

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia and a major contributor to disability and dependency among older people. AD pathogenesis is associated with the accumulation of amyloid-beta protein (Aβ) and/or hyperphosphorylated tau protein in the brain. At present, current therapies provide temporary symptomatic benefit, but do not treat the underlying disease. Recent research has thus focused on investigating the molecular and cellular pathways and processes involved in AD pathogenesis to support the development of effective disease-modifying agents. In accordance with the existing Aβ-cascade hypothesis for AD pathogenesis, immunotherapy has been the most extensively studied approach in Aβ-targeted therapy. Both passive and active immunotherapies have been shown to effectively reduce Aβ accumulation and prevent downstream pathology in preclinical models. Following AN1792, second-generation active immunotherapies have shown promising results in terms of antibody response and safety. Comparatively, tau immunotherapy is not as advanced, but preclinical data support its development into clinical trials. Results from active amyloid-based immunotherapy studies in preclinical models indicate that intervention appears to be more effective in early stages of amyloid accumulation, highlighting the importance of diagnosing AD as early as possible and undertaking clinical trials at this stage. This strategy, combined with improving our understanding of the complex AD pathogenesis, is imperative to the successful development of these disease-modifying agents. This paper will review the active immunotherapies currently in development, including the benefits and challenges associated with this approach.
Review
Introduction
Alzheimer’s disease (AD), the most common cause of dementia [1], is a neuropathological disorder that presents clinically with progressive deterioration in cognitive, memory, and functional capabilities [2]. An estimated 36 million individuals worldwide were burdened by dementia in 2010, and this number is projected to increase to 66 million by 2030 [3,4]. The two major neuropathological hallmarks of AD, first described by Dr Alzheimer in 1907, are extracellular senile plaques and intracellular neurofibrillary tangles (NFTs) [5]. Mutations in the amyloid precursor protein (APP) gene, APP, and the presenilin genes, PSEN1 and PSEN2, are strongly associated with early-onset, familial AD and increased accumulation of amyloid-beta protein (Aβ) [6]. In the more common sporadic or late-onset AD, the genetic risk factor gene ApoE epsilon 4 increases the risk of developing the disease [6]. These genetic lines of evidence, in combination with neuropathological findings, have given rise to the Aβ-cascade hypothesis of AD pathogenesis [7]. Although an imbalance between the production and clearance of Aβ40/42 is thought to be the key initiating pathology in AD, other contributing disease mechanisms remain to be resolved.
The Aβ cascade is thought to be initiated by an elevated Aβ concentration, in particular Aβ42, which aggregates to form soluble dimers, trimers, and the low-ordered oligomers. Further aggregation forms insoluble and proteolysis-resistant fibrils, which accumulate as beta-amyloid deposits. This toxic Aβ cascade is associated with various neuropathological processes such as tau hyperphosphorylation, paired helical filament accumulation, neuritic dystrophy, astrocytosis, altered ionic homeostasis, oxidative stress, and synaptic failure leading to progressive loss of neuronal function. Furthermore, evidence from transgenic mice models showed that Aβ deposition enhances tangle pathology, consistent with the Aβ cascade hypothesis [8]. The role of tau, a microtubule-associated protein, is based on the second neuropathological hallmark of AD, which is the presence of NFTs. Intraneuronal accumulation of abnormally hyperphosphorylated tau is thought to impair axonal transport, resulting in aggregation of tubules into NFTs within the neuron and subsequent cell death [9].
Therapeutic approaches
Current therapies, such as cholinesterase inhibitors and the N-methyl-d-aspartate receptor antagonist memantine, provide temporary symptomatic benefit. Recently, advances have been made towards developing disease-modifying agents based on the two main hypotheses for AD pathogenesis; that is, Aβ and tau. Immunotherapy via administering Aβ antibodies (passive immunotherapy) or inducing a humoral immune response (active immunotherapy) has been the most extensively studied approach in Aβ-targeted therapy (Figure 1). Both passive and active immunotherapies have been shown to reduce Aβ accumulation in transgenic mice [10-12], indicating that interventions which reduce Aβ aggregation are promising therapeutic options.
Figure 1.
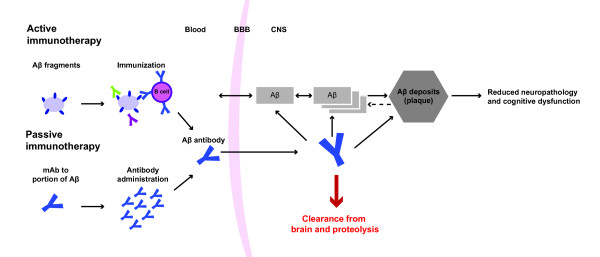
Immunotherapy approach to beta-amyloid clearance. Aβ, amyloid-beta protein; BBB, blood–brain barrier; CNS, central nervous system; mAb, monoclonal antibody.
Passive immunotherapies with monoclonal antibodies against Aβ are in late clinical development, but recently bapineuzumab and solanezumab, targeting the Aβ N-terminal and mid-domain respectively, failed to meet their primary endpoints in cognition and activities of daily living in phase 3 trials [13]. The clinical development of bapineuzumab was halted in August 2012 [14]. One reason for these negative results could be that these antibodies were administered in patients with mild-to-moderate AD, in which amyloid pathology may be too advanced. Secondary analysis of solanezumab phase 3 data indicated potential clinical benefits in patients with mild AD [13]. An additional phase 3 study is currently underway to confirm these findings (ClinicalTrials.gov NCT01900665).
Preclinical evidence suggests that peripherally administered antibodies can enter the central nervous system and bind to Aβ, where it is eliminated via Fc receptor-mediated clearance by microglial cells [11]. However, Aβ antibodies in brain parenchyma only reached 0.1% of the antibody concentration in serum due to the low passage of antibodies across the blood–brain barrier. Antibodies also have to pass through further barriers to reach intracellular compartments. Consequently, intraneuronal Aβ antibody concentrations may not be sufficient to reduce intracellular Aβ, impacting efficacy. This also makes it difficult to optimize dosing. Other passive immunotherapy compounds currently being investigated in clinical trials include gantenerumab, crenezumab, BAN2401, GSK933776, AAB-003, SAR228810, and BIIB037/BART [13].
The concept of active Aβ immunotherapy for AD was first introduced in 1999 [12]. Vaccination of APP transgenic mice with an Aβ1–42 peptide prior to amyloid deposits effectively blocked amyloid accumulation as mice aged, and treatment in the older mice reduced the progression of AD-like neuropathologies [12]. AN1792, a synthetic Aβ1–42 peptide co-administered with a QS21 adjuvant, was the first active Aβ immunotherapy tested in clinical trials. However, a phase 2 trial demonstrated that only 19.7% of patients with mild-to-moderate AD developed a predetermined antibody response [15], and this compound was subsequently discontinued because meningoencephalitis was observed in 6% of patients [16]. An Aβ-specific T-cell response (T-helper type 1 CD4+) is believed to have occurred, given that AN1792 consists of full-length Aβ1–42 that carries T-cell epitopes [17,18]. Results from this study showed that patients with an antibody response demonstrated significantly better scores in certain memory functions of the neuropsychological test battery, but no difference in cognitive and disability scores compared with the placebo group [15]. As T-cell epitopes mainly reside in the central proportion of Aβ1–42[19], a number of approaches have been developed comprising the Aβ N-terminus (B-cell epitopes) to avoid a T-cell response and have shown promise as second-generation active immunotherapeutic options for modifying AD. These compounds involve administering short Aβ peptides, fragmented peptides, or peptide mimetics to activate the patient’s immune system. Aβ peptides are usually conjugated to a carrier, such as a virus-like particle or keyhole limpet hemocyanin (KLH), and administered with an adjuvant to encourage stimulation of the immune response [20]. Therefore, due to the component differences between the active immunotherapies, no direct comparisons can be made with respect to doses or adjuvant effects. The antibody affinity achieved may also be different across vaccines, and may improve on repeated injections demonstrating affinity and avidity maturation.
In comparison, tau immunotherapy is a relatively underexplored therapeutic target for AD. This review will thus mainly focus on the second-generation active immunotherapy options for targeting the Aβ peptide in AD. Table 1 presents the Aβ-targeted active immunotherapy approaches currently being investigated and describes their mechanism of action and key clinical data.
Table 1.
Active amyloid-beta immunotherapies in development
| Compound | Sponsor | Epitope/carrier/adjuvant | Route of administration | Phase of development | Population |
|---|---|---|---|---|---|
| CAD106 |
Novartis (Basel, Switzerland) |
Aβ1–6/bacteriophage Qβ coat protein |
i.m./s.c. |
2 |
Mild-to-moderate AD |
| ACC-001 (vanutide cridificar) |
Pfizer (New York, USA)/Janssen Research & Development, LLC (Raritan and Titusville, NJ, USA) |
Aβ1–7/nontoxic diphtheria toxin (CRM197)/Qs21 adjuvant |
i.m. |
2 |
Mild-to-moderate AD, early AD |
| AD02 |
AFFiRiS (Vienna, Austria)/GlaxoSmithKline (Brentford, UK) |
Aβ1–6 mimetic/keyhole limpet hemocyanin/aluminum adjuvant |
s.c. |
2 |
Mild-to-moderate AD, early AD |
| ACI-24 |
AC Immune (Lausanne, Switzerland) |
Tetra-palmitoylated Aβ1–15/reconstituted in liposome |
s.c. |
1/2 |
Mild-to-moderate AD |
| V950 |
Merck & Co. (Whitehouse Station, NJ, USA) |
Multivalent Aβ peptide/ISCOMATRIX™ adjuvant |
i.m. |
1 (discontinued) |
Mild-to-moderate AD |
| UB-311 |
United Biochemical, Inc. (Hauppauge, NY, USA) |
Two UBITh® synthetic peptides coupled to Aβ1–14 peptide/CpG oligonucleotide |
i.m. |
2 |
Mild-to-moderate AD |
| Lu AF20513 | Lundbeck A/S (Valby, Denmark) | Aβ1–12 peptide replaced with two foreign T-helper epitopes from tetanus toxoid | N/A | Preclinical | Early AD |
Aβ, amyloid-beta protein; AD, Alzheimer’s disease; i.m., intramuscular; s.c., subcutaneous.
Amyloid-beta protein immunotherapy
CAD106
CAD106 is a second-generation, Aβ-based active immunotherapy comprising multiple copies of Aβ1–6 peptide coupled to a carrier that contains 180 copies of the bacteriophage Qβ coat protein [21]. CAD106 is designed to stimulate a strong B-cell response and carrier-induced T-cell help, without activating an Aβ-specific T-cell response [10,21]. In animal models, CAD106 effectively induces Aβ antibodies without potential mechanism-related side effects caused by stimulation of Aβ-specific T cells [10]. As all of the main IgG subclasses were generated, CAD106 has the potential to stimulate the entire range of effector functions. In APP23/24 transgenic mice, CAD106 effectively reduced amyloid accumulation (Figure 2) [10]. CAD106 was more effective when administered in the early stages of amyloid accumulation, with the greatest effect being when administered before amyloid deposition starts. The elevation of vascular Aβ observed in some mice studies did not lead to an increase in microhemorrhages [10]. Of note, CAD106-induced antibodies from rhesus monkeys were also shown to protect from Aβ toxicity in vitro[10].
Figure 2.
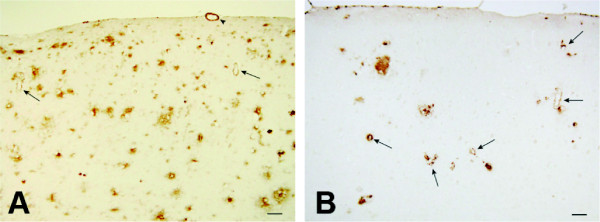
Amyloid deposition in the neocortex of APP24 mice after treatment with CAD106 and vehicle. (A) Treatment with vehicle. (B) Treatment with CAD106. Reprinted from [10]. © 2011, with permission from Society for Neuroscience.
A phase 1, 52-week, placebo-controlled study (study number 2101; ClinicalTrials.gov NCT00411580) in patients with mild-to-moderate AD (Mini-mental State Examination 16 to 26) showed that three subcutaneous (s.c.) injections of CAD106 (50 μg, n = 24; or 150 μg, n = 22) had a favorable safety profile, without an Aβ-specific T-cell response, and an acceptable antibody response (Figure 3) [21]. The proportion of patients treated with CAD106 who developed an Aβ antibody response that met the prespecified IgG titer responder threshold was higher in the 150 μg group versus the 50 μg group (82% vs. 67%) [21]. Results from selected plasma samples showed that free Aβ decreased in parallel with an increase in total Aβ concentration [21]. The binding of CAD106-induced Aβ antibodies from patients to amyloid plaque cores on brain sections from APP23 transgenic mice and from a patient with AD was increased at week 8 compared with baseline and correlated with Aβ IgG titers [21]. No significant differences were observed between CAD106 and placebo for cerebrospinal fluid (CSF) total tau, phospho-tau, Aβ40 and Aβ42 biomarkers [21]. The lack of significance could be attributed to the small sample size, or the antibody exposure of 100 days may have been too short to show a clinical effect [21].
Figure 3.

Mean amyloid-beta protein-specific antibody response with CAD106, by IgM and IgG titers. (A) IgM titers. (B) IgG titers. Aβ, amyloid-beta protein. Reprinted from [21]. © 2012, with permission from Elsevier.
In two 52-week, phase 2a studies in patients with mild AD (Mini-mental State Examination 20 to 26), 150 μg CAD106 was administered subcutaneously at weeks 0, 6, and 12 (study 2201; ClinicalTrials.gov NCT00733863), or either subcutaneously or intramuscularly at weeks 0, 2, and 6 (study 2202; ClinicalTrials.gov NCT00795418) [22]. In both studies, approximately 90% of patients developed an antibody response [23], with the highest total plasma Aβ concentrations being observed in patients with a strong antibody response [24]. Results from these studies showed that the total plasma Aβ concentration increased in parallel to Aβ-specific IgG development [24]. Consistent with results from the phase 1 study [21], CSF sampling at 8 weeks after the third injection did not show significant differences in tau, phospho-tau, isoprostane [24], or Aβ40 and Aβ42 biomarkers (unpublished observations). However, the timing of CSF sampling was driven by safety monitoring rather than optimized biomarker detection. In open-label extensions to these studies (ClinicalTrials.gov NCT00956410; NCT01023685), patients received four additional injections of CAD106 at 12-week intervals (weeks 56, 68, 80, and 92) by s.c. or intramuscular (i.m.) routes. Results from the core studies showed that the mean total plasma Aβ concentration increased, probably due to the longer half-life of Aβ in the periphery upon binding to antibodies [24]. The additional four injections induced a similar antibody titer to the initial three injections, but with a higher increase in total plasma Aβ. The increase of levels over time is consistent with improved affinity of IgG to the target, and confirms that CAD106 is suitable for long-term chronic treatment in AD [22]. Study 2202 data also suggested that i.m. administration of CAD106 generates a more robust IgG response than s.c. administration [23]. An additional phase 2 study investigating up to seven repeated i.m. injections of CAD106 (150 or 450 μg with an adjuvant vs. placebo) in 121 patients with mild AD (Mini-mental State Examination 20 to 26) has recently been completed (study 2203; ClinicalTrials.gov NCT01097096) [25].
Safety and tolerability of CAD106 immunotherapy
Results from previous studies have shown that CAD106 has a favorable safety profile, with no unexpected findings, and does not stimulate an Aβ-specific T-cell response [21,22]. In the phase 1 study, nearly all patients (97%, n = 56/58) experienced an adverse event (Table 2) [21]. Nasopharyngitis (10/24 CAD106-treated patients) and injection site erythema (14/22 CAD106-treated patients) were the most frequently reported adverse events in the 50 μg and 150 μg cohorts, respectively [21]. Nine patients reported serious adverse events, but none were thought to be related to CAD106. No clinical or subclinical cases of meningoencephalitis, aseptic meningoencephalitis, or vasogenic edema were reported [21], and no differences in vital signs, hematological tests, blood chemistry, urinalysis, or laboratory parameters were seen between the CAD106-treated patients versus placebo-treated patients [21]. Moreover, data from the phase 2 studies demonstrated that tolerability was slightly improved with i.m. administration, which was selected for further ongoing studies [26]. Brief self-limiting injection-related reactions – either local (for example, erythema) or systemic (for example, fatigue) – were observed in the majority of patients [25], as commonly expected with successful vaccinations. There was one incidence of intracerebral hemorrhage that did not appear to be related to the level of Aβ titers, but could be potentially related to cerebral amyloid angiopathy [26].
Table 2.
Adverse events (>10% of patients in any group) from a phase 1 study of CAD106 in patients with mild-to-moderate Alzheimer’s disease during the 52-week study period
| |
Cohort 1 |
Cohort 2 |
||
|---|---|---|---|---|
| Event | CAD106 50 μg (n = 24) | Placebo (n = 7) | CAD106 150 μg (n = 22) | Placebo (n = 5) |
| Any serious adverse event |
4 (17) |
1 (14) |
4 (18) |
0 |
| Any adverse events |
23 (96) |
6 (86) |
22 (100) |
5 (100) |
| Injection site erythema |
1 (4) |
1 (14) |
14 (64) |
0 |
| Nasopharyngitis |
10 (42) |
2 (29) |
3 (14) |
0 |
| Fatigue |
7 (29) |
0 |
4 (18) |
1 (20) |
| Nausea |
4 (17) |
2 (29) |
3 (14) |
0 |
| Chills |
1 (4) |
0 |
6 (27) |
0 |
| Headache |
5 (21) |
0 |
4 (18) |
1 (20) |
| Diarrhea |
3 (13) |
1 (14) |
0 |
1 (20) |
| Vomiting |
2 (8) |
1 (14) |
3 (14) |
1 (20) |
| Fever |
1 (4) |
0 |
4 (18) |
0 |
| Injection site pain |
0 |
0 |
4 (18) |
0 |
| Myalgia |
4 (17) |
0 |
0 |
0 |
| Fall |
3 (13) |
1 (14) |
1 (5) |
0 |
| Back pain |
3 (13) |
0 |
3 (14) |
0 |
| Rhinitis | 3 (13) | 0 | 0 | 0 |
Data presented as n (%). Reprinted from [21]. © 2012, with permission from Elsevier.
ACC-001
ACC-001 (vanutide cridificar) is a conjugate of multiple copies of Aβ1-7 peptide linked to a nontoxic variant of diphtheria toxin (CRM197), which is administered intramuscularly [27,28]. Data from preclinical studies in nonhuman primates showed that ACC-001 generates N-terminal Aβ antibodies without inducing an Aβ-directed T-cell response [27]. Ongoing ACC-001 phase 2 clinical trials in mild-to-moderate AD and early AD are investigating dose-ranging, safety, immunogenicity, and long-term treatment in western and Japanese patients (ClinicalTrials.gov NCT01284387; NCT01227564; NCT00955409; NCT00960531; NCT01238991 (Japanese)). Some clinical trials have already been completed (ClinicalTrials.gov NCT00479557; NCT00498602; NCT00752232 (Japanese); NCT00959192 (Japanese)).
Data from a study in Japanese patients with mild-to-moderate AD (ClinicalTrials.gov NCT00752232) demonstrated that repeated i.m. administration of ACC-001 (3 μg, 10 μg, and 30 μg) with adjuvant QS-21 (50 μg) at 3-monthly intervals up to 1 year elicited high-titer and sustained Aβ IgG antibody responses, with no difference among the three ACC-001 doses tested. The addition of QS-21 was essential to stimulate high-titer responses. ACC-001 at all dose levels with/without QS-21 was generally safe and well tolerated, and the safety profile was similar to that of western populations [28]. Adverse events observed in this trial are presented in Table 3. Similar to phase 1 results of CAD106 [29], no significant differences were observed in cognitive evaluations, but this may be due to the small sample size (n = 40) and interpatient variability [28].
Table 3.
Adverse events (>2% of patients overall) from a phase 2a study of ACC-001 in Japanese patients with mild-to-moderate Alzheimer’s disease [[28]]
|
Event |
ACC-001 + QS-21 |
ACC-001 |
QS-21 (n = 6) |
PBS (n = 4) |
|||
|---|---|---|---|---|---|---|---|
| 3 μg (n = 6) | 10 μg (n = 6) | 30 μg (n = 6) | 10 μg (n = 6) | 30 μg (n = 6) | |||
| Any serious adverse eventa |
1 (16.7) |
0 |
1 (16.7) |
0 |
0 |
0 |
0 |
| Any adverse events |
6 (100.0) |
4 (66.7) |
5 (83.3) |
6 (100.0) |
6 (100.0) |
6 (100.0) |
3 (75.0) |
| Nasopharyngitis |
2 (33.3) |
2 (33.3) |
2 (33.3) |
0 |
3 (50.0) |
1 (16.7) |
2 (50.0) |
| Injection site erythema |
0 |
2 (33.3) |
0 |
0 |
1 (16.7) |
0 |
0 |
| Dental caries |
0 |
1 (16.7) |
0 |
1 (16.7) |
1 (16.7) |
0 |
1 (25.0) |
| Protein urine present |
1 (16.7) |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
0 |
| Glucose urine |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
0 |
1 (25.0) |
| Cataract |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
0 |
1 (25.0) |
| Contusion |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
1 (16.7) |
0 |
| Back pain |
0 |
0 |
1 (16.7) |
0 |
1 (16.7) |
0 |
0 |
| Blood triglyceride increased |
0 |
1 (16.7) |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
| Injection site pain |
0 |
1 (16.7) |
1 (16.7) |
0 |
0 |
0 |
0 |
| Pyrexia |
0 |
1 (16.7) |
0 |
0 |
1 (16.7) |
0 |
0 |
| Delirium |
1 (16.7) |
0 |
0 |
0 |
0 |
1 (16.7) |
0 |
| Blood pressure increased |
0 |
0 |
0 |
1 (16.7) |
0 |
1 (16.7) |
0 |
| Eczema | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (25.0) |
Data presented as n (%). PBS, phosphate-buffered saline. aAll patients.
The ongoing phase 2 ACCTION study (ClinicalTrials.gov NCT01284387) is among the first AD studies to use amyloid positron emission tomography as an enrichment strategy to increase diagnostic certainty. The authors concluded that, despite its challenges, amyloid positron emission tomography is an effective tool for sample enrichment in mild-to-moderate AD trials, and CSF sampling is also feasible. Baseline and longitudinal amyloid positron emission tomography, volumetric magnetic resonance imaging, and CSF data may thus provide valuable data for AD trials and may support treatment response determination [30].
Affitope
AD01 and AD02 (Affitope) are KLH vaccines with short (six amino acids) peptides that mimic the N-terminus of Aβ [31]. These compounds were designed to exhibit a favorable safety profile because they are non-endogenous, and will avoid development of tolerance. Moreover, the small size averts autoreactive T-cell activation, and the controlled specificity prevents cross-reactivity with APP [31]. There are limited data available for this compound, but results from a phase 1 study showed a favorable safety profile with both AD02 and AD01 [32]. AD02 has been selected for development in a phase 2, dose-finding trial in patients with early AD to investigate clinical/immunological activity and tolerability (ClinicalTrials.gov NCT01117818). AD03 (MimoVax, Vienna, Austria), a KLH vaccine that additionally targets modified Aβ peptides, is currently in phase 1b development (ClinicalTrials.gov NCT01568086) and has previously been shown to significantly reduce amyloid plaque load in APP mice [33].
ACI-24
ACI-24 is a tetra-palmitoylated Aβ1-15 peptide reconstituted in a liposome [34]. After two intraperitoneal inoculations of ACI-24 in double-transgenic APP × PS-1 mice, significant levels of systemic Aβ1-42 antibodies were detected that were predominantly of the IgG class (mainly IgG1, IgG2b, and IgG3), indicating a preferential T-helper type 2 response. Complete restoration of cognitive, nonspatial memory as measured by a novel object recognition test was observed after six inoculations at 2-week intervals. Aβ1-42-specific IgG antibody titers were positively correlated to the object recognition test index. In addition, ACI-24 led to a significant decrease in insoluble, plaque-related Aβ1-40 and Aβ1-42, and soluble Aβ1-42, and to a lesser extent soluble Aβ1-40. No significant signs of inflammation – that is, absence of proinflammatory cytokines (tumor necrosis factor alpha, interleukin-1β, interleukin-6, and interferon gamma), microglia activation or astrogliosis – were detected [34]. A phase 1/2a clinical trial investigating the safety and efficacy of ACI-24 in patients with mild-to-moderate AD is currently ongoing (EudraCT 2008-006257-40).
V950
V950 is a multivalent Aβ peptide vaccine [35]. Data from preclinical studies have shown that V950 results in the production of Aβ antibodies in the serum and CSF that recognize pyroglutamate-modified and other N-terminally truncated Aβ fragments [35]. A phase 1 dose-escalating study of V950 in patients with mild-to-moderate AD to evaluate the safety, tolerability, and immunogenicity of i.m. V950 with ISCOMATRIX™ adjuvant (ClinicalTrials.gov NCT00464334) at 0, 2, and 6 months has been completed. Results are available online [36] and no further studies have been initiated.
UB-311
UB-311 is an equimolar mixture of two synthetic peptides, consisting of highly active UBITh® helper T-cell epitopes, coupled to the Aβ1–14 peptide. The vaccine is designed to stimulate the T-helper type 2 regulatory response over the T-helper type 1 proinflammatory response using a proprietary vaccine delivery system (CpG oligonucleotide), and is likely to prevent cross-reactivity with different but similar antigens [37]. A phase 1 clinical trial of intramuscularly administered UB-311 at weeks 0, 4, and 12 in Taiwanese patients with mild-to-moderate AD has been completed (ClinicalTrials.gov NCT00965588), demonstrating safety and tolerability; however, results have not yet been published. In addition, United Biochemical, Inc. (Hauppauge, NY, USA) is currently initiating a phase 2 study.
Lu AF20513
Lu AF20513 is an Aβ1-12 peptide in which the T-helper cell epitopes of Aβ42 are replaced with two foreign T-helper epitopes from the tetanus toxin, which stimulate existing memory T-helper cells to promote Aβ antibody production from B cells [38]. Most adults have memory T cells that recognize the tetanus toxin, as they are inoculated against the bacterium earlier in life. In transgenic mice with early-stage AD-like pathology, Lu AF20513 produced Aβ antibodies and induced robust nonself T-cell responses that reduced AD-like pathology without inducing microglial activation and enhancing astrocytosis or cerebral amyloid angiopathy [38]. Strong humoral immunity was induced in mice, guinea pigs, and monkeys. Interestingly, a single injection of Lu AF20513 activated pre-existing CD4+ T cells specific to foreign T-helper epitopes, inducing a rapid and strong T-helper cell and Aβ response, therefore presenting a potential solution to overcoming the limited ability of older people to respond to vaccinations by activating pre-existing anti-P30/P2 memory T-helper cells [38]. In addition, Lu AF20513 suppressed amyloid plaque formation and accumulation of soluble forms of Aβ40/42. Furthermore, Aβ antibodies also protected neuronal cells from Aβ42 oligomer-mediated and fibril-mediated toxicity, and prevented the development of cored and diffuse plaques. The number of amyloid-containing blood vessels did not increase and no microhemorrhages were reported. Preclinical data from this study support the transition of this vaccine to human clinical trials.
DNA amyloid-beta protein immunotherapy
DNA Aβ immunotherapy is currently being investigated in preclinical studies [39-42]. This approach involves using a gene gun to inject DNA encoding Aβ42, which is subsequently translated to the Aβ peptide triggering a respective immune response. Data from transgenic mouse models have demonstrated that DNA Aβ42 immunotherapy effectively reduces Aβ42 levels in the brain by 41% and in plaques by 50% [41]. DNA Aβ immunotherapy induces a strong polarized T-helper type 2 cellular immune response [39,41]. In vitro, the absence of T-cell proliferation in B6SJLF1/J mice immunized with a full-length DNA42 trimer compared with B6SJLF1/J mice immunized with an Aβ42 peptide supports the safety of this approach [40]. As full-length DNA Aβ42 immunotherapies contain B-cell and T-cell epitopes, a wider antibody response against Aβ with a broader variety of antibody responses may be observed compared with Aβ-peptide immunotherapies, especially as T-cell help is required to maintain and further the humoral immune response. However, the antibody response to DNA immunotherapy is much lower compared with peptide immunotherapy, but this can be improved with prime-boost regimens [39]. Moreover, a DNA vaccine targeting a wide range of Aβ species including AβpE3-42, Aβ oligomers, and Aβ fibrils has shown promising results in preclinical studies [43]. These data support investigating DNA Aβ immunotherapy in clinical trials.
Tau immunotherapy
At present, tau-directed therapies are not as advanced as other options currently in development for disease modification of AD. The success of a tau-targeted therapy is complicated by the fact that tau protein is intracellular [44]; however, numerous studies have shown neuronal uptake of antibodies [45-47]. Moreover, inflammatory changes and cellular stress in response to tau pathology may facilitate antibody uptake primarily into diseased neurons, potentially reducing unwanted side effects. Extracellular spreading of tau pathology may also be blocked by antibodies. Recent data have demonstrated that tau monoclonal antibodies blocked development of tau seeding activity detected in brain lysates, reduced microglial activation, and improved cognitive deficits in P301S tau transgenic mice [48]. Preclinical data in various mouse models suggest that tau-targeted immunotherapies with phosphorylated peptides reduce levels of tau phosphorylation and NFT burden when treatment is started before or around the onset of NFT pathology [49,50], indicating that clearance of early-stage pathological conformers may be of therapeutic benefit [49]. Furthermore, in a htau/PS1 tangle mouse model, tau-targeted immunotherapy has been shown to prevent severe cognitive impairment that correlated with extensive clearance of abnormal tau [51]. In another study, suppression of tau expression in a different transgenic P301L mouse model was seen to improve memory, although NFTs remained [52]. The lack of clarity regarding which conformer of tau should be targeted therefore remains an issue [53]. Overall, results from these studies support the feasibility of targeting pathological tau conformers in patients with AD.
AADvac1
AADvac1 (Axon Neuroscience, Bratislava, Slovak Republic), a tau peptide conjugated to a KLH that is administered with an aluminum hydroxide (Alhydrogel) adjuvant, is the first vaccine targeting misfolded tau protein that has been clinically developed [54]. Vaccination of transgenic rats and mice with passive and active forms of AADvac1 significantly improved neurobehavioral deficits, and reduced neurofibrillary degeneration and mortality [54]. The active AADvac1 vaccine is currently being investigated in a 3-month, phase 1, randomized, placebo-controlled clinical trial (ClinicalTrials.gov NCT01850238) to assess tolerability, safety, and efficacy in patients with mild-to-moderate AD.
ACI-35
ACI-35 (AC Immune, Lausanne, Switzerland) is a liposomal vaccine containing a synthetic peptide (16 amino acids) corresponding to human protein tau sequence 393 to 408, with phosphorylated residues S396 and S404, using the same technology as ACI-24. In wild-type and tau.P301L transgenic mice, ACI-35 elicited rapid and robust polyclonal antibody responses specific for phosphorylated tau [55]. Long-term safety of the vaccine was also demonstrated by the improvement of clinical characteristics and lack of inflammation in the brain. These data indicate ACI-35 could be an effective and safe treatment for patients with AD.
Benefits and challenges with active immunotherapy
Benefits
Contrary to passive immunotherapy, which requires frequent re-administrations, active immunotherapy stimulates a natural immune response that can achieve persistent Aβ antibody titer levels with a low antigen dose and a minimal number of administrations. The steady antibody titers may be beneficial in achieving sufficient intraneuronal antibody concentrations targeting intracellular Aβ. This approach also has the potential to induce a polyclonal response against multiple epitopes, which may be relevant for improved efficacy. In addition, peak titers are reached gradually with a lower maximum plasma concentration compared with intravenous infusion of monoclonal antibodies, which may be important for safety. The risk of anaphylaxis reactions is also reduced with s.c. or i.m. administration of active immunotherapies. Furthermore, fewer injections may make treatment suitable for long-term therapy in primary care or in the home setting, promote improved compliance, and significantly reduce cost. Lastly, affinity maturation with repeated injections over time is also expected to lead to better quality antibodies and the possibility of an improved therapeutic response.
Challenges
There are some challenges to overcome with active immunotherapy. First, the mechanism of action relies on the patient’s own immune response, which varies amongst individuals. This may be particularly important in older patients, who quite often have weakened immune systems resulting in a diminished serological response to the antigen. Although achieved with current active immunotherapies in development, avoidance of Aβ-specific T cells to prevent pathological autoreactive T-cell responses is still an important safety consideration that must be assessed through long-term clinical follow-up. Given the complexity and interindividual variability of the immune response involved, a more complete understanding of the relationship of this response to active immunotherapy dose, adjuvants, regimen, route of administration, and impact on clinical outcomes will need to be explored during phase 2 clinical trials.
With repeated administrations, tolerance may develop over time and should be monitored especially with a self-antigen. Another concern to consider when using a self-antigen is the potential to develop an autoimmune response. Although no such incidences have been reported with active amyloid immunotherapy, amyloid-related imaging abnormalities have been observed in patients with AD treated with passive amyloid immunotherapy. The etiology of amyloid-related imaging abnormalities remains unclear but the prevailing data support vascular amyloid as a common pathophysiological mechanism leading to increased vascular permeability. Patients should thus be carefully monitored with brain imaging for amyloid-related imaging abnormalities and any associated clinical features [56]. The benefits and challenges with active immunotherapy are summarized in Table 4.
Table 4.
Benefits and challenges of the active immunotherapies for alzheimer’s disease
| Advantages |
Natural immune response |
| Persistent amyloid-beta protein antibody titer levels | |
| Infrequent re-administrations | |
| Suitable for long-term therapy | |
| Good compliance | |
| Reduced cost | |
| Low antigen dose | |
| Potential polyclonal response | |
| Peak titers are reached gradually | |
| Avidity maturation of antibodies | |
| Reduced risk of anaphylaxis reactions | |
| Disadvantages | Challenges |
| Relies on patients’ own immune response | |
| Autoreactive T-cell responses need to be avoided | |
| An understanding of the relationship of responses to clinical outcomes is required | |
| Generation of antibody responses in older patients | |
| Development of tolerance to be avoided | |
| Risk of developing an autoimmune response | |
| Risk of developing amyloid-related imaging abnormalities |
Conclusion
The need to develop disease-modifying drugs that stabilize or slow the AD pathological process leading to neurodegeneration is recognized worldwide, and most initial drug development programs have largely focused on compounds targeting the Aβ pathway. Active immunotherapies may play an important future role in long-term treatment, and several of these experimental therapies are showing promising early results in terms of antibody response and safety. Recent clinical trial failures with passive immunotherapy could be because the interventions might have been administered too late in the disease course, suggesting that active immunotherapy trials should be directed to individuals with earlier-stage disease. The idea that treatment initiation prior to dementia onset in humans may be most likely to succeed is supported by the observation that active immunotherapies appear to be most effective in early stages of amyloid accumulation in preclinical models. Whether early intervention studies are best conducted in sporadic or familial forms of AD remains an open question because different Aβ antibody concentrations and treatment durations may be needed for sporadic and familial AD due to possible differences in the pathological cascade leading to dementia. In addition, therapies targeting a single pathway may not be able to demonstrate optimal efficacy, and combination therapies targeting both Aβ and tau aggregates may ultimately be required. Once the interplay between AD pathologies is better understood, active immunotherapies may prove to be the ideal therapeutic platform for delivering cost-effective and long-term treatments that slow or halt progression in AD.
Abbreviations
AD: Alzheimer’s disease; APP: Amyloid precursor protein; Aβ: Amyloid-beta protein; CSF: Cerebrospinal fluid; i.m.: intramuscular; KLH: Keyhole limpet hemocyanin; NFT: Neurofibrillary tangle; s.c.: subcutaneous.
Competing interests
Karolinska University Hospital (NA) received research funding for this study from Novartis. AG, M-ER, and JMR are employees of Novartis Pharma AG and hold stock/stock options with Novartis Pharma AG. BW declares no competing interests.
Contributor Information
Bengt Winblad, Email: bengt-winblad-swedishbrainpower@ki.se.
Ana Graf, Email: ana.graf@novartis.com.
Marie-Emmanuelle Riviere, Email: marie-emmanuelle.riviere@novartis.com.
Niels Andreasen, Email: niels.andreasen@karolinska.se.
J Michael Ryan, Email: jmichael.ryan@novartis.com.
Acknowledgements
This study was supported by the Swedish Brain Power and the Karolinska Institutet Strategic Neuroscience Program. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. The authors thank Emma Burke, of iMed Comms, who provided medical writing assistance with this review. This work was supported by Novartis Pharmaceuticals.
References
- Dementia Fact Sheet No. 362. http://www.who.int/mediacentre/factsheets/fs362/en/
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of alzheimer’s disease: report of the NINCDS-adrda work group under the auspices of department of health and human services task force on alzheimer’s disease. Neurology. 1984;6:939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;6:63–75. doi: 10.1016/j.jalz.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Wimo A, Jonsson L, Bond J, Prince M, Winblad B. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;6:1–11. doi: 10.1016/j.jalz.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An english translation of alzheimer’s 1907 paper, 'Uber eine eigenartige Erkankung der Hirnrinde’. Clin Anat. 1995;6:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetics of alzheimer’s disease. Prog Mol Biol Transl Sci. 2012;6:79–100. doi: 10.1016/B978-0-12-385883-2.00008-4. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;6:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;6:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;6:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiessner C, Wiederhold KH, Tissot AC, Frey P, Danner S, Jacobson LH, Jennings GT, Luond R, Ortmann R, Reichwald J, Zurini M, Mir A, Bachmann MF, Staufenbiel M. The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J Neurosci. 2011;6:9323–9331. doi: 10.1523/JNEUROSCI.0293-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;6:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in alzheimer’s disease: what are the most promising targets? Immun Ageing. 2013;6:18. doi: 10.1186/1742-4933-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson & Johnson Announces Discontinuation of Phase 3 Development of Bapineuzumab Intravenous (IV) in Mild-to-moderate Alzheimer’s Disease. http://www.investor.jnj.com/releasedetail.cfm?ReleaseID=698466.
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;6:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;6:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;6:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;6:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL. Increased T cell reactivity to amyloid beta protein in older humans and patients with alzheimer disease. J Clin Invest. 2003;6:415–422. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobello K, Ryan JM, Liu E, Rippon G, Black R. Targeting beta amyloid: a clinical review of immunotherapeutic approaches in alzheimer’s disease. Int J Alzheimers Dis. 2012;6:628070. doi: 10.1155/2012/628070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM, Graf A. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;6:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- Graf A, Riviere ME, Farlow M, Andreasen N, Quarg P, Caputo A, Vostiar I, Winblad B, Orgogozo JM. Poster at the International Conference on Alzheimer’s Disease (ICAD) Vancouver, Canada; 2012. Long-term administration of active immunotherapy CAD106 in phase IIa open-label extension studies in Alzheimer patients. [Google Scholar]
- Imbert G, Andreasen N, Riviere ME, Ros J, Moreau J, Sevigny J, Caputo A, Finelli LA, Winblad B, Graf A. Poster at the International Conference on Alzheimer’s Disease (ICAD) Thessaloniki, Greece; 2010. Optimization of the treatment regimen with active Aβ immunotherapy CAD106 in Alzheimer patients. [Google Scholar]
- Winblad B, Farlow M, Blennow K, Vostiar I, Imbert G, Tomovic A, Quarg P, Riviere ME, Andreasen N, Graf A. Poster presented at the International Conference on Alzheimer’s Disease (ICAD) Paris, France; 2011. Aβ-specific antibodies induced by active immunotherapy CAD106 engage Aβ in plasma in Alzheimer patients. [Google Scholar]
- Graf A. Oral presentation at the International Conference on Alzheimer’s Disease (ICAD) Vancouver, Canada; 2012. Current phase II results from active vaccination with CAD106 in Alzheimer’s disease. [Google Scholar]
- Orgogozo JM, Andreasen N, Farlow M, Caputo A, Moreau J, Collober C, Winblad B, Graf A. Poster at the International Conference on Alzheimer’s Disease (ICAD) Paris, France; 2011. Safety and tolerability of active immunotherapy CAD106 in two phase II studies in Alzheimer patients. [Google Scholar]
- Hagen M, Seubert P, Jacobsen S, Schenk D, Pride M, Arumugham R, Warner G, Kinney G. The Aβ peptide conjugate vaccine, ACC-001, generates N-terminal anti-Aβ antibodies in the absence of Aβ directed T-cell responses [abstract] Alzheimers Dement. 2011;6:S460–S461. [Google Scholar]
- Arai H, Hideo S, Tamotsu Y, Lobello K, Yahong P, Enchi L, Nzeera K, Margolin R, Jackson N, Fujimoto Y. Presented at the International Conference on Alzheimer’s Disease (ICAD) Boston, MA, USA; 2013. Safety, tolerability, and immunogenicity of an immunotherapeutic vaccine (vanutide cridificar [ACC-001]) and the QS-21 adjuvant in Japanese subjects with mild to moderate Alzheimer’s disease: a phase 2a, multicenter, randomized, adjuvant- and placebo-controlled, multiple-ascending-dose study [abstract P1-338] [Google Scholar]
- Winblad B, Minthon L, Floesser A, Imbert G, Dumortier T, He Y, Maguire P, Karlsson M, Ostlund H, Lundmark J, Orgogozo JM, Graf A, Andreasen N. Oral presentation at the International Conference on Alzheimer’s Disease (ICAD) Vienna, Austria; 2009. Results of the first-in-man study with the active Aβ immunotherapy CAD106 in Alzheimer patients. [Google Scholar]
- Margolin R, Ketter N, Gutherie S, Deegan D, Baher L, Tschopp R, Marek K, Seibyl J, Novak G, Streffer JR, Pachai C, Wyman B, Booth K, Yuen E, Gennevois D, Lui E. Presented at the International Conference on Alzheimer’s Disease (ICAD) Boston, MA, USA; 2013. Biomarker strategy for enrichment and monitoring of biological effect in ACCTION, a phase 2 study of ACC-001 (vanutide cridificar) for Alzheimer’s disease [abstract P1-357] [Google Scholar]
- Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W. Development of AFFITOPE vaccines for Alzheimer’s disease (AD) – from concept to clinical testing. J Nutr Health Aging. 2009;6:264–267. doi: 10.1007/s12603-009-0070-5. [DOI] [PubMed] [Google Scholar]
- Mandler M, Schmidt W, Mattner F. Development of AFFITOPE alzheimer vaccines: results of phase I studies with AD01 and AD02 [abstract] Alzheimers Dement. 2011;6:S793. [Google Scholar]
- Mandler M, Santic R, Weninger H, Kopinits E, Schmidt W, Mattner F. The MimoVax vaccine: a novel alzheimer treatment strategy targeting truncated Aβ40/42 by active immunization [abstract] Alzheimers Dement. 2009;6:114. doi: 10.1016/j.jalz.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Muhs A, Hickman DT, Pihlgren M, Chuard N, Giriens V, Meerschman C, van der Auwera I, van Leuven F, Sugawara M, Weingertner MC, Bechinger B, Greferath R, Kolonko N, Nagel-Steger L, Riesner D, Brady RO, Pfeifer A, Nicolau C. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Natl Acad Sci U S A. 2007;6:9810–9815. doi: 10.1073/pnas.0703137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JM, Wu G, McCampbell A, Wessner RK, Citron M, Liang X, Hsieh S, Wolfe AL, Kinney GG, Rosen BL, Renger JJ. A novel multivalent Aβ peptide vaccine with preclinical evidence of a central immune response that generates antisera recognizing a wide range of abeta peptide species [abstract] Alzheimers Dement. 2010;6:S142. [Google Scholar]
- ClinicalTrials.gov. http://www.clinicaltrials.gov.
- Wang CY, Finstad CL, Walfield AM, Sia C, Sokoll KK, Chang TY, Fang XD, Hung CH, Hutter-Paier B, Windisch M. Site-specific UBITh amyloid-beta vaccine for immunotherapy of Alzheimer’s disease. Vaccine. 2007;6:3041–3052. doi: 10.1016/j.vaccine.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Davtyan H, Ghochikyan A, Petrushina I, Hovakimyan A, Davtyan A, Poghosyan A, Marleau AM, Movsesyan N, Kiyatkin A, Rasool S, Larsen AK, Madsen PJ, Wegener KM, Ditlevsen DK, Cribbs DH, Pedersen LO, Agadjanyan MG. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci. 2013;6:4923–4934. doi: 10.1523/JNEUROSCI.4672-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan H, Mkrtichyan M, Movsesyan N, Petrushina I, Mamikonyan G, Cribbs DH, Agadjanyan MG, Ghochikyan A. DNA prime-protein boost increased the titer, avidity and persistence of anti-Aβ antibodies in wild-type mice. Gene Ther. 2010;6:261–271. doi: 10.1038/gt.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Anderson LD Jr, Stuve O, Eagar TN, Rosenberg RN. DNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer’s disease as it diminishes antigen-specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol. 2011;6:867–874. doi: 10.1007/s10571-011-9680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu BX, Xiang Q, Li L, Johnston SA, Hynan LS, Rosenberg RN. Aβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic mice. J Neurol Sci. 2007;6:204–213. doi: 10.1016/j.jns.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CF, Davtyan H, Petrushina I, Hovakimyan A, Davtyan A, Hannaman D, Cribbs DH, Agadjanyan MG, Ghochikyan A. Epitope-based DNA vaccine for Alzheimer’s disease: translational study in macaques. Alzheimers Dement. 2013. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Matsumoto Y, Niimi N, Kohyama K. Development of a new DNA vaccine for Alzheimer disease targeting a wide range of abeta species and amyloidogenic peptides. PLoS One. 2013;6:e75203. doi: 10.1371/journal.pone.0075203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Scarpini E. Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2011;6:203–216. doi: 10.1177/1756285611404470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian RH, Petroff G. Intraneuronal IgG in the central nervous system: uptake by retrograde axonal transport. Neurology. 1987;6:1780–1784. doi: 10.1212/WNL.37.11.1780. [DOI] [PubMed] [Google Scholar]
- Greenlee JE, Burns JB, Rose JW, Jaeckle KA, Clawson S. Uptake of systemically administered human anticerebellar antibody by rat Purkinje cells following blood–brain barrier disruption. Acta Neuropathol. 1995;6:341–345. doi: 10.1007/BF00309627. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008;6:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;6:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;6:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;6:472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;6:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;6:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Petrucelli L, Lewis J. Targeting Aβ and tau in Alzheimer’s disease, an early interim report. Exp Neurol. 2010;6:252–266. doi: 10.1016/j.expneurol.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak M. Presented at the International Conference on Alzheimer’s Disease (ICAD) Boston, MA, USA; 2013. Tau immunotherapy – the way how to crack the immune code of misfolded protein tau [abstract S4-02-02] [Google Scholar]
- Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, Hickman DT, Adolfsson O, Chuard N, Ndao DM, Borghgraef P, Devijver H, Van Leuven F, Pfeifer A, Muhs A. Efficacy and safety of a liposome-based vaccine against protein tau, assessed in Tau.P301L mice that model tauopathy. PLoS One. 2013;6:e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the alzheimer’s association research roundtable workgroup. Alzheimers Dement. 2011;6:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]