetics of protein folding can be represented by a three-dimensional reaction coordinate system [49]. Typically, these surfaces comprise dimensions of energy, entropy, and number concentration. For example, a simple energy surface is illustrated in Figure 2. Nascent proteins (that is, those being extruded from the ribosome) enter this surface in a high-energy state characterized by substantial entropy and by a large number of conformers. If these proteins have native folds (for example if they are enzymes or structural proteins) then they descend this energy surface through different routes, the choice of route being determined probabilistically. Pathways exist for these proteins in which the energy barriers between two positions are relatively small (intrinsically or through the action of chaperones) so that the proteins are able to move rapidly (favorable kinetics) from their initial high-energy unfolded states to their final low-energy natively folded states. In other cases, folding is more complex (Figure 3), and kinetic traps (low-energy wells or high-energy barriers) must be overcome before folding is complete. In each case, the outcome of successful folding is the native state, characterized by low energy, low entropy, and low conformational complexity.
Figure 2.
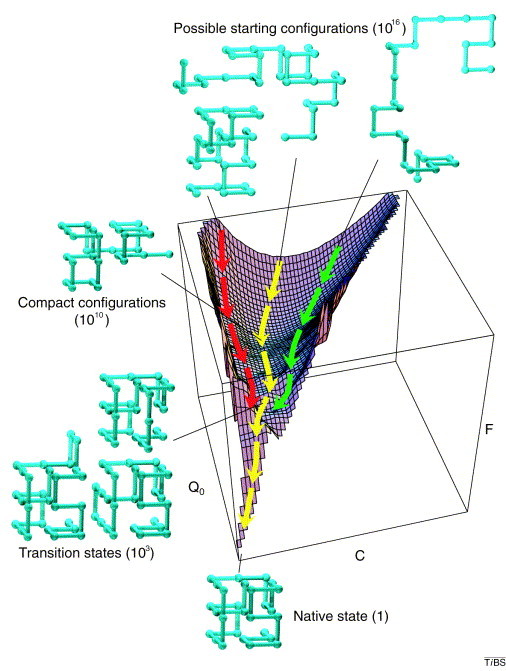
Simple folding surface. Free-energy (F) surface of a 27-residue model protein. The axes are the number of native contacts (Q0) and the total number of (native and non-native) contacts (C). The yellow trajectory shows the average path traced by the unfolded protein. The green and red trajectories lie two standard deviations from the average; thus 95% of all trajectories would be expected to exist within this range. The green structures outside the axes illustrate the various stages of the reaction. A folding peptide collapses rapidly from one of its 1,016 possible random starting conformations to a disordered globule. It then makes a slow, non-directed search among the 1,010 semi-compact conformations for one of the approximately 103 transition states that lead rapidly to the unique native state. Adapted from [51].
Figure 3.

Complex folding surface. Schematic free-energy (F) surface representing features of the folding of hen lysozyme (a protein of 129 residues whose structure consists of two domains denoted α and β). Qα and Qβ are the numbers of native contacts in the α and β domains. The yellow trajectory is a ‘fast track’ in which the α and β domains form concurrently and populate the intermediate (labeled α/β) only transiently. The red trajectory is a ‘slow track’ in which the chain becomes trapped in a long-lived intermediate with persistent structure in only the α domain. Further folding requires either a transition over a higher energy barrier or partial unfolding to enable the remainder of the folding process to occur along the fast track. Residues whose amide hydrogens are protected from solvent exchange in the native structure (as assessed by NMR) are colored red (α domain) or yellow (β domain). All others are blue. Adapted from [51].
Aβ is different from proteins with stable native folds. Its folding landscape is complex and contains deep energy wells and tortuous routes to low-energy states [52-55]. Large parts of the surface are relatively flat, which means that in these areas the peptide can exist in many different conformations with essentially equal probability. These areas may also correspond to the normal intrinsically disordered states of Aβ. However, many positions also exist from which Aβ can reach the limited number of low-energy states occupied by fibrils. Classical amyloid fibrils share a diameter of approximately 10 nm and a cross-β core secondary structure (for a review of Aβ fibril structure, see [56]). However, the existence of multiple low-energy states explains why fibril morphology is still plastic and can vary depending, for example, on primary structure and on conditions of formation (agitated or quiescent) [57-61].
The intermediate positions between high-energy disordered monomer and low-energy highly ordered fibrils are populated by oligomers. The oligomer states are diverse. The conformations of the oligomers vary substantially, so much so that no obvious precursor-product relationship is obvious between many of these assemblies. The free energies of formation of oligomers are similar enough (a thermodynamic consideration) and the energy barriers between states are low enough (a kinetic consideration) that rapid inter-conversions can occur among the different oligomers (they are metastable). Oligomer order (the number of monomers per oligomer) can vary significantly (from dimer to tens or hundreds of monomers). In addition, the conformation of an oligomer state of specific order (for example, dimers) also is populated by many different conformers. These facts mean that the most accurate definition of ‘oligomer’ is a statistical distribution of structures. Importantly, this distribution depends on the establishment of an equilibrium under specific conditions. Any change in conditions can produce a different distribution of oligomer states.
The frequency distribution of specific oligomer states is affected by the Aβ primary structure and by the environment of the peptide, the latter factor encompassing solvent composition (in vitro and in vivo), pH, temperature, and macromolecular components (proteins, lipids, and carbohydrates). The sensitivity of the distribution to environmental perturbations presents to the AD researcher the same problem faced by the physicist trying simultaneously to determine the position and momentum of an electron; namely, that in trying to determine one or another of these parameters, the investigator perturbs the system, making such determination impossible [62]. In the AD field, the most common example of this is the use of SDS in experiments designed to determine oligomer order in populations of non-covalent complexes [63]. Non-covalent complexes of Aβ dissociate in SDS, and in addition SDS can cause adventitious self-association of natively monomeric Aβ42 [64].
An obligatory lexicographical diversion
You like potato and I like potahto
You like tomato and I like tomahto
Potato, potahto, Tomato, tomahto
Let’s call the whole thing off
– George & Ira Gershwin (From ‘Let’s Call The Whole Thing Off’)
A major factor inhibiting progress in the oligomer field is the lack of adherence to an accepted lexicon. When Fred Astaire sang the verse above to Ginger Rogers in the 1936 film Shall We Dance? he was not simply commenting on their pronunciation differences. He was commenting on the incommensurability of their two perspectives on life, which is why the suggestion of ‘calling the whole thing off’ followed. A single lexicon must exist among scientists in the AD field if the field is to advance understanding of AD, its causative agent(s), and effective therapeutic approaches. It is obligatory that when anyone says ‘oligomer’ that all others know precisely to what the speaker is referring. We all must mean the same thing if we use the same term (or we should ‘call the whole thing off’).
Let us first consider some terms that often are misused. This misusage leads to misunderstanding and to the promulgation of concepts that are erroneous. These terms have definitions, which should be used as they were specified when they first were published or as they are presented in textbooks. Even if common usage differs from formal usage, only one definition of each term should exist. I discuss one set of usage standards below.
Protein ‘misfolding’
No such phenomenon exists in nature. As discussed above, protein folding involves the stochastic exploration of conformational space, either by a protein alone or with the assistance of chaperones. Misfolding is a relative term that is meant to contrast the folding, or assembly, of a protein into a non-native state(s) as opposed to a physiologically normal native fold/assembly. Two problems with the term are: (1) it suggests implicitly that two folding systems exist, a folding system and a misfolding system (as discussed immediately above, this cannot be true); and (2) it directs research towards the irrelevant questions of misfolding versus folding, instead of toward the relevant goal of elucidating the folding landscape. The same type of ‘normal–abnormal’ philosophical perspective substantially delayed the discovery that the amyloid β-protein was a ‘normal’ protein and not an ‘abnormal’ protein [9].
Conformation
‘Conformation,’ in the context of protein structure and immunology, means a three-dimensional shape. All proteins, peptides, amino acids, and other macromolecules are three dimensional. No one-dimensional worlds exist, and only a single two-dimensional world has ever been reported [65]. All epitopes are three dimensional. All antibody combining sites are three dimensional. All antibodies recognize three-dimensional epitopes. To prepend the word ‘conformation’ and its derivatives to epitope, specific, dependent, or antibody is redundant and incorrect.
Order
In the context of the discussion of Aβ assemblies, ‘order’ specifies the number of Aβ monomers comprising a particular assembly. The term does not refer to conformation.
Polymer
The word ‘polymer’ is from the Greek polus (πoλυσ, meaning heavy, much, many, large) and meros (μϵρoσ, meaning parts). Polymers (fibrils) are assemblies composed of large numbers of a basic structural unit. It is this repetitive structure that distinguishes polymers from lower-order assemblies. For Aβ, monomer [66], dimer [67], trimer [68], and pentamer [66] ‘unit cells’ have been reported. Amyloid fibrils possess cores organized in a cross-β conformation [69], but can display a variety of morphologies [58] and organizations of their cross-β cores [70].
Protofibril
The word ‘protofibril’ is from the Greek proto (πρoτo, meaning first) and the Latin fibrila (meaning fiber). When the discovery of Aβ protofibrils was published [34,35,71], protofibrils were described as ‘predominantly curved fibrils, 6 to 8 nm in diameter and <200 nm in length’. Protofibrils are unbranched, flexible, and distinct from fibrils, oligomers, or monomers. Their secondary structure comprises a substantial β-sheet [34]. Protofibrils have been shown to be in equilibrium with Aβ monomers and fibrils [34]. Protofibrils are the penultimate fibril precursor. Thus far, structure determination of protofibrils has not reached the level of resolution of fibrils.
Oligomer
The word ‘oligomer’ is from the Greek oligos (oλιγoσ, meaning little, few). The class of oligomers begins with dimers and includes higher-order structures that do not have the characteristics of protofibrils or fibrils. ‘Fibrillar oligomers’ cannot exist because if they display the characteristics of fibrils (repeating basic structural units, tinctorial properties, spectroscopic properties, cross-β secondary structure, 10 nm diameter, and high aspect ratio) they simply are ‘short fibrils.’ If they do not display these characteristics, they simply are ‘oligomers.’ Short oligomers can be distinguished from short fibrils by exclusion; namely, by the fact that they are not straight, unbranched assemblies with substantial aspect ratios. Oligomers comprise a broad range of assemblies, varying in primary, secondary, tertiary, and quaternary structure. The structures of various oligomers, like protofibrils, have not been determined at high resolution.
Intrinsically disordered
Intrinsically disordered proteins, or protein regions, are not random in structure because they do not populate all regions of their potential conformational space with equal probability. However, they also do not appreciably populate α-helix, β-sheet, or β-turn regions of conformational space. Intrinsically disordered protein regions are thought to exist in approximately 15% to 45% of eukaryotic proteins (for recent reviews, see [72-74]). These regions are involved in a myriad of regulatory and signaling functions involving protein binding, kinase activity, DNA replication, and regulation of enzyme activity and substrate binding [75].
Pathways (on/off)
Like ‘misfolding,’ this is a relative term. Researchers commonly consider whether particular assemblies of Aβ are ‘on-pathway’ or ‘off-pathway,’ but to what pathway do they refer? Typically, the pathway is fibril formation. From a strictly academic perspective, these considerations are meritorious. However, they contribute little to the quest of understanding and treating AD, and for the same reason that folding/misfolding is an irrelevant concept. Aβ assembly comprises a conformational ‘space,’ not a pathway per se. This space comprises many pathways that result in equilibria among Aβ monomers and higher-order structures. Fibril formation simply is one of many different ‘random walks’ [76] (stochastic events, although with different occurrence frequencies) that nascent Aβ monomers may take. The therapeutically relevant question is: ‘what positions in Aβ conformational space are occupied by neurotoxic structures?’
Generic structure
When myoglobin was shown to form amyloid fibrils in 50 mM sodium borate, pH 9.0, at 65°C [77], conditions known to destabilize the protein’s native fold, the concept emerged that amyloid formation could be a default process through which many, if not all, proteins might proceed [78]. The result of this process, both structurally and immunologically, was the formation of a ‘generic’ amyloid structure [79]. Many, or even most, proteins may form amyloid, but the process is not generic per se[80]. The process is simply one pathway through assembly space that leads to a particularly low-energy state.c The actual amyloid conformers formed by different proteins are not ‘generic.’ They are structurally distinct, one from the other, because their primary structures are not identical. If we stipulate this fact, then we can discuss amyloid structural motifs, which are non-identical in structure but do share some structural characteristics. The most compelling example of this is the steric zipper [81]. X-ray crystallographic studies have shown how this steric zipper can constitute the cross-β structure of amyloid fibrils and how side-chain and peptide backbone interactions stabilize the structure. The steric zipper is a structural motif, but it is not generic per se. At least eight different structural forms of the steric zipper exist [69].
Generic oligomer epitope
None exist. The antigen binding site of an antibody has a defined three-dimensional structure. This means that its binding affinity for various antigens (epitopes) varies depending on the specific inter-atomic interactions occurring between its idiotope and the antigen epitope. Polyclonal antibody reagents cannot reveal different epitopes existing in oligomers formed by different proteins. The different immunoglobulins comprising the antibody mixture will bind to different epitopes and the result will be a positive dot blot or Western blot for each of many different proteins. The deduction that a common epitope exists is illogical and fallacious. The explanation lies in antibody cross-reactivity. However, even if a monoclonal antibody, which by definition expresses a single idiotope, binds to oligomers formed by distinct proteins, this does not support the conjecture that a common ‘oligomer-specific’ epitope exists. The correct conclusion is that the antibody binds to the protein under study, nothing more. Competition assays and X-ray crystallographic analyses are necessary to determine the structure of the epitope. Such studies will elucidate the binding modes of different oligomers and, for oligomers of different primary structure, will show the atomic basis for antibody affinity and cross-reactivity. These studies also can determine dissociation constants for each antigen:antibody complex. It is important to note, as I discussed immediately above, that structural motifs may exist, as in the examples of cross-β or steric zipper structures. The point to remember is that ‘similar’ is not equivalent to ‘identical,’ and especially that ‘generic’ is not equivalent to ‘identical.’
Antibody ‘X’-reactive (positive)
We should be particularly aware of the uncertainties of immunological approaches to defining structure, especially oligomer structure. This is particularly true if our nomenclature incorporates such terms as ‘antibody X-reactive.’ The watchwords of immunology are specificity and sensitivity. Co-crystallization of a number of Aβ-specific monoclonal antibodies with Aβ has revealed that different secondary structure elements may exist within the same N-terminal peptide segment [82,83]. If one uses one such monoclonal as an ‘N-terminal antibody,’ it is obvious that the antibody may or may not bind to Aβ, depending on the distribution of N-terminal conformations within each Aβ preparation. Specificity then becomes a nebulous and useless term as binding or non-binding may not represent the actual distribution of Aβ structures within a particular isolate or preparation. Similarly, if antibody titers are low (that is, if affinity or avidity is poor) cross-reactivity becomes a significant problem. A ‘positive’ result should not be interpreted as an antibody:antigen interaction involving a single epitope. Instead, as discussed above, we are merely observing a distribution of species that are immunoreactive, preventing accurate structure–activity determinations. Finally, a very important caveat, and one that has been largely ignored by the community, is the issue of heterophilic antibody interference (for a useful editorial, see [84]). Here, the presence of endogenous antibodies recognizing species-specific epitopes on primary or secondary antigen-specific (for example, Aβ-specific) antibodies produces false positive reactions. For example, heterophile antibodies that recognize immunoglobulins from other species, and are present in human plasma and cerebrospinal fluid (CSF), may cause interference in sandwich immunoassays like ELISAs by cross-binding the capture and detection antibodies of the assay (in the absence of an antigen recognized by the capture antibody) [85].
The conundrum of ‘biological relevance’
Pure scientific enquiry requires no consideration of relevance. However, the practical issue of funding biomedical research and producing new insights into biological systems does require consideration of the relevance question. Is this possible for the Aβ system?
The disconnect between the in vivo and in vitro worlds
We often say that we study Aβ ‘under physiologic conditions’. This typically means studies in phosphate-buffered saline, pH 7.4. We then argue that the data derived from our studies are biologically relevant because the system is ‘physiologic.’ However, the milieu of the human body is undefined. It is too complex to understand with the scientific tools currently available. This is the reason we turn to in vitro systems, which reduce questions to forms answerable using the scientific method. The problem then becomes one of establishing the true value of inferences obtained in our simple, yet profoundly incomplete (vis-à-vis the brain in the case of AD), experimental systems.
I argue that the milieu of Aβ in situ in vivo cannot be defined precisely, and if we cannot do so, then what constant of proportionality can be employed to relate our in vitro findings to the structure and behavior of Aβ in the human brain? How does the milieu change between different sub-cellular, cellular, or anatomical locations? How do purification procedures designed to yield Aβ assemblies alter the native states of such assemblies? It has been argued, for example, that when fibrillar Aβ is isolated, a native proteoglycan component is lost [86]. ‘Native state’ assemblies could comprise such complexes, or complexes involving other biomolecules. Importantly, these states could display unique primary, secondary, tertiary, or quaternary structures of the Aβ component. These arguments apply both to studies of Aβ pre-existent in biological milieus and to studies in which Aβ is introduced exogenously.
We also are faced with the dilemma of time. How do Aβ assemblies change over time in a particular milieu? What are the equilibrium relationships involved? Work by Bateman and colleagues [87] has shown that biological half-lives for newly produced Aβ measured in CSF are of the order of 6 hours. From where do the Aβ species we seek to study come? Do subsets of these species traffic to different cell types, organelles, or extra-cellular sites (for example, plaques) within which their states and catabolic characteristics differ substantially? Are the post-translational modifications of Aβ identified in vivo disease-related or irrelevant (representing an epiphenomenon of long-term tissue deposition)?
Iatrogenesis in studies of amyloid β-protein structure and activity
What may be the most intractable issue is that of ‘iatrogenesis’; that is, how do we, in the process of isolating and studying Aβ derived from cellular or organismal sources, alter the native state of the peptide? Such alteration(s) has the capacity to invalidate the determination of structure–activity relationships (SARs). We may know that a particular assembly that we have isolated from the brain, characterized structurally, and studied in vitro has certain activities, but we do not know that this assembly actually existed in the biological milieu prior to its isolation.
The most obvious examples of iatrogenesis are the use of SDS in Aβ isolation procedures and in SDS-PAGE. The theory of SDS-PAGE requires the presence of SDS to denature proteins. Oligomers are predominately non-covalent Aβ complexes. SDS dissociates such complexes well. In addition, and less well understood, there is the ability of SDS to induce Aβ self-association [88], especially into the trimer and tetramer states [64,89]. The use of SDS is but one caveat in our approaches to establishing SARs. Other compounds that can affect oligomerization state and that have been used in Aβ isolation procedures from human brains include 1% Triton X-100, 5 M guanidine HCl, and 70% or 88% formic acid [90-94].
Conclusions
The prior discussion has emphasized the complexity of the Aβ system, both in vitro and in vivo. It has focused on principles governing our study of Aβ oligomers, on weaknesses and caveats in our approaches to these studies, on the nebulous nature of our conception of oligomers and our use of the term itself, and on the problems of establishing the biological relevance of our work. It would be easier if Aβ possessed a native fold and if changes to this fold could be treated in the same way that some cancers can be cured by production of antibodies specific for kinase structural elements [95]. Unfortunately, this is not the case, which leaves us the question of how to handle the non-ideal system in which we work.
Be precise in defining structures and in referring to structures
A first suggestion is to encourage researchers to be far more diligent in defining the assemblies that they study. This is the nomenclature question. How do we work together as a scientific community if we all use the same terms but they mean different things? We work on non-covalent complexes (with the possible exception of covalently cross-linked Aβ oligomers [96,97]). This fact must be foremost in our minds as we consider experimental design and interpret our experimental results. We cannot know, at least not yet, the structural state of the Aβ assemblies that exist in the CSF, plasma, or brain tissues that are the sources of the Aβ used in experiments. We should recall our study, as undergraduate chemistry students, of Le Châtelier’s principle [98] and understand that our manipulation of Aβ, whether from tissue isolates or using chemically synthesized peptides, will change the distribution of oligomeric states if non-equilibrium methods are used (detergent extraction, SDS-PAGE, size exclusion chromatography, dialysis, or filtration). We can speak about what we derive following these procedures, but we cannot infer that what we observe exists in our original isolates or preparations. We can, however, accurately and precisely describe the process(es) through which these assemblies are isolated and characterized. This enables others to compare their results with ours or to reproduce our findings in a different laboratory.
Do relevant experiments that advance the field towards effective therapies
Relevance questions always require an object, namely: ‘to what is a question relevant?’ In the case of AD, the majority of work in the field has had relevance to our basic understanding of cell and molecular biology, but not necessarily to AD. For example, the search for and study of γ-secretase have contributed substantially to our general understanding of regulated intra-membrane proteolysis. This knowledge has been of great value in understanding organismal development and homeostasis, in addition to its potential (though not yet realized) for drug development. Similarly, and especially importantly, structure–neurotoxicity relationships among different Aβ assemblies and various types of cells, especially primary rodent neurons, are leading to an increased understanding of mechanisms of Aβ-mediated toxicity. If the assemblies that we study are poorly defined structurally, our understanding of their mechanisms of action will be poor. However, even if assembly structure is defined at atomic resolution, we may not know formally whether biological studies recapitulate what occurs in the human brain. Nevertheless, I argue that: (1) in well-controlled experimental systems, the data derived from studies of Aβ biological activity tell us something about pathways of Aβ-induced cellular stress, injury, and death per se; and (2) the value of the information with respect to understanding AD pathobiology can be validated if in future work it can be shown that interventions based upon such information successfully modify or prevent the AD state. Again, formally, we cannot know whether our interventions operate in situ in vivo according to the mechanisms we promulgate. However, if our endpoints are preventing, ameliorating, decelerating progression of, or curing AD, and we reach one or more of these endpoints, then the question of validated mechanism becomes a question of an almost entirely academic nature. The fact that patients are helped is what matters.
Coda
A journey of a thousand miles begins with a single step.
– Lao-Tzu (604–531 BC)
Biomedical science is a difficult thing to do well, and within the field, the Aβ system is particularly problematic and vexing. It is a non-ideal system that demands an unusually rigorous approach if its secrets are to be uncovered and effective AD therapeutics are to be developed. Let us strive to follow the advice of Lao-Tzu by making our first step in the right direction!
Endnotes
aThe reader is encouraged to examine an excellent recent treatment of the oligomer field by Benilova et al.[99].
bTwo recent reports do suggest that AD pathology can be propagated through prion-like cell-to-cell spread in mice following injection of brain homogenates from AD transgenic animals [100] or an AD patient [101].
cIt should be emphasized once more that the thermodynamic arguments here refer to the final equilibrium state of a system. However, this state may not be reached (at least in our lifetimes) because of kinetic traps that, in practical terms, take a particular assembly out of a pathway. Protein precipitation, for example, is one process that can lead to a system state in which an insurmountable activation energy (under physiologic conditions) would be required to place the precipitated proteins back ‘on-pathway.’
Abbreviations
AD: Alzheimer’s disease; APP: Amyloid β-protein precursor; Aβ: Amyloid β-protein; CSF: Cerebrospinal fluid; ELISA: Enzyme-linked immunosorbent assay; NMR: Nuclear magnetic resonance; SAR: Structure–activity relationship.
Competing interests
The author declares that he has no competing interests.
Acknowledgements
The author gratefully acknowledges helpful discussions and editorial suggestions from Drs Eric Hayden and Robin Roychaudhuri, Mr Michael Hughes, Mr Joseph Conovaloff, and Ms Shiela Beroukhim. The author also acknowledges the work of others that could not be cited within the abstract [14,46,102,103]. The support of NIH grants NS038328, AG041295, and AG016570 is acknowledged.
References
- Schwartz P. Amyloidosis – Cause and Manifestations of Senile Dementia. Springfield: Charles C Thomas; 1970. [Google Scholar]
- Alzheimer A. On an extraordinary, severe disease process of the cerebral cortex [in German] Neurologisches Centralblatt. 1906;5:1129–1136. [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s Disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;5:1131–1135. doi: 10.1016/0006-291X(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;5:885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;5:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GAB, St George-Hyslop PH, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid β-protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;5:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;5:4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek D. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;5:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;5:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Calkins E. Electron microscopic observation on a fibrous component in amyloid of diverse origins. Nature. 1959;5:1202–1203. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- Kidd M. Alzheimer’s disease – an electron microscopical study. Brain. 1964;5:307–320. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;5:311–314. doi: 10.1016/0006-8993(91)91553-D. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;5:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;5:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Müller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TPJ, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci USA. 2013;5:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA. 1996;5:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc Natl Acad Sci USA. 1997;5:7942–7947. doi: 10.1073/pnas.94.15.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Accounts Chem Res. 2006;5:671–679. doi: 10.1021/ar050069h. [DOI] [PubMed] [Google Scholar]
- Krasnoslobodtsev AV, Peng J, Asiago JM, Hindupur J, Rochet JC, Lyubchenko YL. Effect of spermidine on misfolding and interactions of alpha-synuclein. PLoS ONE. 2012;5:e38099. doi: 10.1371/journal.pone.0038099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P, Ganzinger KA, McColl J, Weimann L, Meehan S, Qamar S, Carver JA, Wilson MR, St George-Hyslop P, Dobson CM, Klenerman D. Single molecule characterization of the interactions between amyloid-β peptides and the membranes of hippocampal cells. J Am Chem Soc. 2013;5:1491–1498. doi: 10.1021/ja3103567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;5:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- Ly S, Altman R, Petrlova J, Lin Y, Hilt S, Huser T, Laurence TA, Voss JC. Binding of apolipoprotein E inhibits the oligomer growth of amyloid-β peptide in solution as determined by fluorescence cross-correlation spectroscopy. J Biol Chem. 2013;5:11628–11635. doi: 10.1074/jbc.M112.411900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Teplow DB, Stanley HE. Computer simulations of Alzheimer’s amyloid β-protein folding and assembly. Curr Alzheimer Res. 2006;5:493–504. doi: 10.2174/156720506779025170. [DOI] [PubMed] [Google Scholar]
- Fawzi NL, Yap EH, Okabe Y, Kohlstedt KL, Brown SP, Head-Gordon T. Contrasting disease and nondisease protein aggregation by molecular simulation. Accounts Chem Res. 2008;5:1037–1047. doi: 10.1021/ar800062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Ding F, Dokholyan NV. Probing protein aggregation using simplified models and discrete molecular dynamics. Frontiers Biosci. 2008;5:4795. doi: 10.2741/3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosawa F, Asakura S, Hotta K, Imai N, Ooi T. G-F transformation of actin as a fibrous condensation. J Polymer Sci. 1959;5:323–336. doi: 10.1002/pol.1959.1203713202. [DOI] [Google Scholar]
- Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. J Biol Chem. 2009;5:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;5:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Lee H, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA. Reexamining Alzheimer’s disease: evidence for a protective role for amyloid-β protein precursor and amyloid-β. J Alzheimer’s Disease. 2009;5:447–452. doi: 10.3233/JAD-2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;5:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ(1–42) in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;5:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;5:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;5:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;5:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;5:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;5:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby L, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;5:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black R, Jenkins L, Griffith S, Fox N, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. AN1792(QS-21)-201 study team: clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;5:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Pollack A. Alzheimer’s drug fails its first big clinical trial. New York Times. 2012.
- Eli L. Eli Lilly and company announces top-line results on solanezumab phase 3 clinical trials in patients with Alzheimer’s disease. 2012. http://newsroom.lilly.com/releasedetail.cfm?releaseid=702211.
- Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nature Rev Neurol. 2010;5:108–119. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J Neurosci. 2010;5:12676–12689. doi: 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch R. Aetiology of Tuberculosis. New York, NY: William R Jenkins; 1890. [Google Scholar]
- Carter KC. Koch’s postulates in relation to the work of Jacob Henle and Edwin Klebs. Med Hist. 1985;5:353–374. doi: 10.1017/S0025727300044689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimer’s Dementia. 2010;5:420–424. doi: 10.1016/j.jalz.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkadhi K, Eriksen J. The complex and multifactorial nature of Alzheimer’s disease. Cur Neuropharmacol. 2011;5:586. doi: 10.2174/157015911798376235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz GP, Legleiter J. The role of amyloidogenic protein oligomerization in neurodegenerative disease. J Molecular Medicine. 2013;5:653–664. doi: 10.1007/s00109-013-1025-1. [DOI] [PubMed] [Google Scholar]
- Weissman JS. All roads lead to Rome? The multiple pathways of protein folding. Chem Biol. 1995;5:255–260. doi: 10.1016/1074-5521(95)90044-6. [DOI] [PubMed] [Google Scholar]
- Dill KA. Polymer principles and protein folding. Protein Sci. 1999;5:1166–1180. doi: 10.1110/ps.8.6.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udgaonkar JB. Multiple routes and structural heterogeneity in protein folding. Annu Rev Plant Physiol Plant Mol Biol. 2008;5:489–510. doi: 10.1146/annurev.biophys.37.032807.125920. [DOI] [PubMed] [Google Scholar]
- Dobson CM. The structural basis of protein folding and its links with human disease. Phil Trans R Soc Lond B. 2001;5:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Teplow DB. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol. 2008;5:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumketner A, Shea JE. The structure of the Alzheimer amyloid β 10–35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J Mole Biol. 2007;5:275–285. doi: 10.1016/j.jmb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Mitomo D, Shea JE, Higo J. Folding of the 25 residue Aβ (12–36) peptide in TFE/water: temperature-dependent transition from a funneled free-energy landscape to a rugged one. J Phys Chem B. 2007;5:5351–5356. doi: 10.1021/jp067075v. [DOI] [PubMed] [Google Scholar]
- Ni CL, Shi HP, Yu HM, Chang YC, Chen YR. Folding stability of amyloid-β 40 monomer is an important determinant of the nucleation kinetics in fibrillization. FASEB J. 2011;5:1390–1401. doi: 10.1096/fj.10-175539. [DOI] [PubMed] [Google Scholar]
- Teplow DB. Structural and kinetic features of amyloid β-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- Lee S, Fernandez EJ, Good TA. Role of aggregation conditions in structure, stability, and toxicity of intermediates in the Aβ fibril formation pathway. Protein Sci. 2007;5:723–732. doi: 10.1110/ps.062514807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of β-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci USA. 2009;5:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitternacht S, Staneva I, Hard T, Irback A. Comparing the folding free-energy landscapes of Aβ42 variants with different aggregation properties. Proteins Struct Funct Bioinform. 2010;5:2600–2608. doi: 10.1002/prot.22775. [DOI] [PubMed] [Google Scholar]
- Wu C, Bowers MT, Shea JE. Molecular structures of quiescently grown and brain-derived polymorphic fibrils of the Alzheimer amyloid Aβ9 − 40 peptide: a comparison to agitated fibrils. PLoS Comput Biol. 2010;5:e1000693. doi: 10.1371/journal.pcbi.1000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;5:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg W. Quantum-theoretical re-interpretation of kinematic and mechanical relations [in German] Zeitschrift fur Physik. 1927;5:172–198. doi: 10.1007/BF01397280. [DOI] [Google Scholar]
- Bitan G, Fradinger EA, Spring SM, Teplow DB. Neurotoxic protein oligomers – what you see is not always what you get. Amyloid. 2005;5:88–95. doi: 10.1080/13506120500106958. [DOI] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;5:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Square A. Flatland: A Romance of Many Dimensions. London: Seeley & Co; 1884. [Google Scholar]
- Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;5:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc Natl Acad Sci USA. 2005;5:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci USA. 2008;5:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes AJ, Parker KD, Atkins ED, Beighton E. Cross-β conformation in proteins. J Mol Biol. 1968;5:343–358. doi: 10.1016/0022-2836(68)90014-4. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;5:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;5:119–125. doi: 10.1016/S1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;5:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- Gsponer J, Babu MM. The rules of disorder or why disorder rules. Prog Biophys Molecular Biol. 2009;5:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;5:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Fong JH, Shoemaker BA, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV, Panchenko AR. Intrinsic disorder in protein interactions: insights from a comprehensive structural analysis. PLoS Comp Biol. 2009;5:e1000316. doi: 10.1371/journal.pcbi.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikipedia: Random walk. http://en.wikipedia.org/wiki/Random_walk.
- Fandrich M, Fletcher MA, Dobson CM. Amyloid fibrils from muscle myoglobin. Nature. 2001;5:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;5:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nature Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;5:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;5:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardberg AS, Dice LT, Ou S, Rich RL, Helmbrecht E, Ko J, Wetzel R, Myszka DG, Patterson PH, Dealwis C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proc Natl Acad Sci USA. 2007;5:15659–15664. doi: 10.1073/pnas.0705888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basi GS, Feinberg H, Oshidari F, Anderson J, Barbour R, Baker J, Comery TA, Diep L, Gill D, Johnson-Wood K, Goel A, Grantcharova K, Lee M, Li J, Partridge A, Griswold-Prenner I, Piot N, Walker D, Widom A, Pangalos MN, Seubert P, Jacobsen JS, Schenk D, Weis WI. Structural correlates of antibodies associated with acute reversal of amyloid β-related behavioral deficits in a mouse model of Alzheimer disease. J Biol Chem. 2010;5:3417–3427. doi: 10.1074/jbc.M109.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerner J, Børmer OP, Nustad K. The war on heterophilic antibody interference. Clin Chem. 2005;5:9–11. doi: 10.1373/clinchem.2004.042994. [DOI] [PubMed] [Google Scholar]
- Sehlin D, Sollvander S, Paulie S, Brundin R, Ingelsson M, Lannfelt L, Pettersson FE, Englund H. Interference from heterophilic antibodies in amyloid-beta oligomer ELISAs. J Alzheimers Dis. 2010;5:1295–1301. doi: 10.3233/jad-2010-100609. [DOI] [PubMed] [Google Scholar]
- Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struc Biol. 2000;5:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nature Med. 2006;5:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangachari V, Moore BD, Reed DK, Sonoda LK, Bridges AW, Conboy E, Hartigan D, Rosenberry TL. Amyloid-β (1–42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochem. 2007;5:12451–12462. doi: 10.1021/bi701213s. [DOI] [PubMed] [Google Scholar]
- Watt AD, Perez KA, Rembach A, Sherrat NA, Hung LW, Johanssen T, McLean CA, Kok WM, Hutton CA, Fodero-Tavoletti M, Masters CL, Villemagne VL, Barnham KJ. Oligomers, fact or artefact? SDS-PAGE induces dimerization of β-amyloid in human brain samples. Acta Neuropathol. 2013;5:549–564. doi: 10.1007/s00401-013-1083-z. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Med. 2008;5:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Welzel AT, McDonald JM, Selkoe DJ, Walsh DM. Isolation of low-n amyloid β-protein oligomers from cultured cells, CSF, and brain. Alzheimer Dis Frontotemporal Dementia Meth Mole Biol. 2011;5:33–44. doi: 10.1007/978-1-60761-744-0_3. [DOI] [PubMed] [Google Scholar]
- Moore B, Chakrabarty P, Levites Y, Kukar T, Baine A, Moroni T, Ladd T, Das P, Dickson D, Golde T. Overlapping profiles of Aβ peptides in the Alzheimer’s disease and pathological aging brains. Alzheimer’s Res Therapy. 2012;5:18. doi: 10.1186/alzrt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;5:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, Akioka M, Kikuchi S, Sato M, Ideno S, Noda M, Fukunari A, Muramatsu S, Itokazu Y, Sato K, Takahashi H, Teplow DB, Nabeshima Y, Katika A, Imahori K, Hoshi M. Isolation and characterization of patient-derived, toxic, high mass amyloid β-protein (Aβ) assembly from Alzheimer disease brains. J Biol Chem. 2009;5:32895–32905. doi: 10.1074/jbc.M109.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nature Rev Cancer. 2006;5:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Kato Y, Maruyama W, Naoi M, Hashizume Y, Osawa T. Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. FEBS Lett. 1998;5:231–234. doi: 10.1016/S0014-5793(98)01372-6. [DOI] [PubMed] [Google Scholar]
- Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neuroscience. 1998;5:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chatelier HL. A general statement of the laws of chemical equilibria [in French] Comptes Rendus Académie des Sci. 1884;5:786–789. [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nature Neurosci. 2012;5:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci USA. 2012;5:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-β deposition in vivo. Mol Psychiatry. 2012;5:1347–1353. doi: 10.1038/mp.2011.120. [DOI] [PubMed] [Google Scholar]
- Kuhn TS. The Structure of Scientific Revolutions. Chicago and London: University of Chicago Press; 1970. [Google Scholar]
- Ono K, Condron MM, Teplow DB. Structure–neurotoxicity relationships of amyloid β-protein oligomers. Proc Natl Acad Sci USA. 2009;5:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]