

Abstract
Whole-exome sequencing provides a cost-effective means to sequence protein coding regions within the genome, which are significantly enriched for etiological variants. We describe a panel of single nucleotide polymorphisms (SNPs) to facilitate the validation of data provenance in whole-exome sequencing studies. This is particularly significant where multiple processing steps necessitate transfer of sample custody between clinical, laboratory and bioinformatics facilities. SNPs captured by all commonly used exome enrichment kits were identified, and filtered for possible confounding properties. The optimised panel provides a simple, yet powerful, method for the assignment of intrinsic, highly discriminatory identifiers to genetic samples.
Background
Whole-exome sequencing (WES) is presently one of the most efficient means of identifying aetiological genetic mutations [1], minimising some of the challenges associated with whole-genome sequencing, such as high cost and data processing burden, analysis and interpretation. In WES, protein-coding regions of the genome are targeted and enriched via specific hybridisation of genomic fragments with complementary oligonucleotides, or 'baits’. These targeted regions are then sequenced using high throughput next-generation sequencing (NGS) technologies [2].
The high start-up investment required for in-house WES is currently prohibitive to many groups so sample preparation and/or sequencing is commonly outsourced. This transference of sample custody, combined with the complex sample preparation workflow, makes sample mix-ups possible, and difficult to detect. In both clinical and research contexts, ensuring provenance of data is essential to allow the accurate assignment of clinical details to sequence data. It is possible that samples may be misidentified at any stage of the analytical process, both in vitro and in silico. Therefore, sample tracking must be contiguous throughout both data generation and analysis.
Consequent to sample mix-ups in a research setting, erroneous data and sample matching may result in a loss of power for identification of causal variants [3]. In a clinical setting, this may lead to delayed or inaccurate reporting of results to patients. Whilst good practice in the handling of samples and increased laboratory automation minimises potential for error, additional checkpoints are still required to support quality control [4]. A method for post hoc confirmation of sample identity is therefore highly desirable.
Genetic sample identification methods have an advantage over alternative sample management systems in that the genetic 'label’ is intrinsic to the biological sample itself, removing the possibility of manual labelling errors. Single nucleotide polymorphisms (SNPs) are increasingly utilised for DNA-based identification of human samples, with several benefits compared to standard forensic methods [5-7]. Existing SNP panels for human forensic identification and commercial SNP panels for sample identification, such as the iPLEX Sample ID Plus panel (Sequenom, San Diego, CA, USA), utilise pan-genome SNPs, the majority of which are non-exonic, and are therefore not useful for WES studies, as the majority of markers will not lie within the enriched regions of the genome. In addition to existing SNP panels, short tandem repeat markers can be used for genetic sample tracking. However, again, markers applied are frequently outside exomic regions and, if captured, will be prone to erroneous NGS genotyping using standard pipelines due to the repetitive nature of the markers [8].
Several methods for genetic tracking of human biological samples have been previously described, some of which are application specific - for example, for transcriptome microarray studies [3,9,10]. Although software for the validation of NGS (including WES) sample identity, such as verifyBamID, exist [11], for the detection of sample misidentifications external array-based genotypes of the samples are required, without which only contamination of the samples can be assessed.
Here we describe an optimised panel of SNPs for which WES data are typically informative, the genotypic profile of which can be utilised to extract intrinsic identifiers from human genomic DNA. These SNP profiles have high discriminatory power, even in large datasets. The profile derived from this panel can be compared to an independently genotyped profile for the same individual, allowing accurate validation of data and sample pairings, at a modest cost per sample.
Methods
Candidate identification and panel selection
Regions of overlap between three current commonly used whole-exome enrichment kits, (namely Agilent SureSelect Human All Exon V4, Illumina TruSeq Exome Enrichment and Nimblegen SeqCap EZ Human Exome Library V3.0 kits), and common SNPs (dbSNP 137, [12]) were established using BEDTools [13]. SNPs were further filtered for inclusion based upon their presence in genes targeted by the Illumina TruSight Exome kit, which targets only genes of clinical interest.
Primary candidate selection criteria required SNPs to: 1) represent bi-allelic substitutions, excluding substitutions of complementary bases, that is, A↔T and G↔C transversions; 2) be technically amenable to both accurate WES and orthogonal genotyping, that is, not present in large-scale genomic repeats [14], or homopolymeric tracts of ≥5 bp, GC content for the flanking 250 bp was restricted to a range of between 40% and 55% and no other variant within 50 bp with an alternative-allele frequency (AF) ≥0.01 was permitted; 3) conform to desirable phase 3 HapMap AFs across several populations, explicitly AFs of between 0.2 and 0.8 in: CEPH (Utah residents with ancestry from northern and western Europe; CEU), Japanese in Tokyo, Japan (JPT), Han Chinese in Beijing, China (CHB) and Yoruba in Ibidan, Nigeria (YRI) [15] and; 4) not alter the primary sequence of the encoded protein or have an associated Online Mendelian Inheritance in Man (OMIM) record [16].
Following primary candidate identification steps, SNPs were further optimised by the following requirements: 1) be located at least 10 bp from exon boundaries; 2) not be situated in regions with a high sequence similarity to non-target regions, that is, no non-target BLAT score >100 [17], as this could result in non-specific genotyping; and 4) be outside of linkage disequilibrium with all other selected SNPs.
Finally, SNPs were prioritised for inclusion in the panel by proximity of the AFs to 0.5, across HapMap populations, in order to maximise discriminatory power.
SNP coverage in whole-exome sequencing data
A set of 91 in-house exome samples was evaluated for depth of sequence coverage for the candidate SNPs. Exome capture was performed using Agilent SureSelect Human All Exon V3 (n = 22) and V4 (n = 55), Illumina TruSeq Exome Enrichment (n = 9) and Nimblegen SeqCap EZ Human Exome Library V3.0 (n = 5). Exome enrichment, sequencing and in silico analysis of samples was performed as previously described [18,19].
Optimised panel validation
The power of sample resolution for the panel was validated using data from phase 1 of the 1000 Genomes Project (n = 1,092) [20] and the UK10K project (n = 2,688; 2,432 of which are whole-genome data) [21]. Genotypes were extracted from data using custom scripts and Tabix [22]. Quantification of mismatches between samples was performed using MEGA5 [23].
Simulated datasets were generated by taking individual population AFs for each SNP as input and generating random SNP profiles in accordance with Hardy-Weinberg equilibrium based upon this; the randomisation of each SNP was independent of all other SNPs. We then quantified the rate of non-unique profiles per simulated dataset. We performed 20,000 independent replicates of dataset generation in all cases.
Panel application
We applied the panel to a batch of 48 samples exome sequenced by an external service provider, for which orthogonal genotypes were obtained concurrently through an independent genotyping provider using KASP genotyping (LGC Genomics, Hoddeston, UK). Following plating of DNA samples for dispatch, a replicate plate was made directly from the primary plate, to be dispatched for the orthogonal genotyping. Genotypes derived from exome data and orthogonal genotyping assays were compared using PLINK [24] and custom scripts.
Ethics
This study was approved by the Southampton and South West Hampshire Research Ethics Committee (09/H0504/125). Informed consent was obtained for all participants.
Results
In total, 26.2 Mbp of genome sequence was found to overlap all three commonly applied whole-exome capture kits, containing 9,493 common SNPs (Figure 1A,B). Of these, 1,662 SNPs are additionally covered by the Illumina TruSight Exome kit. Within this subset, following the filtering for all primary candidate criteria, 117 candidate SNPs were identified (Figure 1C; Additional file 1), from which the optimised panel of 24 SNPs was selected (Table 1). Within the set of 91 in-house exome samples, all 24 SNPs were sequenced at sufficient read-depth for accurate genotype calling, across all capture kits.
Figure 1.

Venn diagrams showing commonality of targeting between capture kits (A,B) and properties of encompassed SNPs (C). Overlap between exome capture kits is presented in Mbp (A) and number of SNPs with an AF ≥0.3 (B). Agilent - SureSelect Human All Exon V4; Illumina - TruSeq Exome Enrichment; Nimblegen - SeqCap EZ Human Exome Library V3.0. For a subset of SNPs present in both the intersection of the three kits shown, and the Illumina TruSight Exome kit, a breakdown of fulfilment of the four classes of candidate filtering criteria is shown (C) (see the main text for details of filtering criteria); 117 SNPs exhibited all desired characteristic; 74 SNPs exhibited none of the desired characteristics.
Table 1.
Optimised panel of identifying SNPs
|
Chromosome |
Positiona |
dbSNP rsID |
Gene |
Alleles |
HapMap 3 AF |
|||
|---|---|---|---|---|---|---|---|---|
| CEU | CHB | JPT | YRI | |||||
| 1 |
179520506 |
rs1410592 |
NPHS2 |
A/C |
0.59 |
0.62 |
0.54 |
0.53 |
| 1 |
67861520 |
rs2229546 |
IL12RB2 |
A/G |
0.64 |
0.36 |
0.44 |
0.58 |
| 2 |
169789016 |
rs497692 |
ABCB11 |
A/Gb |
0.55 |
0.65 |
0.51 |
0.22 |
| 2 |
227896976 |
rs10203363 |
COL4A4 |
C/T |
0.46 |
0.44 |
0.36 |
0.57 |
| 3 |
4403767 |
rs2819561 |
SUMF1 |
A/Gb |
0.56 |
0.73 |
0.73 |
0.72 |
| 4 |
5749904 |
rs4688963 |
EVC |
A/Gb |
0.33 |
0.65 |
0.67 |
0.52 |
| 5 |
82834630 |
rs309557 |
VCAN |
A/Gb |
0.49 |
0.34 |
0.52 |
0.50 |
| 6 |
146755140 |
rs2942 |
GRM1 |
C/T |
0.54 |
0.49 |
0.55 |
0.47 |
| 7 |
48450157 |
rs17548783 |
ABCA13 |
C/T |
0.46 |
0.72 |
0.53 |
0.48 |
| 8 |
94935937 |
rs4735258 |
PDP1 |
C/T |
0.40 |
0.64 |
0.66 |
0.46 |
| 9 |
100190780 |
rs1381532 |
TDRD7 |
A/Gb |
0.48 |
0.59 |
0.50 |
0.58 |
| 10 |
100219314 |
rs10883099 |
HPSE2 |
A/G |
0.52 |
0.52 |
0.53 |
0.62 |
| 11 |
16133413 |
rs4617548 |
SOX6 |
C/T |
0.52 |
0.65 |
0.61 |
0.51 |
| 12 |
993930 |
rs7300444 |
WNK1 |
A/G |
0.46 |
0.55 |
0.48 |
0.28 |
| 13 |
39433606 |
rs9532292 |
FREM2 |
A/G |
0.29 |
0.41 |
0.44 |
0.54 |
| 14 |
50769717 |
rs2297995 |
L2HGDH |
A/G |
0.55 |
0.65 |
0.67 |
0.59 |
| 15 |
34528948 |
rs4577050 |
SLC12A6 |
C/T |
0.68 |
0.75 |
0.63 |
0.32 |
| 16 |
70303580 |
rs2070203 |
AARS |
A/Gb |
0.53 |
0.28 |
0.51 |
0.49 |
| 17 |
71197748 |
rs1037256 |
COG1 |
C/T |
0.50 |
0.67 |
0.65 |
0.56 |
| 18 |
21413869 |
rs9962023 |
LAMA3 |
A/G |
0.67 |
0.81c |
0.75 |
0.51 |
| 19 |
10267077 |
rs2228611 |
DNMT1 |
C/Tb |
0.47 |
0.73 |
0.56 |
0.48 |
| 20 |
6100088 |
rs10373 |
FERMT1 |
G/Tb |
0.54 |
0.31 |
0.35 |
0.58 |
| 21 |
44323590 |
rs4148973 |
NDUFV3 |
C/T |
0.65 |
0.33 |
0.38 |
0.73 |
| 22 | 21141300 | rs4675 | SERPIND1 | A/C | 0.46 | 0.62 | 0.51 | 0.57 |
aPosition as defined in genome reference assembly GRCh37 (hg19).
bSNP is defined on the negative strand.
cAF marginally outside target range for candidate selection. Selected due to paucity of candidates on chromosome 18.
The 24 biallelic SNPs afford 48 points of allelic comparison. Testing the optimised panel in the 1000 Genomes Project data (n = 1,092) [20], an average of 18.0 (standard deviation = 3.3) allelic differences between all pairwise combinations was observed, with a range of 3 to 34. As such, there will be, on average, 18 differential alleles between any two samples, enabling discrimination.
On addition of the UK10K data (n = 2,688) to the 1000 Genomes Project data (ncombined = 3,780), there remained an average of 17.8 allele mismatches across the profiles. Eighteen UK10K sample pairs produced duplicate profiles. On investigation of these pairs, they were found to share >98% genotypic concordance across an extended panel of 1,662 SNPs in all cases, compared to an average of 42%, with a range of 27 to 77%, for all sample pairs with unique SNP profiles (Additional file 2). As such, these pairs represent extreme outliers, and are derived from genetically identical biological samples, either from the same individual or monozygotic twins, and were therefore excluded from the mismatch average.
Simulated data
The discriminatory power of the panel was evaluated by dataset simulation. We simulated datasets of 10,000 individuals, that conformed to AF distributions for investigated HapMap populations (CEU, CHB, JPT and YRI), 1000 Genomes Project pilot average [25], as well as for a hypothetical perfect allele distribution (AF = 0.5 for all SNPs) (Table 2). In all simulated populations, <2.5% of simulated datasets of 10,000 contained any repeat SNP profiles (henceforth termed 'collisions’). This translates approximately into less than 1 in every 40 independent datasets of 10,000 individuals containing a single matching pair of profiles.
Table 2.
Profile collisions per simulated dataset of 10,000 individuals with various population AFs
| AF source | Average collisions per dataset (±SD) |
|---|---|
|
1000 Genomes average |
0.0039 (0.062) |
|
HapMap phase 3: |
|
| CEU |
0.0064 (0.079) |
| CHB |
0.0239 (0.154) |
| JPT |
0.0082 (0.090) |
| YRI |
0.0076 (0.086) |
| Theoretical perfect a | 0.0031 (0.056) |
aAll 24 SNPs assigned an AF of 0.5, which will give the most even trifurcation per SNP, and thus discriminatory power. SD, standard deviation.
The effect of dataset size on the frequency of collisions was investigated for populations present in 1000 Genomes Project phase 1 data [20]. An exponential increase in the frequency of collisions was observed with increasing dataset size, though the panel continued to have high power for the discrimination of samples. For instance, were we to have 85,000 Southern Han Chinese (CHS) samples, (the worst performing 1000 Genomes population evaluated, due to the AF distribution for SNPs within this panel), we would expect the dataset to contain, on average, a single duplicate SNP profile (Figure 2). In addition, total SNP absence - for example, through technical failure of orthogonal genotyping - was modelled. For each SNP that entirely failed to provide data, a less than three-fold drop in discriminatory power was observed in all cases (data not shown). This suggests that our approach is robust against technical failure.
Figure 2.
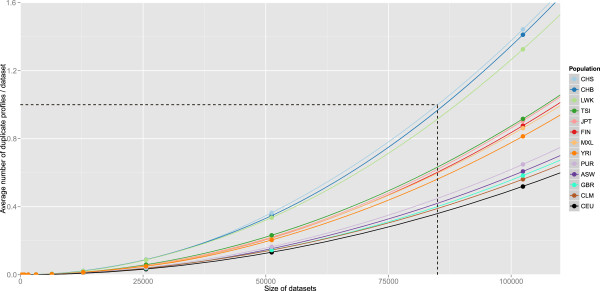
Relationship between size of simulated datasets and the occurrence of non-unique profiles. Thirteen 1000 Genomes Project populations were simulated [20]. Datasets were simulated as described in Methods. With increasing dataset size, the probability of repeat profiles increases. Only populations with a sample size of >50 individuals in the dataset were simulated. Additional populations are Americans of African ancestry in Southwest USA (ASW), Columbians from Medellin, Colombia (CLM), Finnish in Finland (FIN), British in England and Scotland (GBR), Luhya in Webuye, Kenya (LWK), Mexican ancestry from Los Angeles, USA (MXL), Puerto Ricans from Puerto Rico (PUR) and Toscany in Italia (TSI).
Application of the SNP panel to our batch of 48 samples revealed a discrepancy between exome and orthogonal genotypes for two samples dispatched in adjacent wells, suggesting a reciprocal transposition (Figure 3). The occurrence of this error in the exome data was also supported by interrogation of X-chromosome heterozygosity to confirm sample gender. In addition to the identification of the switch, the panel allowed for expeditious resolution of the error, permitting the continued use of the data in downstream analyses.
Figure 3.
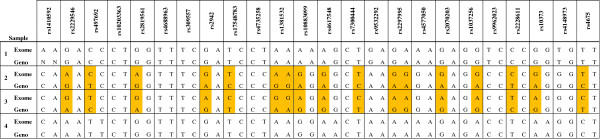
Exome derived and orthogonal genotypes (Geno) for four samples, showing a sample-switch between samples 2 and 3. Informative markers for the resolution of this switch are highlighted in yellow.
Discussion
Validation of sample identity is essential in order to ensure data integrity and validity of conclusions drawn from data. We have described a powerful tool for the identification and validation of data provenance throughout the workflow of WES data collection and analysis. The power of discrimination, that is, the precision with which samples can be uniquely identifiable, is sufficient and robust for most projects on the current scale of up to 10,000 samples, with inbuilt redundancy of SNPs to protect against technical failures. In WES, the exome enrichment process provides the limiting step for the availability of data on SNPs for use in sample identification. As such, this panel will also be of utility for whole-genome sequencing data, where there is no such limitation on SNP coverage. This will be beneficial where there are mixed datasets of both whole-genome sequence and WES data.
NGS is now developing as the diagnostic methodology of choice across a range of applications, including mutation scanning in targeted gene panels and WES for congenital disorders, as well as high depth analysis for tumour profiling. Whilst the service model for delivery of these tests is not fully resolved at this stage, there will certainly be economic arguments for centralising certain tests. This will have the effect of increasing the throughput requirements as well as physically moving samples between labs. Both of these factors will increase the opportunity for sample misidentification.
Even for testing within a single lab, the use of inherent sample and data identification methods, as described in this study, seems a robust approach to fulfil the regulatory requirement for providing a full audit trail and ensuring data provenance [26,27]. The SNP panel presented here is immediately usable across all commonly used exome capture kits, and would be equally applicable to any gene panel by incorporating, or 'spiking’, the SNP regions into the custom capture kit at the design stage. Where it can be shown that there are no expected repeat profiles (that is, no paired samples from the same individual are being analysed), it may even be beneficial from a process perspective to use the SNP profile as the primary method for sample tracking.
The discriminatory power of the panel may be reduced for various reasons, such as geographically localised variation in AFs, and degradation of DNA samples, resulting in incomplete data. We have shown our panel to have a high discriminatory power across a diverse range of populations. Additionally, the discriminatory power will be marginally reduced where many relatives are sequenced. In the case of highly consanguineous families, sample tracking methods such as barcoding will afford optimal certainty in these particular cases. Should concerns over insufficient discriminatory power arise, additional SNPs may be added to the panel from the existing list of candidates (Additional file 1), also allowing the tailoring of an enhanced panel to the population(s) of interest, should this be desired. Nevertheless, we have demonstrated our panel to be sufficiently robust to withstand power reductions without loss of utility for most purposes.
We have also presented a recent case in which use of this panel has allowed us to identify, confirm, and resolve a sample switch, highlighting the importance of using such a tool. Monetary cost will vary with the technology used for orthogonal genotyping and sample throughput. We have intentionally designed the panel to be platform non-specific, allowing for the establishment of in-house assays using preferred genotyping methodology or outsourced where required. Our own chosen methodology costs approximately £5 GBP per sample, representing a small fraction of the cost of exome data generation.
Conclusions
The size of held NGS datasets continues to increase, with the UK Government recently committing to the sequencing of 100,000 samples as part of healthcare provisions [28]. As such, the demand for the development of effective tools for bioinformatic analysis, data compression, mutation effect prediction and quality control is high. We have described a panel of SNPs for the discrimination of human biological samples on the basis of data intrinsic to WES data derived from samples processed using common capture kits. We recommend the routine use of this panel to maintain data integrity and protect sample provenance.
Abbreviations
AF: alternative-allele frequency; bp: base pair; CEU: CEPH (Utah residents with ancestry from northern and western Europe); CHB: Han Chinese in Beijing, China; CHS: Southern Han Chinese; JPT: Japanese in Tokyo, Japan; Mbp: megabase pair; NGS: next-generation sequencing; SNP: single nucleotide polymorphism; WES: whole-exome sequencing; YRI: Yoruba in Ibidan, Nigeria.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RJP performed analysis and interpretation of data, and drafted the manuscript, JG, GA and AC contributed to analysis, CJM contributed to data interpretation and manuscript preparation and SE conceived and supervised the project, and contributed to manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
List of all candidate SNPs with evaluated properties.
Distribution of pairwise genotype concordance between samples. Pairs resulting in duplicate SNP profiles (n = 18) and pairs between samples with unique SNP profiles (n = 7,142,293) within the combined dataset of 3,780 samples are shown. Concordance across the 1,662 SNPs detailed in Figure 1C was evaluated. All pairs resulting in duplicate profiles have >98% concordance, well separated from the distribution of samples with unique profiles. Note the logarithmic scale.
Contributor Information
Reuben J Pengelly, Email: r.j.pengelly@soton.ac.uk.
Jane Gibson, Email: j.gibson@soton.ac.uk.
Gaia Andreoletti, Email: ga4g12@soton.ac.uk.
Andrew Collins, Email: a.r.collins@soton.ac.uk.
Christopher J Mattocks, Email: chris.mattocks@nhs.net.
Sarah Ennis, Email: s.ennis@soton.ac.uk.
Acknowledgements
The authors thank the Technology Strategy Board and the University of Southampton for funding, and Dr Dietrich Lueerssen for discussion. The authors also acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work. This study makes use of data generated by the UK10K Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.UK10K.org. Funding for UK10K was provided by the Wellcome Trust under award WT091310.
References
- Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH, Goldstein DB. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;5:353–361. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;5:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Westra H-J, Jansen RC, Fehrmann RSN, te Meerman GJ, van Heel D, Wijmenga C, Franke L. MixupMapper: correcting sample mix-ups in genome-wide datasets increases power to detect small genetic effects. Bioinformatics. 2011;5:2104–2111. doi: 10.1093/bioinformatics/btr323. [DOI] [PubMed] [Google Scholar]
- Lam CW, Jacob E. Implementing a laboratory automation system: experience of a large clinical laboratory. J Lab Autom. 2012;5:16–23. doi: 10.1177/2211068211430186. [DOI] [PubMed] [Google Scholar]
- Pakstis AJ, Speed WC, Fang R, Hyland FC, Furtado MR, Kidd JR, Kidd KK. SNPs for a universal individual identification panel. Hum Genet. 2010;5:315–324. doi: 10.1007/s00439-009-0771-1. [DOI] [PubMed] [Google Scholar]
- Zietkiewicz E, Witt M, Daca P, Zebracka-Gala J, Goniewicz M, Jarzab B, Witt M. Current genetic methodologies in the identification of disaster victims and in forensic analysis. J Appl Genet. 2012;5:41–60. doi: 10.1007/s13353-011-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire-Aradas A, Fondevila M, Kriegel AK, Phillips C, Gill P, Prieto L, Schneider PM, Carracedo A, Lareu MV. A new SNP assay for identification of highly degraded human DNA. Forensic Sci Int Genet. 2012;5:341–349. doi: 10.1016/j.fsigen.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Highnam G, Franck C, Martin A, Stephens C, Puthige A, Mittelman D. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2013;5:e32. doi: 10.1093/nar/gks981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro F, Dirks WG, Fähnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer. 2013;5:308–314. doi: 10.1002/ijc.27675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gao H, Seok J, Wilhelmy J, Mindrinos MN, Davis RW, Xiao W. Coding SNPs as intrinsic markers for sample tracking in large-scale transcriptome studies. Biotechniques. 2012;5:386–388. doi: 10.2144/0000113879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, Kang HM. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet. 2012;5:839–848. doi: 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;5:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;5:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repeat Masker. http://www.repeatmasker.org/
- International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;5:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMIM - Online Mendelian Inheritance in Man. http://www.omim.org/
- Kent WJ. BLAT–The BLAST-Like Alignment Tool. Genome Res. 2002;5:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou K, Wiskin AE, Gibson J, Tapper W, Willis C, Afzal NA, Upstill-Goddard R, Holloway JW, Simpson MA, Beattie RM, Collins A, Ennis S. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut. 2013;5:977–984. doi: 10.1136/gutjnl-2011-301833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J, Tapper W, Ennis S, Collins A. Exome-based linkage disequilibrium maps of individual genes: functional clustering and relationship to disease. Hum Genet. 2013;5:233–243. doi: 10.1007/s00439-012-1243-6. [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;5:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK10K Study Samples. http://www.uk10k.org/studies/
- Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011;5:718–719. doi: 10.1093/bioinformatics/btq671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;5:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;5:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;5:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Organization for Standardization. Medical Laboratories - Requirements for Quality and Competence. 2012. ISO 15189:2012. [DOI] [PubMed]
- Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, Friez MJ, Funke BH, Hegde MR, Lyon E. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;5:733–747. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DNA tests to revolutionise fight against cancer and help 100,000 NHS patients. https://www.gov.uk/government/news/dna-tests-to-revolutionise-fight-against-cancer-and-help-100000-nhs-patients.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of all candidate SNPs with evaluated properties.
Distribution of pairwise genotype concordance between samples. Pairs resulting in duplicate SNP profiles (n = 18) and pairs between samples with unique SNP profiles (n = 7,142,293) within the combined dataset of 3,780 samples are shown. Concordance across the 1,662 SNPs detailed in Figure 1C was evaluated. All pairs resulting in duplicate profiles have >98% concordance, well separated from the distribution of samples with unique profiles. Note the logarithmic scale.