

Abstract
Recent advances in whole-genome technologies have supplied the field of cancer research with an overwhelming amount of molecular data. Improvements in massively parallel sequencing approaches have led to logarithmic decreases in costs, and so these methods are becoming almost commonplace in the analysis of clinical trials and other cohorts of interest. Furthermore, whole-transcriptome quantification by RNA sequencing is quickly replacing microarrays. However, older chip-based methodologies such as comparative genomic hybridization and single-nucleotide polymorphism arrays have benefited from this technological explosion and are now so accessible that they can be employed in increasingly larger cohorts of patients. The study of breast cancer lends itself particularly well to these technologies. It is the most commonly diagnosed neoplasm in women, giving rise to nearly 230,000 new cases each year. Many patients are given a diagnosis of early-stage disease, for which surgery is the standard of care. These attributes result in excellent availability of tissues for whole-genome/transcriptome analysis. The Cancer Genome Atlas project has generated comprehensive catalogs of publically available genomic breast cancer data. In addition, other studies employing the power of genomic technologies in medium to large cohorts were recently published. These data are now publically available for the generation of novel hypotheses. However, these studies differed in the methods, patient cohorts, and analytical techniques employed and represent complementary snapshots of the molecular underpinnings of breast cancer. Here, we will discuss the convergences and divergences of these reports as well as the scientific and clinical implications of their findings.
Introduction
Breast cancer is the most common malignancy in women, totaling nearly 230,000 new cases in 2012 [1]. Owing to improvements in screening, many patients are identified before disease dissemination and can undergo surgical resection of the primary tumor. Despite early detection, surgery, and adjuvant therapy, a fraction of patients ultimately recur with metastatic disease. There are three clinical subtypes of breast cancer that differ on the basis of the expression of estrogen receptor-alpha (ER-α), progesterone receptor (PR), and the HER2 oncogene. These clinical markers can predict the response to anti-estrogens and anti-HER2 therapies with a reasonable degree of accuracy [2,3]. Via gene expression profiling, several 'molecular' subtypes (termed the PAM50 intrinsic subtypes) of breast cancer have been characterized [4]. These include luminal A (high expression of ER, sensitive to anti-estrogens, and good prognosis), luminal B (lower expression of ER, less responsive to anti-estrogens, high proliferative rate and histological grade, and poor prognosis), HER2-enriched (containing most HER2 gene-amplified tumors and a subset of tumors lacking HER2 amplification), basal-like (mainly tumors not expressing HER2 or ER or PR, high relapse rate, and responsive to chemotherapy), and claudin-low (tumors with a pronounced cancer stem cell-like component, resistant to chemotherapy, and poor prognosis) [4,5]. Although molecular stratification has improved risk prediction and clinical trial design, the genomic alterations and therapeutic targets (in addition to ER, PR, and HER2) underlying these subtypes have not been established.
The high rate of surgical resection in breast cancer has resulted in readily available tissues for high-throughput genomic analyses such as massively parallel sequencing (MPS), expression microarray, and comparative genomic hybridization (CGH). The availability of tissue coupled with the rapid acceleration of these technologies and their dwindling cost has allowed the construction of comprehensive catalogs of the genomic architecture of breast cancer in large sample sizes [6-11]. Combined, these data have provided a catalog of somatic alterations in more than 3,500 breast tumors. These results confirm the somatic mutation landscape, where few genes are mutated in many tumors (for example, TP53) whereas many genes are recurrently mutated in few tumors. However, these diverse mutations can often be organized into several frequently mutated pathways. These data provide novel insights into the pathogenesis and classification of the disease, driving forward molecular analyses to discover new treatments.
Summary of genomic data in breast cancer
The largest of these studies interrogated copy-number alterations (CNAs) by SNP-chip and gene expression profiles by microarray in nearly 2,000 tumors representing all major subtypes of breast cancer [9]. Aside from identifying recurrent CNAs, the authors integrated gene expression and CNA data to determine the impact of inherited and somatic gene copy number on gene expression levels. CNAs and single-nucleotide polymorphisms (SNPs) associated with the genes in the altered region (termed cis loci) as well as those outside the altered region (trans loci) were identified. Approximately 40% of the tumor transcriptome was associated with the presence of SNPs or CNAs, acting either in cis or in trans. Somatic CNAs, as opposed to SNPs and germline CNAs, had the strongest association with gene expression architecture in breast tumors. The authors identified, among other alterations, novel deletions in PPP2R2A among luminal B tumors and MAP2K4 deletions (previously identified in luminal breast cancer cell lines [12] and multiple tumor types, including breast [13]) in ER+ tumors, suggesting that these genes may be tumor suppressors. However, molecular evidence for tumor suppression by either gene has not been demonstrated.
The authors proposed, on the basis of gene expression and CNA data, 10 novel subclassifications of breast cancer [9]. Several of these clusters overlapped considerably with the PAM50 intrinsic subtypes [4]. However, the integration of CNA data coupled with the large sample size allowed more intricate dissection of some heterogeneous subtypes, such as basal-like breast cancer (primarily contained in one cluster, with losses in chromosome 5 and gains at 8q, 10p, and 12p). These subtypes include a previously identified [14,15] poor prognosis luminal group containing amplification of 11q13/14, which includes CCND1 and other potential drivers. Of note, the authors also identified a 'CNA-devoid' subgroup which had good prognosis and was characterized by high genomic stability, a flat copy-number landscape, and signatures of an adaptive immune response [9]. Finally, the authors employed bioinformatic approaches to ascribe biological underpinnings to the profiles observed in each of these subtypes. The categories of molecular processes identified in several of the clusters paralleled those established previously in triple-negative breast cancer (TNBC) [16], reaffirming the diversity of this clinical subtype. This study also identified rare but recurrent CNAs in therapeutic targets such as amplification of IGF1R, KRAS, and EGFR.
Shah and colleagues [7] used MPS to describe the somatic mutational landscape in 104 TNBCs. TNBC accounts for approximately 15% of breast cancers, representing a heterogeneous and highly virulent disease subtype. An advantage of this study, despite its smaller size, was its focus on TNBC, which allowed the authors to make inferences about the diversity of its clonal evolution. TNBCs showed a continuous distribution of numbers of CNAs and point mutations per tumor, which were not associated with one another. This suggests that the mechanisms and environmental factors contributing to the development and progression of breast tumors through the generation of CNAs and somatic mutations are distinct. Also, by integrating copy-number and deep re-sequencing data, the authors calculated allele frequencies for thousands of identified mutations. They examined the mutation-frequency distribution in each tumor, finding that some TNBCs have only a few 'peaks' of allele frequencies but that others have more than 15 peaks. Thus, some tumors consisted of a heterogeneous multi-clonal pool of transformed cells, whereas some consisted of only one or two dominant transformed clones. The nature and number of these clones may have implications for targeted therapies. For instance, therapeutic targeting of lesions present in only one of five theoretical clonal populations may not be sufficient to induce a clinical response. By organizing aberrations into targetable pathways and prioritizing by their clonal frequency, the authors concluded that alterations in several known drivers, including PTEN, PIK3CA, and TP53, demonstrated the highest degree of clonal frequency and are likely the initiating or 'founder' lesions in TNBC. The authors reported that 20% of tumors contained at least one somatic mutation that is currently targetable, but these mutations were not always commonly associated with TNBC or breast cancer in general (for example, one case contained a V600E BRAF mutation). Similar findings supporting the notion of dominant subclones composed of only a handful of driver mutations were reported shortly thereafter by Nik-Zainal and colleagues [17].
In a study that focused on ER+ breast cancer, Ellis and colleagues [8] performed whole- genome sequencing of 46 tumor-normal pairs and an additional 31 exomes, followed by targeted re-sequencing in an additional 240 tumors. This study was unique in that it incorporated therapeutic response into its design. The tumors analyzed were from patients treated for approximately 4 months with aromatase inhibitors (AIs). Drug-induced changes in tumor cell proliferation were measured by Ki67 immunohistochemistry (IHC) before and after therapy. The proportion of Ki67+ cells was used as a biomarker for response to estrogen deprivation as previously established [18-20]. In addition to identifying recurrent mutations in genes not previously implicated in breast cancer, including TBX3, RUNX1, LDLRAP1, MYH9, AGTR2, STMN2, SF3B1, and CBFB, the authors identified molecular markers for response to an AI. For example, mutations in TP53, a hallmark of basal-like breast cancer and TNBC, were enriched in luminal B tumors and were associated with a lower anti-proliferative response. However, it is unclear whether TP53 plays a direct role in resistance to anti-estrogen therapy or whether it is a marker of a less hormone-driven tumor (that is, basal-like). Pathway analysis [21] of the mutation data identified several cell signaling networks associated with anti-proliferative response to an AI, such as the MAPK, FYN (an Src-family kinase), and MYC pathways. Both the MYC and MAPK pathways have been reported to be highly activated in (ER−) basal-like tumors and cell lines [22,23]. Finally, the authors also reported mutations in the HER2 gene in the absence of gene amplification. A recent study documented that these are gain-of-function mutations and are sensitive to small-molecule HER2 tyrosine kinase inhibitors [24].
Banerji and colleagues [10] sequenced the exomes of 103 primary tumors and an additional five cancers by whole-genome sequencing. Seventeen cases were sequenced by both methods. The authors identified six recurrently mutated genes within this cohort: CBFB, TP53, PIK3CA, AKT1, GATA3, and MAP3K1. All of these, with the exception of CBFB, have been previously identified as recurrently mutated in breast cancer. CBFB, encoding core-binding factor beta subunit, was mutated in four ER+ tumors. CBFB is a heterodimeric partner with the RUNX family of transcription factors. This transcriptional complex appears to be frequently inactivated as a hemizygous deletion of RUNX1 at the genomic level co-occurred with CBFB mutations. In addition, several cancers with homozygous deletions of RUNX1 were identified. CBFB and RUNX1 mutations were also identified by Ellis and colleagues [8] and in The Cancer Genome Atlas (TCGA) study (below), suggesting that disruption of CBFB/RUNX1 function is involved in breast cancer progression. Like Ellis and colleagues [8], the authors identified mutations in HER2 gene in tumors without HER2 amplification. Finally, the authors identified an in-frame genomic translocation event between MAGI3 and AKT3 [10]. Among 235 additional tumors, nine cases (3.8%) with MAGI3-AKT3 fusions were identified. These fusions retained the kinase domain of AKT3 while disrupting the pleckstrin homology and PTEN-binding domains of AKT3 and MAGI3, respectively. When expressed in breast cancer cells, the fusion transcript resulted in constitutive AKT phosphorylation and activity, rendering the cells sensitive to an ATP-competitive AKT inhibitor. However, these fusions have not yet been identified in other sequencing studies.
Stephens and colleagues [6] sequenced 100 primary breast cancers by using whole-exome MPS. Similar to the studies by Ellis and colleagues [8] and Curtis and colleagues [9], recurrent inactivating mutations and deletions in the MAP3K1 and MAP2K4 genes were identified. The mutation patterns of ARID1B, CASP8, MAP3K13, NCOR1, SMARCD1, and CDKN1B (p27) suggested that these genes may also be tumor suppressors. Several of these genes, including MAP3K1, CASP8, and TBX3, are recessive cancer genes previously identified in genome wide association studies [25,26]. Interestingly, germline mutation of TBX3 causes ulnar-mammary syndrome, which includes, among other defects, failure of mammary gland development [27].
The TCGA is a comprehensive collection of copy number (SNP-chip), microarray, RNA-sequencing, MPS, clinical, and proteomic data across multiple cancer types [11]. Approximately 450 breast cancers with data from all methods were compiled. The TCGA confirmed the presence of previously identified mutations in TBX3, RUNX1, and CBFB, including subtype-specific associations of PIK3CA, MAP3K1, and GATA3 mutations with ER+ and luminal A tumors. Additionally, AFF2, PIK3R1, PTPN22, PTPRD, NF1, SF3B1, and CCND3 were found to be recurrently mutated. Interestingly, a comparison of the breast cancer and ovarian TCGA data demonstrates that, from a molecular perspective, basal-type breast cancers more closely resemble ovarian serous carcinomas than luminal breast cancers do. Furthermore, basal-type breast cancers, despite having the lowest percentage of PIK3CA mutations, exhibit the highest PI3K pathway activity as measured by phosphoprotein and gene expression signatures. These findings may be due to deletions in tumor suppressors - such as PTEN and INPP4B - that negatively regulate the PI3K pathway. Mutual exclusivity module analyses demonstrated that the receptor tyrosine kinase (RTK)-PI3K and p38-JNK pathways are affected in almost 80% of breast carcinomas, providing a clear set of common targetable pathways despite the enormous genetic heterogeneity. The greatest contributions of the TCGA study are the integration of multiple data types, including clinical annotation and proteomic data, and the accessibility of these data to outside investigators. As such, the TCGA data represent an excellent source for the generation of novel hypotheses and some access to limited retrospective clinical data which can be accessed through the cBio web portal [28]. This data portal has been constructed to allow all investigators, including those not used to handling and analyzing high-dimensional data, to easily interrogate genes and pathways of interest. However, the retrospective nature of the TCGA project and its use of tumors not necessarily linked to specific clinical trials, in which the outcome of therapy can be assessed rigorously, limit the ability of the TCGA to establish whether the genomic data can 'predict' the action of the current treatment standards (that is, anti-estrogens or chemotherapy). Nonetheless, the TCGA project provides a 'blueprint' that in some form should be part of the molecular analyses linked to investigational clinical trials with novel drugs and combinations, particularly where a diagnostic biomarker for patient selection is lacking.
Finally, Nik-Zainal and colleagues [17,29] sequenced the complete genome of 21 breast cancers. These authors identified driver mutations in many of the genes mentioned above, but the major contribution of these articles was to use whole-genome data to elucidate mutational processes in breast cancer. Indeed, they identified at least five different mutational processes, such as C>T mutations at TpCpX tri-nucleotides. Clustering of the samples based on these mutational processes identified two major groups, separated by BRCA1/BRCA2 status. The significant heterogeneity in the mutational processes occurring in breast cancer suggests similar heterogeneity in the defects in repair mechanisms in these tumors. With the development of PARP inhibitors, defective DNA repair mechanisms are emerging as therapeutic targets. Exploitation of such targets in breast cancer will require the identification of the defective processes underlying each of these five categories and the development of drugs that target these alterations.
These landmark studies each catalogued the molecular lesions in breast cancer by using different sets of patients, technologies, and methods of analysis (Table 1). Novel insights gleaned from these data into the pathogenesis of breast cancer will be important for our understanding of the basic biology of this disease. However, what does this mean for breast cancer patients seeking standard care or a clinical trial or both? Do the results of these studies and the massive amount of data generated portend novel therapies that can change the natural history of the disease? It is somewhat reassuring, but not surprising, that striking similarities in recurrently mutated or altered genes were identified among the studies (Figure 1). Furthermore, many commonly altered genes identified were previously implicated in breast or other cancers, attesting to the visionary work of those investigators who previously identified them in the absence of MPS technologies. The power of the studies reviewed here lies not in single gene alterations but in the ability to combine less commonly altered genes into pathways on the basis of known biology and function. From these data, pathways not previously implicated in breast cancer can now be explored to determine whether they are associated with exploitable therapeutic vulnerabilities.
Table 1.
Summary of genomics studies
| Reference | Cohort | Methods of analysis | Summary of key findings |
| Ellis et al. [8] | Biopsies from ER+ breast tumors undergoing neoadjuvant aromatase inhibitor therapy | WGS (n = 46), ES (n = 31), and MRS (n = 240) | Recurrent mutations in MAP3K1 pathway in ER+ disease with good prognosis; anti-proliferative response to aromatase inhibitor associated with mutational activation of FYN, MAPK, and MYC pathways; recurrent mutations in methyl and demethyltransferases and AT-rich interactive domain-containing genes; ERBB2 mutations in non-ERBB2-amplified breast cancer |
| Shah et al. [7] | Triple-negative breast cancer cases | WGS (n = 15), ES (n = 54), CGH (n = 104), and RS (n = 80) | Clonal dominance of PIK3CA, PTEN, and TP53 mutations; intragenic PARK2 deletions in TNBC; 30%-40% of somatic mutations are detectable at the RNA level. |
| Curtis et al. [9] | Unselected primary breast tumors | CGH (n = 1,992) and MA (n = 1,992) | Copy-number alterations influence gene expression in over 40% of the breast cancer genome; deletions in PPP2R2A, MTAP, and MAP2K4; integrated copy-number and microarray data defined 10 unique clusters of breast cancer. |
| Banerji et al. [10] | Unselected primary breast tumors | WGS (22) and ES (103) | Recurrent mutations in CBFB and RUNX1; recurrent MAGI3-AKT3 fusions which are sensitive in vitro to AKT ATP-competitive inhibitors |
| Stephens et al. [6] | Unselected primary breast tumors | ES (n = 100) | Novel recurrent mutations in AKT2, ARID1B, CASP8, CDKN1B, MAP3K1, MAP3K13, NCOR1, SMARCD1, and TBX3 |
| The Cancer Genome Atlas [11] | Unselected primary breast tumors | MA (n = 547), DNA-methylation array (n = 802), CGH (n = 773), MIRS (n = 697), RPPA (n = 403), and ES (n = 507) |
Luminal breast cancer is most heterogeneously mutated subtype; basal-like breast cancer molecularly resembles serous ovarian carcinoma; mutation patterns consistent with the other studies; convergence of genetic and epigenetic alterations into four primary subtypes of breast cancer |
CGH, comparative genomic hybridization; ER, estrogen receptor; ES, exome sequencing; MA, microarray; MIRS, microRNA-sequencing; MRS, mutation recurrence screening; RPPA, reverse-phase proteomic array; RS, RNA sequencing; WGS, whole-genome sequencing.
Figure 1.
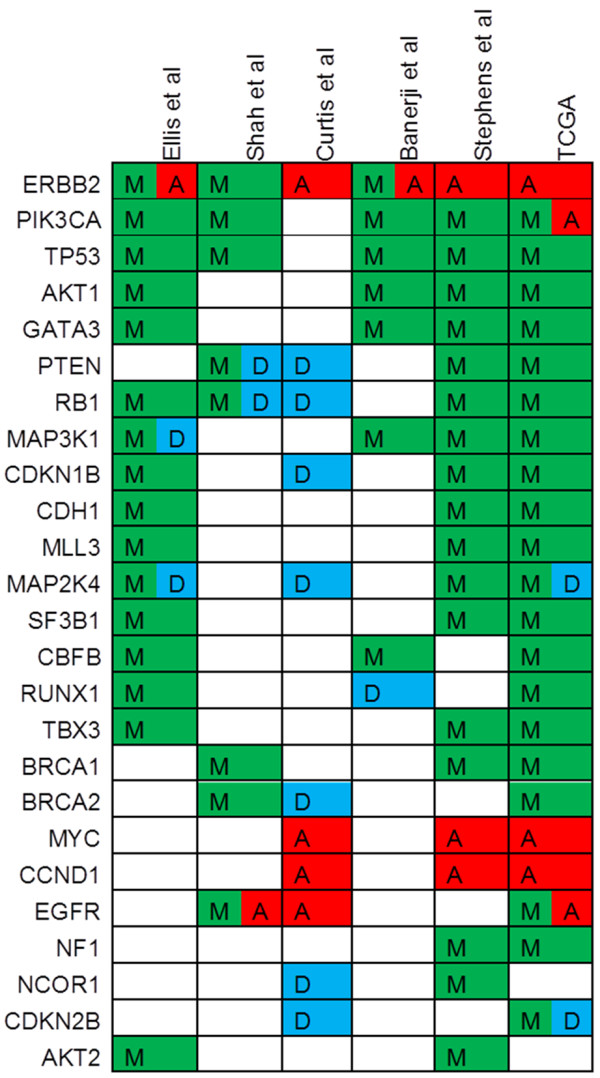
Selected recurrently altered genes identified in breast cancer in recent genomic studies. Selected genes identified as significantly or recurrently altered in breast cancer in six genomic studies: Ellis et al. [8], Shah et al. [7], Curtis et al. [9], Banerji et al. [10], Stephens et al. [6], and The Cancer Genome Atlas (TCGA) [11]. Deleted (D) genes are in blue, mutated (M) in green, and amplified (A) in red.
Implications for breast cancer classification and treatment
A key direction resulting from the studies presented is a more specific and diverse classification of breast cancer into multiple genome-driven subtypes, each potentially with a personalized treatment approach. Breast cancer is currently treated on the basis of three clinical markers: ER, PR, and HER2. Breast cancers expressing ER or PR are more likely to be responsive to anti-estrogen therapy. However, a significant proportion of HR+ tumors exhibit de novo or acquired resistance to endocrine therapy. These tumors, termed 'luminal B', are hyper-proliferative and less well differentiated and benefit from chemotherapy in addition to anti-estrogens. Several approaches have been used to identify luminal B tumors, such as the PAM50 clinical assay [4], the Oncotype DX algorithm [30], and IHC markers such as high Ki67, HER2 over-expression, absence of PR, or low ER [18,31]. This separation of luminal B from the less aggressive luminal A counterpart, though not yet optimal, is important for the selection of patients with ER+ luminal B breast cancer into clinical trials of combinations of anti-estrogens with novel therapies. Significant data support the notion that the distinction between luminal A and B tumors may be more of a continuum of proliferation, differentiation, and accompanying HER2 expression, leading to difficulty in clinical cutoffs and decision-making.
Alterations in several genes and pathways were identified as being linked to the luminal A/B phenotype in multiple studies. These include activating PIK3CA mutations and deletions or inactivating mutations within the JNK pathway (MAP2K4, MAP3K1, and possibly STMN2) in luminal A tumors and PPP2R2A deletions in luminal B tumors. At this time, several prospective randomized clinical trials of anti-estrogens with or without PI3K pathway inhibitors in patients with ER+/ PIK3CA mutant breast cancer are ongoing. The initial phase I study of the p110α-specific inhibitor BYL719 was limited to patients with tumors harboring PIK3CA-activating mutations. Results from this study have already shown clinical activity of the single agent in patients with PIK3CA-mutant metastatic ER+ breast cancer [32]. Results of these trials should shed light on the role of these mutations in anti-estrogen-treated luminal breast cancers. In the case of MAP2K4 and MAP3K1 alterations, which putatively inactivate the JNK pathway, uncovering a therapeutic role for targeting tumors with these lesions could be challenging: they tend to occur in tumors with the best prognosis, many of which are cured with surgery and adjuvant endocrine therapy.
Amplification or high expression (or both) of HER2 is a predictive marker for response to HER2-targeted therapies [33,34]. However, some breast cancers display gene expression patterns consistent with HER2 over-expression (that is, the HER2-enriched molecular subtype of the PAM50 classifier [4]) in the absence of HER2 gene amplification as measured by fluorescence in situ hybridization. Several of these studies have reaffirmed this discrepancy and also have led to the discovery of novel HER2 mutations in the kinase and juxtamembrane regions, which were recently established as possible drivers in non-HER2-amplified breast cancer [24]. Breast cells transformed with these mutations were sensitive to HER2 kinase inhibitors, such as lapatinib and neratinib. Thus, it is increasingly apparent that there may be several subgroups of HER2-driven breast cancer. This could potentially expand the number of patients in whom HER2-targeted combinations are effective.
TNBC, often associated with basal-like gene expression, lacks expression of ER, PR, and HER2 and represents a heterogeneous group of tumors at clinical, histopathological, and molecular levels [11,16,35-37]. Although TNBCs, as a group, are generally more sensitive to conventional chemotherapy, they also demonstrate higher relapse rates and poorer survival compared with other breast cancer subtypes [38]. Clinical studies have shown good responses with poly (ADP-ribose) polymerase (PARP) inhibitors in TNBC with BRCA gene deficiency [39,40]. The DNA cross-linker cisplatin has also shown remarkable activity in patients with BRCA1 mutant TNBC but its role in TNBC with wild-type BRCA1 is less clear [41]. The studies reviewed here did not identify possible therapeutic sensitivities of TNBC or basal-like breast cancer [6-11]. However, the high degree of genomic rearrangement identified in this subtype supports the further examination of agents targeting the DNA damage response and repair mechanisms, which are defective in some TNBCs [7,11]. Nonetheless, many unanswered questions about how future clinical trials should incorporate targeted therapies in TNBC remain.
Gene signatures such as Oncotype DX [30] and genomic grade index [42] have been less useful in identifying different risk categories within TNBC than within ER+ breast cancer. A recent study identified biologically diverse TNBC clusters by using transcriptome datasets from 21 independent studies [16]. These clusters were defined by DNA damage response genes, mesenchymal features, immune-related genes, and androgen receptor (AR) signaling [16]. The 'mesenchymal' cluster overlaps with a previously reported claudin-low breast cancer subtype also characterized by loss of epithelial differentiation, an intense immune infiltrate, and cancer stem-like characteristics [5,16]. Using TNBC cell lines and xenografts, Lehmann and colleagues [16] associated these clusters with particular therapeutic vulnerabilities (that is, anti-androgens in the AR luminal cluster, cisplatin and other DNA damaging agents in the DNA damage response subgroup, and PI3K pathway inhibitors in the mesenchymal genes cluster). The use of these expression signatures for treatment selection and prediction of response remains to be determined. Importantly, many of these subtypes or TNBC clusters appear analogous to those described by Curtis and colleagues [9].
Many alterations identified in the studies reviewed here lie in pathways that have been previously explored for drug development. Perhaps the largest challenge to developing new therapeutics against these commonly altered pathways lies in understanding the biological functionality of these mutations. Is the alteration a driver of an oncogenic phenotype? The presence of an alteration does not imply oncogene dependence or therapeutic sensitivity. Does the mutation confer gain-of-function, loss-of-function, or a new functionality altogether? Support for clinical targeting of these alterations will require extensive in vitro and in vivo experimentation to understand whether and how the mutation functions biologically.
Implications for clinical trials and care
The wealth of data generated in the context of the studies presented should pave the way for additional genotype-driven trials with targeted therapies. Potential markers of sensitivity and resistance to targeted agents can be tested at the bench on the basis of these findings. However, the use of these biomarkers in investigational clinical trials must be approached cautiously. For instance, should only those patients harboring a tumor with mutations in the PI3K pathway be considered for trials with PI3K inhibitors? Although preclinical data support this notion, the complexity of the cancer genome as demonstrated by the reviewed studies may circumvent this simplistic approach. Patients who may benefit from the targeted therapy but lack (known) lesions in the targeted pathway may be missed on the basis of a lack of comprehensive knowledge of all the molecular lesions dysregulating such a pathway. Alternatively, pathway activation without direct mutation may portend a more plastic target, less addicted to the pathway's output and thus more able to evade inhibition.
Another important implication for clinical trials and biomarker studies can be drawn from the study by Shah and colleagues [7], who examined clonal frequency of genomic alterations in TNBC. Given the regional heterogeneity present in the tumors, also demonstrated in other studies [29,43-46], therapeutic target identification becomes challenging. Sampling of the tumor performed from a cross-section of the entire specimen likely gives the best representation of all the lesions and alterations present [46]. This can be best achieved from surgical specimens, which are not always available for research purposes. Use of the primary diagnostic biopsy in patients with metastatic breast cancer enrolled in a clinical trial may yield significant discrepancies between the primary and metastatic lesions [47]. Although core biopsies of metastatic lesions are increasingly being performed over fine-needle aspirates (FNAs), the relatively small region of tumor in a core biopsy may still miss a significant percentage of the clonal lesions present in cancer as a whole. The clonal lesions may be important in overall response, as recently was experimentally demonstrated in colorectal cancer [45]. Such issues may be overcome by multiple biopsies or by simultaneous sampling of circulating tumor DNA [48-50].
In the course of clinical trials of targeted agents, a frequently reported observation is the single patient with an extraordinary response. Examples of such case reports include EGFR inhibitors erlotinib and gefitinib and the ALK inhibitor crizotinib in non-small cell lung cancer [51,52], vemurafenib in melanoma [53], and everolimus in bladder cancer [54]. These cases present a unique opportunity to identify genomic biomarkers of sensitivity and, in principle, should lead to genotype-specific phase II studies in which the initial observation can be validated. Positive genotype-specific trials, like those that led to the approval of crizotinib in EML4-ALK fusion-positive NSCLC [55,56], can rapidly lead to drug approval. For example, trials with the TORC1 inhibitor everolimus limited to patients with mutations in TSC1/2 and mTOR are being developed.
The increased availability of Clinical Laboratory Improvement Amendments (CLIA)-certified MPS at academic centers and commercial sources is presenting patients and oncologists with new treatment options and dilemmas. Previously identified mutations in 'targetable' genes that are known to be functional will be identified, but drugs directed against these lesions may not yet be approved for use in breast cancer. Furthermore, some mutations will be variants of unknown significance (that is, mutations not previously observed in genes of interest). Tumor heterogeneity will lead to identification of mutations that are clearly not in all tumor cells. Cancers will present with multiple 'actionable' targets, but the key driver of tumor progression may not be apparent. A plausible example is that of a patient who has metastatic TNBC and good performance status and who has exhausted all standard of care options. While being considered for enrollment in a chemotherapy trial at a National Cancer Institute (NCI)-designated cancer center, tissue from a recent metastatic biopsy is submitted for DNA extraction and MPS of approximately 300 known oncogenes and tumor suppressors. The assay identifies mutations in TSC1 and PIK3CA as well as EGFR and CDK4 gene amplifications. Encumbered by the molecular data, this patient could consider treatment with an EGFR antibody (for example, cetuximab) or a TORC1 inhibitor (for example, everolimus). However, these drugs have not shown efficacy in TNBC, although clinical evidence suggests that they may be effective against other tumor types with these alterations. In light of the mutation in PIK3CA, a trial with a combination targeting the PI3K pathway could also be considered. At present, many situations like the one described here are happening across cancer care centers in the US. This hypothetical scenario portends that (a) patient needs and patterns of care will play an important role in the incorporation of 'predictive' genomics to clinical investigation and care and (b) the design and sometimes rigid process of clinical trials may have to adapt by incorporating genomic information at the time of patient eligibility and enrollment. Finally, casting a wide net to identify the causative lesions by using whole-genome sequencing may be cost-and labor-intensive and limited by lower depth of coverage. The large genomic studies summarized in this review provide a catalog of genes that can be drawn from to assemble targeted capture assays for higher-depth analysis. However, given that most patients in the presented studies were assayed prior to systemic treatment, it is possible that the majority of alterations mediating acquired resistance to targeted therapies remain to be discovered. Thus, the sampling and analysis of tumors at the time of acquired resistance remain a largely unexplored frontier.
Conclusions
In summary, a wealth of integrated molecular data now exists in primary breast cancer. Although the data available are not exhaustive, they provide a framework for biological validation and experimentation that should guide preclinical studies. For those pathways with therapeutic targets currently under clinical investigation, these studies can be used as a catalog of molecular lesions potentially representing biomarkers of response or resistance to treatment.
Abbreviations
AI: aromatase inhibitor; CAN: copy-number alteration; ER: estrogen receptor; FNA: fine-needle aspirate; IHC: immunohistochemistry; MPS: massively parallel sequencing; PARP: poly (ADP-ribose) polymerase; PR: progesterone receptor; SNP: single-nucleotide polymorphism; TCGA: The Cancer Genome Atlas; TNBC, triple-negative breast cancer.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Justin M Balko, Email: justin.balko@vanderbilt.edu.
Thomas P Stricker, Email: thomas.stricker@vanderbilt.edu.
Carlos L Arteaga, Email: carlos.arteaga@vanderbilt.edu.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;15:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010;15:R245–262. doi: 10.1677/ERC-10-0136. [DOI] [PubMed] [Google Scholar]
- Rakha EA, Reis-Filho JS, Baehner F, Dabbs DJ, Decker T, Eusebi V, Fox SB, Ichihara S, Jacquemier J, Lakhani SR, Palacios J, Richardson AL, Schnitt SJ, Schmitt FC, Tan PH, Tse GM, Badve S, Ellis IO. Breast cancer prognostic classification in the molecular era: the role of histological grade. Breast Cancer Res. 2010;15:207. doi: 10.1186/bcr2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;15:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;15:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;15:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;15:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G. et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;15:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S. METABRIC Group; Langerød A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Børresen-Dale AL, Brenton JD, Tavaré S. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;15:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Piña V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;15:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;15:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, Riaz M, Koopman DG, Ten Hagen TL, de Leeuw BH, Zwarthoff EC, Teunisse A, van der Spek PJ, Klijn JG, Dinjens WN, Ethier SP, Clevers H, Jochemsen AG, den Bakker MA, Foekens JA, Martens JW, Schutte M. Distinct gene mutation profiles among luminal-type and basaltype breast cancer cell lines. Breast Cancer Res Treat. 2010;15:53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, Kern SE. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 1998;15:2339–2342. [PubMed] [Google Scholar]
- Schuuring E, Verhoeven E, Mooi WJ, Michalides RJ. Identification and cloning of two overexpressed genes, U21B31/PRAD1 and EMS1, within the amplified chromosome 11q13 region in human carcinomas. Oncogene. 1992;15:355–361. [PubMed] [Google Scholar]
- Tsuda H, Hirohashi S, Shimosato Y, Hirota T, Tsugane S, Yamamoto H, Miyajima N, Toyoshima K, Yamamoto T, Yokota J, Yoshida T, Sakamoto H, Terada M, Sugimura T. Correlation between long-term survival in breast cancer patients and amplification of two putative oncogene-coamplification units: hst-1/int-2 and c-erbB-2/ear-1. Cancer Res. 1989;15:3104–3108. [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;15:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, Menzies A, Stebbings LA, Leroy C, Jia M, Rance R, Mudie LJ, Gamble SJ, Stephens PJ, McLaren S, Tarpey PS, Papaemmanuil E, Davies HR, Varela I, McBride DJ, Bignell GR, Leung K, Butler AP. et al. The life history of 21 breast cancers. Cell. 2012;15:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;15:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, A'Hern R, Salter J, Detre S, Hills M, Walsh G. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. 2007;15:167–170. doi: 10.1093/jnci/djk020. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ma C. Letrozole in the neoadjuvant setting: the P024 trial. Breast Cancer Res Treat. 2007;15:33–43. doi: 10.1007/s10549-007-9701-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;15:i237–245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, Yaswen P, Mills GB, Esserman LJ, Goga A. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;15:679–696. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, Hu Z, Knight Z, Feiler HS, Gascard P, Parvin B, Spellman PT, Shokat KM, Wyrobek AJ, Bissell MJ, McCormick F, Kuo WL, Mills GB, Gray JW, Korn WM. Basal subtype and MAPK/ERK kinase (MEK)- phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;15:565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, Ellis MJ. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013;15:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoussaini M, Fletc her O, Michailidou K, Turnbull C, Schmidt MK, Dicks E, Dennis J, Wang Q, Humphreys MK, Luccarini C, Baynes C, Conroy D, Maranian M, Ahmed S, Driver K, Johnson N, Orr N, dos Santos Silva I, Waisfisz Q, Meijers-Heijboer H, Uitterlinden AG, Rivadeneira F. Netherlands Collaborative Group on Hereditary Breast and Ovarian Cancer (HEBON); Hall P, Czene K, Irwanto A, Liu J, Nevanlinna H, Aittomäki K, Blomqvist C, Meindl A. et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet. 2012;15:312–318. doi: 10.1038/ng.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese JS, Easton DF. Genome-wide association studies in common cancers-what have we learnt? Curr Opin Genet Dev. 2010;15:201–209. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, Franceschini P, Lala R, Holmes LB, Gebuhr TC, Bruneau BG, Schinzel A, Seidman JG, Seidman CE, Jorde LB. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;15:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- cBioPortal for Cancer Genomics. http://www.cbioportal.org/public-portal/
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E. et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;15:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, nodenegative breast cancer. N Engl J Med. 2004;15:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Bhargava R, Beriwal S, Dabbs D J, Ozbek U, Soran A, Johnson RR, Brufsky AM, Lembersky BC, Ahrendt GM. Immunohistochemical surrogate markers of breast cancer molecular classes predicts response to neoadjuvant chemotherapy: a single institutional experience with 359 cases. Cancer. 2010;15:1431–1439. doi: 10.1002/cncr.24876. [DOI] [PubMed] [Google Scholar]
- Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, MR M, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J. BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2012;15:CT–01. [Google Scholar]
- Murphy CG, Morris PG. Recenta dvances in novel targeted therapies for HER2-positive breast cancer. Anticancer Drugs. 2012;15:765–776. doi: 10.1097/CAD.0b013e328352d292. [DOI] [PubMed] [Google Scholar]
- Stern HM. Improving treatment of HER2-positive cancers: opportunities and challenges. Sci Transl Med. 2012;15:127rv122. doi: 10.1126/scitranslmed.3001539. [DOI] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI, Brown PH, Green JE, Kopelovich L, Furth PA, Palazzo JP, Olopade OI, Bernard PS, Churchill GA, Van Dyke T, Perou CM. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;15:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2010;15:39–48. doi: 10.1634/theoncologist.2010-S5-39. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;15:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;15:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;15:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;15:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, Cybulski C, Kladny J, Gorski B, Lubinski J, Narod SA. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat. 2009;15:359–363. doi: 10.1007/s10549-008-0128-9. [DOI] [PubMed] [Google Scholar]
- Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, Sotiriou C. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;15:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A, Cabral C, Pawar V, Mackay A, A'hern R, Marchiò C, Palacios J, Natrajan R, Weigelt B, Reis-Filho JS. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;15:42–52. doi: 10.1002/path.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing. Nature. 2011;15:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, O'Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, Ma J, Wienholds E, Dunant C, Pollett A, Gallinger S, McPherson J, Mullighan CG, Shibata D, Dick JE. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;15:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;15:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, Koboldt DC, Schmidt H, Kalicki J, Zhang Q, Chen L, Lin L, Wendl MC, McMichael JF, Magrini VJ, Cook L, McGrath SD, Vickery TL, Appelbaum E, Deschryver K, Davies S, Guintoli T, Lin L. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;15:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethdorf S, Müller V, Zhang L, Rau T, Loibl S, Komor M, Roller M, Huober J, Fehm T, Schrader I, Hilfrich J, Holms F, Tesch H, Eidtmann H, Untch M, von Minckwitz G, Pantel K. Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin Cancer Res. 2010;15:2634–2645. doi: 10.1158/1078-0432.CCR-09-2042. [DOI] [PubMed] [Google Scholar]
- Somlo G, Lau SK, Frankel P, Hsieh HB, Liu X, Yang L, Krivacic R, Bruce RH. Multiple biomarker expression on circulating tumor cells in comparison to tumor tissues from primary and metastatic sites in patients with locally advanced/inflammatory, and stage IV breast cancer, using a novel detection technology. Breast Cancer Res Treat. 2011;15:155–163. doi: 10.1007/s10549-011-1508-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, Halsall D, Wallis M, Bentley D, Caldas C, Rosenfeld N. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;15:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- Lai CS, Boshoff C, Falzon M, Lee SM. Complete response to erlotinib treatment in brain metastases from recurrent NSCLC. Thorax. 2006;15:91. doi: 10.1136/thx.2005.052233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Nakajima T, Takeuchi K, Soda M, Mano H, Iizasa T, Matsui Y, Yoshino M, Shingyoji M, Itakura M, Itami M, Ikebe D, Yokoi S, Kageyama H, Ohira M, Nakagawara A. ALK fusion gene positive lung cancer and 3 cases treated with an inhibitor for ALK kinase activity. Lung Cancer. 2012;15:66–72. doi: 10.1016/j.lungcan.2011.05.027. [DOI] [PubMed] [Google Scholar]
- Gautschi O, Pauli C, Strobel K, Hirschmann A, Printzen G, Aebi S, Diebold J. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol. 2012;15:e23–24. doi: 10.1097/JTO.0b013e3182629903. [DOI] [PubMed] [Google Scholar]
- Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;15:221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;15:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, Salgia R, Maki RG, Varella-Garcia M, Doebele RC, Bang YJ, Kulig K, Selaru P, Tang Y, Wilner KD, Kwak EL, Clark JW, Iafrate AJ, Camidge DR. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;15:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]