

Abstract
A-to-I RNA editing is a post-transcriptional modification that converts adenosines to inosines in both coding and noncoding RNA transcripts. It is catalyzed by ADAR (adenosine deaminase acting on RNA) enzymes, which exist throughout the body but are most prevalent in the central nervous system. Inosines exhibit properties that are most similar to those of guanosines. As a result, ADAR-mediated editing can post-transcriptionally alter codons, introduce or remove splice sites, or affect the base pairing of the RNA molecule with itself or with other RNAs. A-to-I editing is a mechanism that regulates and diversifies the transcriptome, but the full biological significance of ADARs is not understood. ADARs are highly conserved across vertebrates and are essential for normal development in mammals. Aberrant ADAR activity has been associated with a wide range of human diseases, including cancer, neurological disorders, metabolic diseases, viral infections and autoimmune disorders. ADARs have been shown to contribute to disease pathologies by editing of glutamate receptors, editing of serotonin receptors, mutations in ADAR genes, and by other mechanisms, including recently identified regulatory roles in microRNA processing. Advances in research into many of these diseases may depend on an improved understanding of the biological functions of ADARs. Here, we review recent studies investigating connections between ADAR-mediated RNA editing and human diseases.
A-to-I editing is widespread and highly conserved
Eukaryotic RNA transcripts can undergo a range of post-transcriptional modifications, which increase the diversity of the transcriptome without requiring increases in genome size. These include alternative splicing and RNA editing. RNA editing refers to post-transcriptional processes that alter the nucleotide sequence of an RNA transcript by insertion, deletion or nucleotide conversion. In mammals, the most prevalent form of RNA editing involves the conversion of adenosine to inosine (A-to-I) by hydrolytic deamination at the C6 position of adenine (Figure 1a) [1]. A-to-I editing, which is catalyzed by enzymes of the adenosine deaminase acting on RNA (ADAR) family, is most prevalent in the central nervous system (CNS) but occurs in many tissues [1-3]. Once an adenosine nucleotide is converted to an inosine, it acts in a manner similar to a guanosine nucleotide, with a number of potential consequences [4]. When this conversion occurs in the coding region of mRNA, it results in an altered nucleotide codon and, therefore, can change the amino acid sequence of the coded protein in what is referred to as a re-coding editing event. A-to-I editing can also result in the creation or elimination of splice sites, potentially altering the portions of the RNA that remain in the final product. Additionally, the A-to-I conversion alters base pairing, because inosine pairs preferentially with cytidine, and this potentially affects the secondary structure of the RNA. In the case of RNA molecules that bind target RNA segments, such as microRNAs (miRNAs), the altered base pairing can change binding specificities. Thus, A-to-I editing in both translated and untranslated regions of RNA can be biologically significant. The consequences of an A-to-I editing event range from the trivial to the critically important, and the cellular functions of the majority of editing events are still unknown [5]. Although RNA editing has long been considered a relatively rare processing event, more recent research suggests that the vast majority of pre-mRNAs are edited [6].
Figure 1.

Adenosine deamination and the ADAR enzyme family. (a) ADAR enzymes catalyze the A-to-I hydrolytic deamination reaction, by which an adenosine loses an amine group and is converted to inosine. (b) There are four main proteins of the ADAR enzyme family: two isoforms of ADAR1 (p110 and p150), ADAR2 and ADAR3. All of these enzymes contain a conserved deaminase domain, shown in blue. The double-stranded (ds)RNA-binding domains, shown in purple, determine substrate specificity. The two ADAR1 isoforms differ in their Z-DNA-binding domains, shown in green. ADAR3 contains an arginine-rich domain, shown in pink, which binds single-stranded RNA.
Three primary members of the ADAR family have been identified in humans: ADAR1, ADAR2 and ADAR3 (Figure 1b) [1]. These proteins are highly conserved across vertebrates [7]. ADAR1 is expressed in both the constitutive p110 isoform and the interferon-inducible p150 isoform [8]. ADAR1 and ADAR2 are present in many tissues, whereas ADAR3 is specifically expressed in brain tissues and is believed to be catalytically inactive [1,9]. ADARs contain a conserved deaminase domain that mediates A-to-I editing, as well as variable double-stranded RNA-binding domains that are required for substrate specificity and binding [1]. Homodimerization of ADARs is required for editing activities, as observed in vitro and confirmed with in vivo studies [10]. A single mutated ADAR subunit affects dimer function in a dominant negative manner, suggesting a degree of cooperativity between ADAR subunits [11]. ADARs can edit both coding and noncoding double-stranded RNA and can act in a site-specific or nonspecific manner [1]. Changes in ADAR activity, caused by mutations or changes in expression, have been associated with a wide range of human diseases, including cancer, neurological disorders, metabolic diseases, viral infections and autoimmune disorders (Table 1). Currently, many of these diseases have limited treatment options as ADAR-specific therapies have not yet been developed.
Table 1.
Key studies: A-to-I RNA editing and human disease
| Disease | Finding | Model | Reference(s) |
|---|---|---|---|
| Amyotrophic lateral sclerosis (ALS) |
Downregulation of ADAR2 expression and GluA2 editing in ALS |
Human patients |
[20,21] |
| |
Conditional ADAR2 knockout produces ALS-like symptoms that are rescued by edited GluA2 |
In vivo, mice |
[23] |
| |
Unedited GluA2 causes mislocation of TDP-43 also observed in sporadic ALS |
In vivo, mice |
[24] |
|
In vitro, mouse cells | |||
|
In vitro, human cells | |||
| Astrocytoma |
Correlation between ADAR2 activity and malignancy |
Pediatric astrocytoma tissue |
[28] |
|
In vitro, human cells | |||
| Constant expression of ADAR2 but upregulation of ADAR1 | |||
| Overexpression of ADAR2 results in decreased proliferation and mobility | |||
| |
Influx of Ca2+ results in an activation of the Akt pathway, which is rescued by edited GluA2 |
In vitro, human cells |
[29] |
| |
ADAR2 edits CDC14B mRNA, promoting its translation. This leads to degradation of Skp2, which upregulates p21 and p27. This prevents S-phase transition and tumor growth |
Mouse xenograft |
[30] |
| Aicardi-Goutières syndrome (AGS) |
Novel Adar1 mutations associated with AGS |
Human patients |
[53] |
| Hepatocellular carcinoma (HCC) |
ADAR1 activity correlated with tumor aggressiveness ADAR1 edits AZIN1, which increases its inhibition of antizyme |
Human tumor specimens |
[62] |
| Mouse xenograft | |||
|
In vitro, human cells | |||
| This upregulates ODC and CCND1, increasing proliferation and mobility | |||
| Computational model | |||
| Measles virus |
Biased hypermutation in matrix gene |
Human patients |
[71] |
| |
Hypermutation promotes viral infection |
In vivo, mouse |
[73] |
|
In vitro, mouse cells | |||
| |
ADAR1 promotes viral infection |
In vitro, human cells |
[74] |
| Metastatic melanoma |
Cancer amplifies genes encoding microRNAs that target ADAR1 |
Mouse xenograft |
[60] |
|
In vitro, human cells | |||
| This interferes with the regulation of >100 microRNAs and promotes tumorigenicity |
Here, we review recent studies investigating connections between A-to-I editing and human diseases, with the aim of highlighting the importance of improving upon our current understanding of RNA editing and ADAR enzymes.
A-to-I editing of the AMPA receptor
Mammalian α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are ionotropic glutamate receptors prevalent in the CNS [12]. Following activation by the excitatory neurotransmitter glutamate, AMPA receptors allow the influx of Na+ and Ca2+ ions into the postsynaptic density [12]. AMPA receptors are tetrameric, composed of combinations of the subunits GluA1, GluA2, GluA3 and GluA4 (also referred to as either GluR1 to GluR4 or GluR-A to GluR-D) [13]. The mRNA that encodes the GluA2 subunit is edited by ADAR2, an event that results in the replacement of a glutamine (Q) residue with an arginine (R) residue (Figure 2a) [12,14]. The larger and positively charged arginine residue significantly reduces the Ca2+ permeability of the receptor (Figure 2b) [13]. This editing event is evolutionarily conserved and has been observed in mammals, amphibians and certain fish [15]. In human brain tissue, nearly all GluA2 mRNA is edited at this Q/R site, and therefore, AMPA receptors containing a GluA2 subunit are Ca2+-impermeable [16]. AMPA receptors that lack the GluA2 subunit or that contain only unedited versions of GluA2 are, however, Ca2+-permeable [12]. Decreased ADAR2-mediated editing of GluA2 mRNA at the Q/R site has been proposed as a component of excitotoxic cell death, which is often attributed to increases in Ca2+ influxes [17].
Figure 2.

ADAR-mediated editing of the GluA2 subunit of the mammalian AMPA receptor. (a) The unedited GluA2 subunit contains a glutamine residue (Q) at the Q/R site (left). ADAR2 can mediate editing of the Q/R site, resulting in a GluA2 subunit that has a positively charged arginine residue (right). (b) When the tetrameric AMPA receptor contains only the unedited form of the GluA2 subunit (light green), the receptor facilitates the flow of calcium ions (left). Incorporation of an edited GluA2 subunit (light green) sharply reduces the calcium permeability of the receptor because of the presence of the large, positive arginine residue (right).
Mice that have a homozygous null mutation in the ADAR2 gene exhibit epileptic seizures and premature death [17,18]. Most importantly, the epileptic phenotype was not observed in mice lacking ADAR2 but expressing edited GluA2 subunits, implying that the lack of ADAR2-mediated GluA2 RNA editing caused the abnormal phenotype [17].
Changes in the editing of the AMPA receptor subunit RNA have been associated with a number of human diseases, primarily neurological disorders.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive deterioration of motor neurons [19]. Familial ALS has been widely studied, and a number of causal genes have been identified, although upwards of 90% of ALS patients have sporadic symptoms [19]. The pathogenesis of this sporadic disease is largely unknown [19].
One of the proposed mechanisms of neuronal death in patients with sporadic ALS is AMPA-mediated excitotoxicity. While GluA2 mRNA levels remain essentially unchanged, GluA2 Q/R site-editing is markedly decreased in the motor neurons of patients who have sporadic ALS compared to that in control subjects [20,21]. The hypoediting of the Q/R site in ALS patients has been attributed to downregulation of ADAR2, which was observed in the motor neurons presenting with under-edited GluA2 mRNA [22].
To further investigate the role of ADAR2 and AMPA receptors in motor neuron death, the Cre/loxP system was used to conditionally target the ADAR2 gene in the motor neurons of mice [23]. These mice developed ALS-like progressive loss of motor neuron function [23]. Additionally, ADAR2 null mice carrying alleles engineered to express edited GluA2 subunits exhibited normal phenotypes [23]. This finding confirms that the motor neuron death observed in ADAR2 null mice is primarily caused by the loss of editing of GluA2.
The human research suggests a connection between the motor neuron death of sporadic ALS and ADAR2-mediated editing of GluA2. The mice studies established a clear connection between ADAR2-mediated editing of GluA2 and motor neuron death. Taken together, AMPA-mediated excitotoxicity related to ADAR2 regulation is one of the most promising hypotheses for the etiology of sporadic ALS. This hypothesis was strengthened by recent research demonstrating that prevention of ADAR2-editing of GluA2 in mouse motor neurons results in the mislocation of transaction response DNA-binding protein 43, an abnormality often observed in sporadic ALS patients [24]. The cause of downregulation of ADAR2 in ALS patients is still unknown, and it remains to be seen if upregulation of ADAR2 is sufficient to slow or reverse the effects of the disease.
Astrocytoma
Astrocytoma is a brain cancer of the astrocytes, which are star-shaped glial cells [25]. Glioblastoma multiforme (GBM) is a grade IV astrocytoma, the most severe grade of malignancy, and is a particularly invasive tumor that is usually fatal within 18 months [26].
Analysis of GBM tissue samples revealed that the GluA2 Q/R site was significantly under-edited compared to that of control samples [27]. In a study focusing on RNA editing in pediatric astrocytomas, a comparison between control tissues and tumor tissues from astrocytomas (grade I to grade IV) found that decreased ADAR2 activity correlated with increased malignancy, although ADAR2 expression levels were unchanged [28]. The same study showed that ADAR1 expression was upregulated as malignancy increased, and that the inactivation of ADAR2 can be explained by the formation of inactive ADAR1-ADAR2 heterodimers resulting from overexpression of ADAR1 [28]. Furthermore, the overexpression of ADAR2 in various human astrocytoma cell lines (U118, A172 and U87) was able to reduce cellular proliferation and migration significantly [28].
Hypoediting of the GluA2 subunit causes an increased influx of Ca2+. In GBM, the Ca2+ influx mediated by AMPA receptors has been shown to activate Akt by phosphorylation, thereby promoting proliferation and mobility [29]. The upregulation of the Akt pathway is reversed when the edited GluA2 is expressed [29]. This research provides a preliminary mechanism by which decreased ADAR2 activity corresponds to the level of astrocytoma malignancy.
A recent study aimed to better characterize the effect of ADAR2-mediated editing on astrocytoma proliferation and mobility [30]. The initial finding was that increased expression of catalytically active ADAR2 inhibits tumor growth in mice when compared to increased expression of inactive ADAR2 or controls [30]. The study identified the phosphatase CDC14B as an important substrate for ADAR2. CDC14B pre-mRNA is edited by ADAR2 at several non-coding sites, and this editing (by an unknown mechanism) appears to promote expression of CDC14B [30]. The upregulation of CDC14B promoted the degradation of the substrate Skp2, which in turn upregulated proteins p21 and p27 [30]. Increased levels of p21 and p27 act to prevent the cell cycle transition to the S phase [30]. The effect of ADAR2 on the CDC14B-Skt2-p21-p27 pathway provides an alternative explanation for both the tumor-suppressor abilities of ADAR2 and the correlation between ADAR2 inactivity and the level of astrocytoma malignancy.
In summary, ADAR2 editing activity exhibits tumor-suppressor capabilities and has been widely observed to be decreased in astrocytoma tumor tissue. The precise mechanism by which ADAR2 influences tumor growth has not been determined, and it is likely that there are multiple editing events that are responsible for the observed effect. ADAR2 should be considered as a target for future research aimed at identifying novel approaches for treating GBM.
Transient forebrain ischemia
Transient forebrain ischemia is characterized by reduced blood flow following an interruption of circulation, such as cardiac arrest. CA1 pyramidal neurons are damaged following ischemia due to increased Ca2+ influx [31]. This increased influx has been attributed to increased Ca2+ permeability of AMPA receptors caused by decreased ADAR2-mediated GluA2 editing [31]. The hypoedited GluA2 is a consequence of a reduction in ADAR2 expression [32]. The Ca2+ influx results in CA1 pyramidal neuron death induced by the phosphorylation of N-methyl-D-aspartate (NMDA) receptors by Cdk5 [31]. Following induced ischemia, transfection with edited GluA2 is sufficient to prevent Cdk5 activation and cell death in CA1 pyramidal neurons in rats [31]. Similarly, overexpression of ADAR2 protects CA1 pyramidal neurons from ischemic injury [32]. Finally, a knockdown of ADAR2 was found to induce ischemic degeneration of CA1 pyramidal neurons, an effect that was rescued by the introduction of a fully edited GluA2 gene [32].
This research provides a convincing explanation for the neuronal damage observed following transient forebrain ischemia. AMPA and NMDA receptors have long been associated with ischemic injury. Understanding the role of ADAR2-mediated RNA editing in the neuronal damage pathway may prove essential in developing a specific therapy that minimizes side effects in uninjured neurons.
A-to-I editing of serotonin receptors
Serotonin receptors are a family of receptors that are considered to be major contributors to many psychological and behavioral processes, as well as to mental illnesses [33]. The hydroxytryptamine subtype 2C receptor (5-HT2CR) is a G-protein-coupled serotonin receptor abundant in the CNS that has been associated with dopamine modulation as well as with appetite, energy consumption and anxiety [34,35]. This receptor is activated by serotonin and subsequently increases phospholipase C activity [35]. The human 5-HT2CR has high sequence homology with its mouse counterpart and is the only serotonin receptor known to undergo A-to-I editing [35]. The 5-HT2CR pre-mRNA is edited by ADARs at five conserved sites, named sites A to E (Figure 3) [36]. Sites A and B are edited preferentially by ADAR1, whereas the editing of site D is mediated specifically by ADAR2 [37]. Sites C and E are edited efficiently by both ADAR1 and ADAR2 [36]. Editing of the five sites can result in amino acid exchanges in three positions, 156, 158 and 160. Theoretically, 24 protein isoforms are possible as the result of varying degrees of ADAR-mediated editing.
Figure 3.
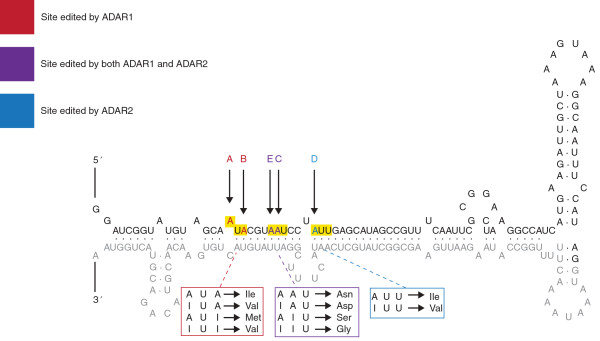
ADAR-mediated editing sites of the mammalian 5-HT2CR, serotonin receptor. In the 5-HT2CR pre-mRNA, exon 5 (black text) is bound to the untranslated intron 5 (gray text) and contains ADAR-mediated editing sites A to E (colored in red, blue or purple). Sites A and B (red) are edited selectively by ADAR1 and can produce four codons that encode three distinct amino acids (red box). Site D (blue) is edited preferentially by ADAR2 and can produce two codons that encode two distinct amino acids (blue box). Sites C and E (purple) can be edited by either ADAR1 or ADAR2 and can produce four codons that encode four distinct amino acids (purple box). Editing of these sites can cause various amino acid substitutions, detailed in boxes below the RNA segment. Editing of these sites affects G-protein coupling and serotonin sensitivity. Modified from [36] with permission (RNA Biology).
In vivo studies on mice have detected 12 distinct isoforms, with eight variants observed at significant levels [38]. Editing of 5-HT2CR significantly reduces serotonin potency and G-protein coupling, demonstrating a level of regulation of receptor sensitivity [39-41]. One study analyzed mice expressing either only the fully unedited form of 5-HT2CR (Ile-Asn-Ile) or only the fully edited form (Val-Gly-Val) [42]. Mice expressing the unedited serotonin receptors developed normally, but mice expressing the edited version presented with several abnormalities, including growth retardation and hyperphagia. Additionally, these mice exhibited significantly decreased fat mass and increased energy expenditure, corresponding to hyperactivation of the sympathetic nervous system [42]. These results suggest a regulatory role for 5-HT2CR RNA editing in certain metabolic pathways.
Recent research has investigated associations between patterns of 5-HT2CR RNA editing and various human diseases.
Prader-Willi syndrome
Prader-Willi syndrome (PWS) is a condition thought to be caused by a deletion or mutation of the paternal chromosome 15q11-q13, and is often characterized by life-threatening obesity and a range of developmental abnormalities [43]. PWS is associated with decreased levels or the complete absence of several small nucleolar RNAs located within the PWS locus [43]. Among these is MBII-52, which has a sequence that complements the C-site of 5-HT2CR RNA and acts to decrease C-site editing [3]. Thus, PWS is often marked by increased editing of 5-HT2CR. Mice expressing fully edited 5-HT2CR isoforms present with PWS-like symptoms, including hyperphagia and hypotonia [44].
This research demonstrated that ADAR-mediated editing of 5-HT2CR contributes to the PWS phenotype, but it is worth noting that the transgenic mice did not exhibit the complete spectrum of PWS-associated symptoms, specifically morbid obesity. In mice, increased levels of 5-HT2CR RNA editing have been associated with increased energy expenditure and decreased body mass [42]. One explanation for this disparity is that changes in the editing of 5-HT2CR is just one of multiple contributors to PWS onset [45]. Additionally, no PWS mouse model has achieved a disease phenotype identical to that seen in human PWS patients, which suggests that there are species-specific differences that prevent mice from exhibiting the full human PWS phenotype [44].
Psychiatric disorders
Despite significantly increased research attention, the pathologies of many psychiatric disorders remain difficult to study and are poorly understood. Editing patterns of 5-HT2CR have been associated with a number of psychiatric disorders, including anxiety, depression, bipolar disorder and schizophrenia [3]. It is often difficult to diagnose and gauge the severity of many of these conditions reliably. This obstacle, combined with the reliance of research on human postmortem brain studies that are plagued by small samples and confounding variables, has resulted in inconclusive evidence regarding the relationship between RNA editing and psychiatric disorders [46].
One study found no differences in 5-HT2CR editing when comparing schizophrenic, depressed and control subjects, but there was a significant correlation between suicidal behavior (independent of psychiatric diagnosis) and hyperediting of 5-HT2CR at the A site [47]. A similar study found no significant differences in the 5-HT2CR editing patterns of individuals diagnosed with depression, schizophrenia, bipolar disorder or substance abuse [48]. This study also found an association between individuals who committed suicide and increased ADAR-mediated editing of 5-HT2CR, reporting hyperediting of sites A, B, C and D [48]. Another study looked at suicide victims who had a history of major depression and found increased editing of the C and E sites, whereas the D site was found to show decreased editing [49]. An interesting finding from this study is that mice treated with the antidepressant drug fluoxetine, marketed as Prozac™, exhibited editing patterns exactly opposite to those exhibited in the suicide victims [49].
There is significant discrepancy among different studies aiming to analyze patterns of ADAR-mediated editing of 5-HT2CR and, as mentioned previously, there are numerous obstacles that may obscure otherwise clearer results. Nevertheless, there appears to be a trend of increased editing of 5-HT2CR, which is associated with decreased sensitivity of the receptor, in the case of suicide victims. Suicide is often a complication of a psychiatric disorder, and thus the research suggests that 5-HT2CR editing and 5-HT2CR sensitivity play a role in the pathology of psychiatric disorders [50].
Mutations in ADAR1
Owing to the large number of ADAR substrates and the high conservation of ADARs, mutations in ADAR genes would be expected to cause various abnormal phenotypes. Proper function of ADAR1, the most prevalent ADAR enzyme, is essential for proper development in mammals. In mice, a homozygous null mutation of the ADAR1 gene results in widespread apoptosis and embryo death [51]. In humans, the ADAR1 gene is located on chromosome 1q21 and at least two diseases associated with mutations in ADAR1 have been identified.
Dyschromatosis symmetrica hereditaria
Dyschromatosis symmetrica hereditaria (DSH) is a rare genetic pigmentation disorder characterized by hyperpigmented and hypopigmented macules on the extremities [52]. The cause of DSH has been identified as mutations in the ADAR1 gene [52]. Over the course of the past decade, at least 131 mutations in the coding portion of the ADAR1 gene have been identified in individuals with DSH (Figure 4) [53-55]. DSH exhibits autosomal dominant inheritance, which is probably explained by a dominant-negative effect of mutant ADAR1 [36]. The dominant-negative effect hypothesis predicts that a mutant ADAR1 can impair wild-type ADAR1 activity by forming a dysfunctional dimer complex. The pathology of DSH is not well understood and relevant ADAR1-editing targets have yet to be identified.
Figure 4.

Mutations in the ADAR1 gene that are associated with human disease. The ADAR1 protein contains several distinct domains (labeled and colored in green, purple, or blue). A number of nucleotide mutations of the ADAR1 gene have been associated with human diseases. There are 130 mutations associated with dyschromatosis symmetrica hereditaria (blue), eight mutations associated with Aicardi-Goutières syndrome (red), and 1 mutation associated with both conditions (black and bold). Many of these mutations occur within key domain regions (indicated by lines from each domain to the first and the last mutation falling within that domain), with the majority of the mutations occurring in the catalytic deaminase domain. dsRNA, double-stranded RNA.
Aicardi-Goutières syndrome
Aicardi-Goutières syndrome (AGS) is a rare hereditary neurological disorder characterized by inflammation of the brain and skin. A number of causative genes have been previously identified, such as RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and TREX1[56-58]. By sequencing ADAR1 in patients diagnosed with AGS but lacking any known AGS-linked mutations, nine mutations of ADAR1 were identified as potential causes of AGS, with eight of the mutations previously unidentified (Figure 4) [53]. AGS is considered an autoimmune disease and has been associated with elevated levels of an interferon immune response initiated by interferon-stimulatory nucleic acid of an unknown source [53]. As ADAR1 has been shown to suppress type I interferon signaling, ADAR1 mutations have been proposed as a cause of AGS and a possible source of interferon-stimulatory nucleic acid [53].
ADAR1 is associated with at least one other interferon-related autoimmune disease. As reviewed by Gallo and Locatelli [45], over-expression of ADAR1, as well as increased ADAR1 activity, is observed in individuals suffering from systemic lupus erythematosus. ADAR1 is considered essential for mitigating the effects of over-activation of the interferon response [59]. Future research is needed to determine whether ADAR1 plays a similar mitigating role in other autoimmune disorders.
Other diseases associated with A-to-I editing
There are many additional diseases that have been studied in connection with ADARs. In this section, we focus on a few of those that are currently being studied that fall outside the mechanisms previously discussed. Specifically, we discuss examples of A-to-I editing in cancer, metabolic diseases and viral infections. Last, we focus on recently established connections between ADARs and microRNAs.
Hepatocellular carcinoma
ADAR enzymes have recently emerged as a target in cancer research. Many cancers are thought to arise as the compounding effect of several minor mutations and abnormalities. Aberrant regulation of ADAR-mediated editing of coding and noncoding RNA has been associated with many types of cancer. In addition to those discussed in this review, breast cancer, bladder cancer, prostate cancer, neuroblastoma, Kaposi’s sarcoma, acute leukemia, metastatic melanoma and chronic myeloid leukemia have all been studied in connection with A-to-I RNA editing [25,60,61]. The role of ADARs in carcinogenesis was recently reviewed by Galeano et al.[25].
Human hepatocellular carcinoma (HCC) is one of the most common causes of cancer-related deaths, and HCC pathogenesis is not well defined. Chen et al.[62] recently made significant progress toward identifying a molecular pathway for HCC pathogenesis. Transcriptome sequencing found that pre-mRNA transcripts of the AZIN1 gene underwent increased levels of A-to-I editing in HCC tissue samples. AZIN1 encodes antizyme inhibitor 1, a specific inhibitor of the growth-regulating protein antizyme. Antizyme induces the degradation of ODC and CCND1, two proteins that promote cell growth. For this reason, antizyme is a tumor-suppressing protein, whereas AZIN1 has the potential to promote tumorigenesis [62].
The level of editing of AZIN1 in HCC tissue samples was roughly correlated with severity of prognosis. ADAR1 was found to be upregulated in HCC tissue and to mediate AZIN1 editing specifically. HCC cells were transduced to express different ratios of edited to unedited AZIN1, and cells expressing higher percentages of edited AZIN1 exhibited higher rates of proliferation and colony formation [62].
ADAR1-mediated editing of AZIN1 results in an amino acid substitution of a glycine residue for a serine residue at position 367. Computational analysis of protein conformations predicted that the amino acid conversion occurred within a ‘switch’ region capable of inducing conformational change. Furthermore, edited AZIN1 was observed to have significantly higher rates of nuclear localization than the unedited form, which is primarily localized in the cytoplasm. In addition to altered localization, edited AZIN1 exhibited a higher binding affinity for antizyme than the unedited form and, consequently, was a more potent inhibitor of antizyme [62].
This study is important for many reasons. Most significantly, the authors presented convincing evidence for a molecular mechanism of HCC that undoubtedly will be the focus of future research aimed at improving HCC therapy. It is important to note that this mechanism suggests that hyperediting can promote tumor growth, and therefore contrasts with the conventional paradigm of problematic hypoediting that has been identified in a number of cancer phenotypes [63]. Additionally, it illustrates the complexity of many RNA editing-mediated processes. It is also indicative of how increased research efforts and improving sequencing technologies will greatly deepen our understanding of RNA-editing pathways.
Diabetes mellitus
Type 2 diabetes mellitus (T2DM) is a metabolic disease characterized by abnormal secretion or uptake of insulin that affects over 330 million people worldwide [64]. The secretion of insulin by pancreatic β cells in response to glucose levels in the blood is understood to play a pivotal role in the pathology of T2DM [64].
It has been demonstrated that ADAR2 mRNA expression and ADAR2-mediated editing in pancreatic β cells are upregulated in response to increases in glucose concentration [65]. It has also been reported that ADAR2 mediates insulin secretion following glucose stimulation, and that selectively knocking down ADAR2 impairs the secretion of insulin, an effect rescued by catalytically active ADAR2 [66]. This suggests that the editing activity of ADAR2 is required for normal secretion of insulin by glucose-stimulated pancreatic β cells.
The molecular pathway that links ADAR2 to glucose-stimulated insulin secretion in pancreatic β cells remains unknown. It has been observed that, in addition to having impaired insulin secretion, ADAR2-deficient pancreatic β cells exhibited decreased levels of expression of Munc18-1 and synaptotagmin-7, two regulators of exocytosis [66]. Alternatively, insulin secretion in pancreatic β cells is known to require increases in intracellular calcium concentrations [64]. ADAR2 mediates A-to-I editing of mRNA transcripts of both glutamate-dependent and voltage-dependent calcium channels. Additional research is required to determine if ADAR2-mediated changes in calcium concentrations play a role in insulin secretion.
Persistent measles virus infection
ADAR1 p150 is induced by interferon activation, which often occurs in response to a pathogen. For this reason, ADAR1 would be expected to exhibit antiviral activity. Indeed, ADAR1 has been implicated as an antiviral factor in a few diseases, such as those caused by the hepatitis C virus, lymphocytic choriomeningitis virus and polyomavirus [67]. Nevertheless, for many viral pathogens, including the influenza virus, measles virus, human immunodeficiency virus-1 and many others, ADAR1 has been shown to enhance viral replication and proliferation [67]. ADARs affect viral replication by either direct editing of viral RNA or inhibiting RNA-activated protein kinase (PKR) [68]. These two mechanisms can operate either independently or in concert. The effect of ADAR-editing on viral replication has been recently reviewed by Gélinas et al. [68] and by Samuel [69].
Measles virus is a highly contagious paramyxovirus that causes a respiratory system infection in humans [70]. In rare cases, the viral infection can develop into a persistent infection of the CNS, presenting as fatal subacute sclerosing panencephalitis (SSPE) [70]. Analysis of the viral genome isolated from patients diagnosed with SSPE revealed that the measles matrix gene, encoding a key protein for viral assembly, had undergone biased hypermutation [71]. Nearly half of the adenosines of this gene were mutated to guanosine, which is indicative of high levels of A-to-I editing [71,72]. This study found that the hypermutation of the matrix gene prevented its translation [71], and the authors concluded that the lack of a matrix gene prolonged the viral infection of the CNS and interfered with immune system recognition [71]. Another study compared wild-type measles virus and the hypermutated measles virus of SSPE in mice and in primary neuron cultures [73]. The authors of this work concluded that the ADAR-mediated mutations actively contributed to the pathogenesis of the CNS disease [73].
HeLa cells have been studied to analyze the impact of ADAR1 on the antiviral response following measles virus infection [74]. Compared to wild-type HeLa cells, ADAR1-knockdown HeLa cells infected with strains of measles virus exhibited a decreased rate of viral growth [74]. Additionally, the knockdown cells had significantly higher rates of apoptosis following viral infection [74]. The increased apoptosis was correlated with increased activation of PKR and interferon regulatory transcription factor-3, proapoptotic proteins associated with innate antiviral responses [74]. This study demonstrated that ADAR1, despite being stimulated by interferon activation, contributes to prolonged measles virus infections through proviral, antiapoptotic activity.
ADARs and microRNAs
miRNAs are approximately 22-nucleotide RNA sequences that regulate gene expression as part of the RNA interference (RNAi) process. miRNAs can bind to a complementary sequence of an untranslated region of target mRNA, and block translation or promote degradation. Over the past two decades, research has shown that miRNAs affect a wide range of mRNA transcripts and has implicated miRNAs in a number of human diseases [60,75-77]. More recently, it was demonstrated that miRNAs can undergo A-to-I editing [78,79]. Editing of a miRNA transcript has a number of potential outcomes. An extensively edited miRNA sequence can be targeted for degradation by ribonucleases that are specific for inosine-containing RNA [80]. The most likely result of A-to-I conversions is interference with miRNA processing, preventing the formation of mature miRNAs and blocking their activity [79]. Also, editing of a miRNA can affect base-pairing interactions and can redirect the miRNA, ultimately changing the target mRNA sequence [5].
At least one example of miRNA redirection by ADAR-mediated editing has been identified [81]. Mice and humans share a similar miR-376 cluster of miRNA genes that is processed into three and six miRNAs in mice and humans, respectively, and all these miRNAs have high sequence homology [81]. ADAR2 is able to edit miR-376a, a miRNA from the mouse miR-376 cluster, at a site located in the 5’-proximal ‘seed’ region, a region critical for miRNA hybridization [81]. The unedited and edited forms of miR-376a exist in different proportions, varying in a tissue-specific manner [81]. Analysis of potential target genes for unedited miR-376a and edited miR-376a identified 78 and 82 matches, respectively, with only two overlapping targets [81]. A switch-of-function was demonstrated in vivo, through which the editing of miR-376a was able to regulate uric acid synthesis [81]. A number of human diseases are associated with aberrant regulation of uric acid levels, and this recently discovered ADAR2 activity may contribute to such conditions [5,81]. It is important to note that the uric acid regulation was just an effect of one of the target genes of the edited miR-376a. It is anticipated that many such examples of RNA editing-mediated redirection of miRNAs have yet to be identified.
A recent study investigated the role of ADAR1 in RNAi pathways [82]. ADAR1 was found to have direct protein-protein interactions with Dicer, an endoribonuclease critical for miRNA processing [82]. Additionally, the presence of ADAR1 was able to significantly increase the rate of miRNA processing [82]. The study also found that miRNA expression was globally inhibited in ADAR1-knockout mouse embryos [82]. The resulting changes in the expression of genes that are targeted by miRNAs may contribute to the lethality of the ADAR1 null phenotype [82].
A study by Nemlich et al.[60] demonstrated both the importance and the complexity of the relation between ADARs and RNAi. The study found that ADAR1 expression is often reduced in metastatic melanomas and that knocking down ADAR1 in vivo enhanced tumorigenicity [60]. By using microarray analysis, the authors were able to identify many miRNAs controlled by ADAR1 that regulate genes that are related to the observed phenotypes [60]. They also demonstrated that ADAR1 regulates miRNA processing by controlling Dicer expression using let-7 miRNAs and by binding to DGCR8, which prevents the formation of a DGCR8-Drosha complex [60]. The Nemlich et al. study concluded that the observed tumor-suppressing effect of ADAR1 in metastatic melanomas results from this editing-independent miRNA processing regulation [60]. They further demonstrated that two ADAR1-targeting miRNAs, miR-17 and miR-432, are overexpressed in melanoma and serve as a mechanism through which ADAR1 is silenced in cancer cells [60]. These findings can likely be applied to other types of cancer in order to demonstrate the significance of the recently established connection between ADAR1 and miRNA processing.
Taken together, these findings show that ADARs are key enzymes in the RNAi pathway, and highlight the extent of the regulatory roles of ADARs.
Conclusions and future directions
It has been established that changes in A-to-I editing are involved in a range of human diseases. We have discussed disease pathologies associated with altered editing of specific ADAR-substrates, changes in ADAR expression, and mutations of ADAR genes. Many of these ADAR-disease connections have been made recently, and the causes of the changes in ADAR expression or ADAR activity are generally not well understood. Even where the pathological pathway appears to be identified clearly, such as in the study on ADAR1 and HCC, it is expected that countless other ADAR-related pathways play lesser but still important roles [62]. In addition, there are several diseases for which interesting correlations with aberrant ADAR editing have been demonstrated but no proposed mechanism exists. One such example is a recent study that observed that patients who had been diagnosed with autism were more likely to exhibit abnormally high or low A-to-I editing of the analyzed synaptic genes [83].
ADAR editing of coding RNA regions can be studied in a relatively straightforward manner. As a result, much of the research associating ADARs with human diseases involves editing events in coding regions. Nevertheless, the majority of A-to-I editing events occur in noncoding regions, which are far more difficult to study in vivo. ADARs have been shown to edit miRNAs and are expected to contribute to the regulation of various cellular processes by influencing RNAi pathways. Editing events that affect repetitive RNA sequences or other noncoding regions generally serve an unknown function, and it remains to be seen if they play a role in the pathology of human diseases. Research investigating A-to-I editing of noncoding RNAs has benefited and will continue to benefit from improving high-throughput sequencing technologies. It will be interesting to see if this research yields insight into the regulatory capabilities of ADARs that might explain why ADARs are highly conserved and essential for life in mammals.
ADARs edit such a large number of RNA substrates that ADAR activity seems likely to be involved, directly or indirectly, in many cellular processes. The implications of this are two-fold. First, much more research is needed to provide a better understanding of the pathways mediated by ADAR editing, some of which play crucial roles in the pathology of human diseases. Second, potential therapies that specifically target ADAR enzymes may not be feasible because of unintended side effects, as the functions of the vast majority of such editing events have not been determined. Conversely, viable therapies are more likely to target specific ADAR substrates. A novel study recently demonstrated the potential of specific RNA-interacting proteins to act as substrate-specific inhibitors of ADAR enzymes [84]. This highlights the importance of improving our understanding of the molecular pathways involved in ADAR-related human diseases. Continued research efforts and advances in sequencing technology are necessary for such improvement, and ultimately for the development of viable therapeutic strategies.
Abbreviations
5-HT2CR: 5-hydroxytryptamine subtype 2C receptor; ADAR: Adenosine deaminase acting on RNA; AGS: Aicardi-goutières syndrome; ALS: Amyotrophic lateral sclerosis; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; A-to-I: Adenosine to inosine; CNS: Central nervous system; DSH: Dyschromatosis symmetrica hereditaria; GBM: Glioblastoma multiforme; HCC: Hepatocellular carcinoma; miRNA: microRNA; NMDA: N-methyl-D-aspartate; PKR: RNA-activated protein kinase; PWS: Prader-Willi syndrome; RNAi: RNA interference; SSPE: Subacute sclerosing panencephalitis; T2DM: Type 2 diabetes mellitus.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
William Slotkin, Email: wslotkin@alumni.upenn.edu.
Kazuko Nishikura, Email: kazuko@wistar.org.
Acknowledgements
We thank Ellyn Barkley for her assistance with figures. We are grateful for grant support provided by the National Institutes of Health (GM040536, HL009342 and P30 CA010815), the Ellison Medical Foundation (AG-55-2281-09), and the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health to KN and by the Vagelos Scholars Program in Molecular Life Sciences to WS.
References
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;5:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;5:437–449. doi: 10.1016/S0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Tariq A, Jantsch MF. Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front Neurosci. 2012;5:99. doi: 10.3389/fnins.2012.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA. The ADAR protein family. Genome Biol. 2012;5:252. doi: 10.1186/gb-2012-13-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. The Biomedical and Life Sciences Collection. Henry Stewart Talks Ltd; 2013. A-to-I RNA editing in human disease. http://hstalks.com/main/view_talk.php?t=2475&c=252. [Google Scholar]
- Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;5:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov D, Crnogorac-Jurcević T, Clark M, Gardiner K. Comparative analysis of the DRADA A-to-I RNA editing gene from mammals, pufferfish and zebrafish. Gene. 2000;5:53–60. doi: 10.1016/S0378-1119(00)00175-X. [DOI] [PubMed] [Google Scholar]
- Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;5:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;5:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- Cho D-SC, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem. 2003;5:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J Biol Chem. 2007;5:16054–16061. doi: 10.1074/jbc.M611392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A, Vissel B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci. 2012;5:34. doi: 10.3389/fnmol.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;5:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;5:11–19. doi: 10.1016/0092-8674(91)90568-J. [DOI] [PubMed] [Google Scholar]
- Kung SS, Chen YC, Lin WH, Chen CC, Chow WY. Q/R RNA editing of the AMPA receptor subunit 2 (GRIA2) transcript evolves no later than the appearance of cartilaginous fishes. FEBS Lett. 2001;5:277–281. doi: 10.1016/S0014-5793(01)03183-0. [DOI] [PubMed] [Google Scholar]
- Kwak S, Hideyama T, Yamashita T, Aizawa H. AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology. 2010;5:182–188. doi: 10.1111/j.1440-1789.2009.01090.x. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;5:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh D-S, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;5:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;5:S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Kwak S, Sun H, Ito K, Hashida H, Aizawa H, Jeong S-Y, Kanazawa I. Human spinal motoneurons express low relative abundance of GluR2 mRNA: an implication for excitotoxicity in ALS. J Neurochem. 2003;5:680–689. doi: 10.1046/j.1471-4159.2003.01703.x. [DOI] [PubMed] [Google Scholar]
- Kwak S, Kawahara Y. Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J Mol Med. 2005;5:110–120. doi: 10.1007/s00109-004-0599-z. [DOI] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, Kwak S. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012;5:1121–1128. doi: 10.1016/j.nbd.2011.12.033. [DOI] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci. 2010;5:11917–11925. doi: 10.1523/JNEUROSCI.2021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, Saido TC, Kwak S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012;5:1307. doi: 10.1038/ncomms2303. [DOI] [PubMed] [Google Scholar]
- Galeano F, Tomaselli S, Locatelli F, Gallo A. A-to-I RNA editing: The ‘ADAR’ side of human cancer. Semin Cell Dev Biol. 2012;5:244–250. doi: 10.1016/j.semcdb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Stupp R, Bent M, Hegi M. Optimal role of temozolomide in the treatment of malignant gliomas. Curr Neurol Neurosci Rep. 2005;5:198–206. doi: 10.1007/s11910-005-0047-7. [DOI] [PubMed] [Google Scholar]
- Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. 2001;5:14687–14692. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Rocco CD, O’Connell MA, Gallo A. Down-regulation of RNA editing in pediatric astrocytomas ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;5:7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T, Saito N, Tsuzuki K, Okado H, Miwa A, Nakazato Y, Ozawa S. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;5:7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M, Boldrini R, Massimi L, Di Rocco CM, Locatelli F, Gallo A. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene. 2013;5:998–1009. doi: 10.1038/onc.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Lau L, Wei J, Zhu D, Zou S, Sun H-S, Fu Y, Liu F, Lu Y. Expression of Ca2+-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron. 2004;5:43–55. doi: 10.1016/j.neuron.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Peng PL, Zhong X, Tu W, Soundarapandian MM, Molner P, Zhu D, Lau L, Liu S, Liu F, Lu Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;5:719–733. doi: 10.1016/j.neuron.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Hood JL, Emeson RB. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr Top Microbiol Immunol. 2012;5:61–90. doi: 10.1007/82_2011_157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti M, Tecott LH. Contributions of 5-HT2C receptors to multiple actions of central serotonin systems. Eur J Pharmacol. 2004;5:1–9. doi: 10.1016/j.ejphar.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacol. 1999;5:1083–1152. doi: 10.1016/S0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006;5:1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;5:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Ensterö M, Öhman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;5:978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;5:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem. 1999;5:9472–9478. doi: 10.1074/jbc.274.14.9472. [DOI] [PubMed] [Google Scholar]
- Price RD, Weiner DM, Chang MSS, Sanders-Bush E. RNA editing of the human serotonin 5-HT2C receptor alters receptor-mediated activation of G13 protein. J Biol Chem. 2001;5:44663–44668. doi: 10.1074/jbc.M106745200. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Grimberg A, Teegarden S, Mombereau C, Liu S, Bale TL, Blendy JA, Nishikura K. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci. 2008;5:12834–12844. doi: 10.1523/JNEUROSCI.3896-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG. Prader-Willi syndrome: obesity due to genomic imprinting. Curr Genomics. 2011;5:204–215. doi: 10.2174/138920211795677877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, Hachey DL, Roth BL, Emeson RB. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol Dis. 2010;5:169–180. doi: 10.1016/j.nbd.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Locatelli F. ADARs: allies or enemies? The importance of A-to-I RNA editing in human disease: from cancer to HIV-1. Biol Rev. 2012;5:95–110. doi: 10.1111/j.1469-185X.2011.00186.x. [DOI] [PubMed] [Google Scholar]
- O’Neil RT, Emeson RB. Quantitative analysis of 5HT2C receptor RNA editing patterns in psychiatric disorders. Neurobiol Dis. 2012;5:8–13. doi: 10.1016/j.nbd.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-HT2C receptor: alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacol. 2001;5:478–491. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- Dracheva S, Patel N, Woo DA, Marcus SM, Siever LJ, Haroutunian V. Increased serotonin 2C receptor mRNA editing: a possible risk factor for suicide. Mol Psychiatry. 2007;5:1001–1010. doi: 10.1038/sj.mp.4002081. [DOI] [PubMed] [Google Scholar]
- Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;5:349–356. doi: 10.1016/S0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- Mann JJ. Neurobiology of suicidal behaviour. Nat Rev Neurosci. 2003;5:819–828. doi: 10.1038/nrn1220. [DOI] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;5:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;5:693–699. doi: 10.1086/378209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, Laet CD, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla À, Heiberg A, Kawaguchi M, Kumar R, Lin J-PS-M, Lourenco CM, Male AM, Marques W Jr, Mignot C. et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;5:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai ML, Yang LJ, Zhu XH, Li M. A novel mutation of the DSRAD gene in a Chinese family with dyschromatosis symmetrica hereditaria. Genet Mol Res. 2012;5:1731–1737. doi: 10.4238/2012.June.29.5. [DOI] [PubMed] [Google Scholar]
- Kono M, Ito Y, Ujiie H, Morimoto K, Akiyama M. Novel ADAR1 mutations including single amino acid deletion in the deaminase domain underly dyschromatosis symmetrica hereditaria. J Dermatol Sci. 2013;5:e57. doi: 10.1111/j.1365-4632.2012.05765.x. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Déry C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martínez-Frías ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW. et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;5:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the I locus. Nat Genet. 2006;5:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BCJ, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L. et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;5:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;5:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, Jacoby E, Eyal E, Rivkin L, Prieto VG, Chakravarti N, Duncan LM, Kallenberg DM, Galun E, Bennett DC, Amariglio N, Bar-Eli M, Schachter J, Rechavi G, Markel G. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest. 2013;5:2703–2718. doi: 10.1172/JCI62980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Barrett CL, Chun H-J, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao K-HT, Morris SR, Goldstein LSB, Marra MA, Frazer KA, Jamieson CHM. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;5:1041–1046. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li Y, Lin CH, Chan THM, Chow RKK, Song Y, Liu M, Yuan Y-F, Fu L, Kong KL, Qi L, Li Y, Zhang N, Tong AHY, Kwong DL-W, Man K, Lo CM, Lok S, Tenen DG, Guan X-Y. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;5:209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, Barbash ZS, Adamsky K, Safran M, Hirschberg A, Krupsky M, Ben-Dov I, Cazacu S, Mikkelsen T, Brodie C, Eisenberg E, Rechavi G. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;5:1586–1595. doi: 10.1101/gr.6493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Diabetes mellitus and the β Cell: the last ten years. Cell. 2012;5:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z, Zhao L, Yang L, Huang P, Zhao F, Li W, Liu Y. RNA editing by ADAR2 is metabolically regulated in pancreatic islets and β-cells. J Biol Chem. 2006;5:33386–33394. doi: 10.1074/jbc.M604484200. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao L, Gan Z, He Z, Xu J, Gao X, Wang X, Han W, Chen L, Xu T, Li W, Liu Y. Deficiency in RNA editing enzyme ADAR2 impairs regulated exocytosis. FASEB J. 2010;5:3720–3732. doi: 10.1096/fj.09-152363. [DOI] [PubMed] [Google Scholar]
- Samuel CE. ADARs: viruses and innate immunity. Curr Top Microbiol Immunol. 2012;5:163–195. doi: 10.1007/82_2011_148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gélinas J-F, Clerzius G, Shaw E, Gatignol A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J Virol. 2011;5:8460–8466. doi: 10.1128/JVI.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;5:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R, Billeter MA. In: Genetic Diversity of RNA Viruses. Holland JJ, editor. Berlin, Heidelberg: Springer; 1992. Mutations and A/I hypermutations in measles virus persistent infections; pp. 63–74. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, Billeter MA. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;5:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H, Cattaneo R, Billeter MA. Biased hypermutation of viral RNA genomes could be due to unwinding/modification of double-stranded RNA. Cell. 1989;5:331. doi: 10.1016/0092-8674(89)90234-1. [DOI] [PubMed] [Google Scholar]
- Patterson JB, Cornu TI, Redwine J, Dales S, Lewicki H, Holz A, Thomas D, Billeter MA, Oldstone MBA. Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology. 2001;5:215–225. doi: 10.1006/viro.2001.1182. [DOI] [PubMed] [Google Scholar]
- Toth AM, Li Z, Cattaneo R, Samuel CE. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem. 2009;5:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the Human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;5:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;5:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu C-G, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;5:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. RNA editing of human microRNAs. Genome Biol. 2006;5:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;5:5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López J, Hourcade Jde D, del Mazo J. Reprogramming of microRNAs by adenosine-to-inosine editing and the selective elimination of edited microRNA precursors in mouse oocytes and preimplantation embryos. Nucleic Acids Res. 2013;5:5483–5493. doi: 10.1093/nar/gkt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by Adenosine-to-Inosine editing of miRNAs. Science. 2007;5:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff B-E, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;5:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eran A, Li JB, Vatalaro K, McCarthy J, Rahimov F, Collins C, Markianos K, Margulies DM, Brown EN, Calvo SE, Kohane IS, Kunkel LM. Comparative RNA editing in autistic and neurotypical cerebella. Mol Psychiatry. 2013;5:1041–1048. doi: 10.1038/mp.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res. 2013;5:2581–2593. doi: 10.1093/nar/gks1353. [DOI] [PMC free article] [PubMed] [Google Scholar]