

Abstract
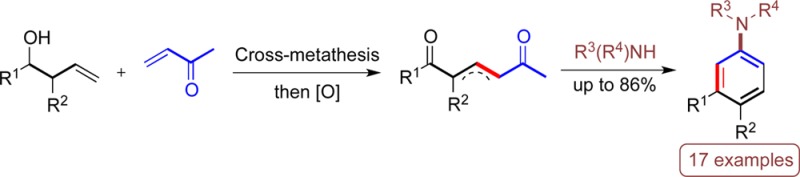
The olefin cross-metathesis reaction allows rapid access to 1,5-dicarbonyl intermediates which, upon treatment with a primary or secondary amine, allow the synthesis of a range of multisubstituted carbocyclic aryl amines. This de novo arene synthesis yields nonclassical substitution patterns in a regioselective and predictable approach that is compatible with several functional groups.
Efficient and selective routes to functionalized aryl amines are of continued interest due to the presence of these molecules in medicines, materials, and natural products and also because of their importance as intermediates in the synthesis of heterocycles. The most widely utilized route to prepare aryl amines is via metal-catalyzed coupling reactions epitomized by the palladium-catalyzed Buchwald–Hartwig amination1 and Chan–Lam type coupling2 or the copper-catalyzed Ullmann reaction.3 There are also some interesting methods that utilize C–H activation as a means for aminating arenes.4 In addition to cross-coupling chemistry, a number of useful dehydrogenative aromatization strategies have been developed.5 There have also been reports on the synthesis of substituted aryl amines using de novo methods.6
However, it is still important to develop novel routes to aryl amine targets with strategies avoiding the need for prefunctionalized arene rings as starting materials and allowing for greater functional group tolerance. Approaches that allow rapid access to ring systems with nonclassical substitution patterns with predictable regioselectivity are also highly valued.
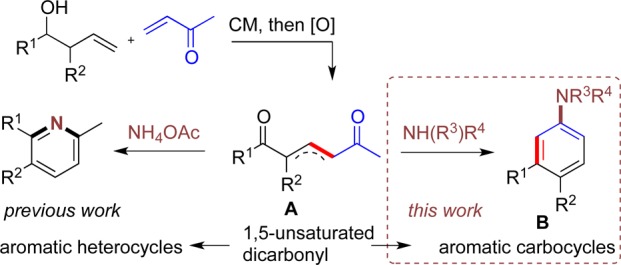
Recent studies in our laboratories have enabled advances in the de novo synthesis of heteroarenes using catalytic reactions to construct acyclic precursors. In particular, the ring closing metathesis reaction7 and the cross metathesis (CM) reaction8 have both been employed as lynchpins in the synthesis of multisubstituted pyridines, pyridazines, furans, and pyrroles.
We envisaged that unsaturated 1,5-dicarbonyl A, synthesized via a one-pot CM/oxidation of a homoallylic alcohol and vinylketone, could be utilized in a de novo synthesis of substituted carbocyclic aryl amines B (Scheme 1). Previously, we have used intermediates A as precursors to pyridines,8d obtained upon reaction with ammonia and a mild acid. In this study we explore the suitability and scope of the same intermediates for the formation of carbocyclic rings, brought about by a reaction with a primary or secondary amine.
Scheme 1. A General Approach to Aromatic Carbocycles.
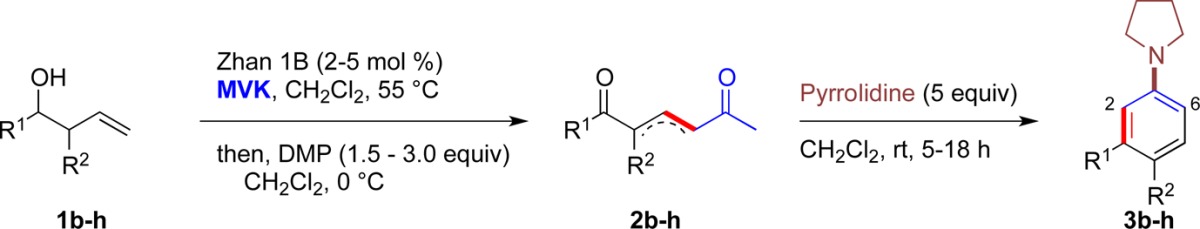
Earlier work showed that 1,5-dicarbonyl 2a could be easily prepared from a CM reaction between alcohol 1a and methyl vinyl ketone catalyzed by the Zhan-1B catalyst,9 followed by oxidation with Dess-Martin periodinane10 (DMP) in the same pot to give product 2a in 78% yield (Table 1).
Table 1. Optimization of the Cyclization.

| entry | amine equiv | temp (°C) | 3aa | 4a |
|---|---|---|---|---|
| 1 | 2.0 | –78 | 74 (63b) | 26 |
| 2 | 2.0 | 0 | 71 | 29 |
| 3 | 2.0 | rt | 57 (50b) | 43 |
| 4 | 2.0 | 55 | 41 | 59 |
| 5 | 1.5 | 0 | 53 | 57 |
| 6 | 3.0 | 0 | 79 | 21 |
| 7 | 5.0 | 0 | 89 (83b) | 11 |
| 8 | 5.0 | 0 | 67c | 33 |
| 9 | 5.0 | 55 | 70 | 30 |
Ratio of products shown (high conversion in each case).
Isolated yield.
ZnCl2 (1 equiv) was added.
Treatment of 1,5-dicarbonyl 2a with pyrrolidine (2 equiv) in CH2Cl2 at a range of temperatures between −78 and 55 °C allowed a cyclization to take place forming 3a in good yields (Table 1, entries 1–4). In each case, the only other major component in the reaction mixture was enamine 4 (note, compound 4 was not converted into 3a upon resubjection to the reaction conditions, vide infra). However, reducing the reaction temperature was clearly effective at minimizing this compound and optimizing the cyclization.
Variation in the equivalents of amine was also investigated. Reducing the amount of amine favored the formation of unwanted enamine 4 (Table 1, entry 5) while increasing the equivalents of amine gave more of the desired product 3a; with 5 equiv, nearly complete conversion was observed at 0 °C (83% isolated yield of 3a, Table 1, entry 7).
Finally, we discovered that the addition of a Lewis acid (ZnCl2 shown) was also compatible with the reaction conditions (Table 1, entry 8) and that the addition of 5 equiv of amine was sufficient to allow good conversion to the desired product 3a at higher temperatures (compare Table 1, entries 4 and 9). Both of these observations were to become useful when examining more substituted systems.
Next we investigated the scope of the substitution that was tolerated by this method. Keeping the amine portion constant, we found that it was possible to easily vary the substitution at positions C3 and C4 on the arene ring by altering the components used in the synthesis of the 1,5-dicarbonyl (Table 2) providing a rapid and regioselective route to a range of multisubstituted aryl amines. As the systems became more substituted we found that the reaction at lower temperatures was slow, and therefore the new examples shown in Table 2 were performed at rt or 55 °C. In particular, substrates 2e and 2f bearing electron-donating groups gave increasing amounts of the enamine side product at lower temperatures. It was hypothesized that electron-donating groups deactivated the adjacent carbonyl group, shutting down the cyclization. Consequently, we found that in these cases heating the reaction to 55 °C was sufficient to allow good conversion to aryl amines 3e and 3f.
Table 2. Scope of the Aryl Amine Substitution Pattern.
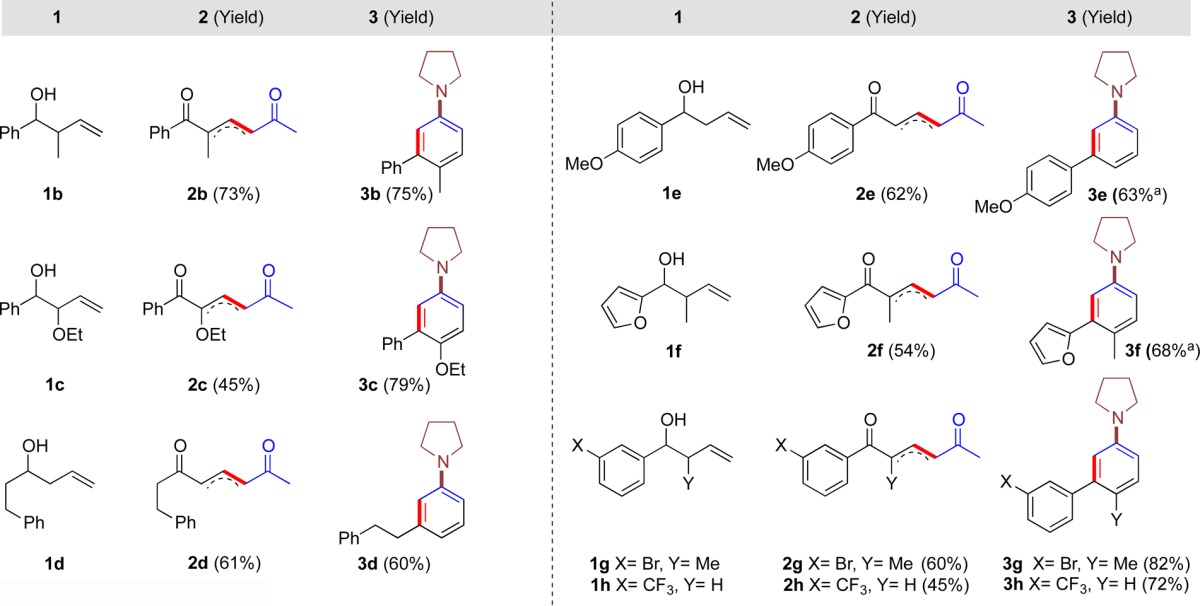
Reaction carried out at 55 °C.
Using this methodology, we were able to synthesize 3,4-disubstitued aryl amines incorporating both a methyl 3b and 3f–g and an ethoxy group 3c at C4 in good yields. It was also possible to install both electron-rich and -deficient aryl groups 3e, 3g, and 3h and a heteroaryl group at C3 3f. We were aware that the cyclization of dicarbonyl 2d may result in two different isomers being formed because of the presence of two enolizable C=O centers; however, only the regioisomer 3d was observed.
Note also that this methodology proved tolerant of halogen substituents as demonstrated in 3g (this halogen could be used for further functionalization using orthogonal palladium chemistry if necessary). Unfortunately, substituting methyl vinyl ketone for any other vinyl ketone in the cross metathesis reaction gave substrates that did not participate in the cyclization, thus restricting access to the C2 and C6 positions in the product, adjacent to the nitrogen.
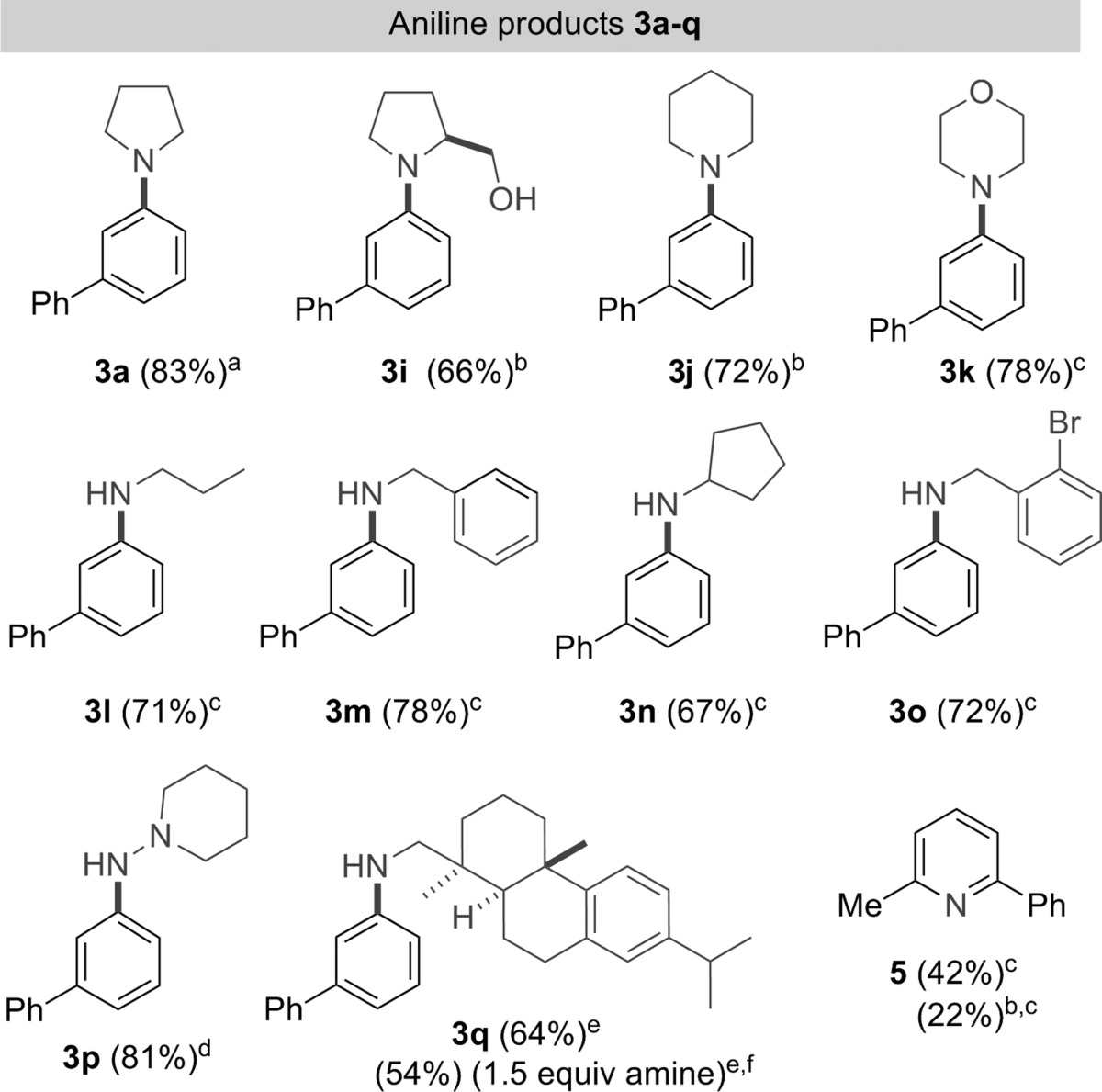
We also decided to investigate the range of amines tolerated in the cyclization. Pyrrolidine is the most active amine that we have studied, and every other derivative that was used required more vigorous conditions. Keeping the amine equivalents high, we examined the temperature of reaction and the addition of zinc chloride, Table 3. Unfortunately, a coherent picture of the effect of each variable did not emerge and the conditions for each amine were optimized individually.
Table 3. Scope of the Amine.
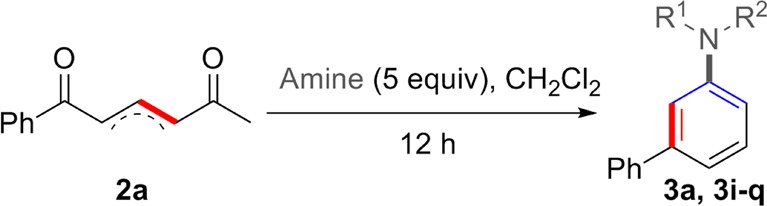
Reaction carried out 0 °C.
Reaction carried out at rt with 1 equiv ZnCl2.
Reaction carried out 55 °C.
Reaction carried out 85 °C.
Reaction carried out at 55 °C with 1 equiv of ZnCl2.
Reaction carried out in C2H4Cl2 and 2a added over 12 h.
For example, utilizing piperidine or prolinol in a condensation with 2a necessitated an increase in temperature from 0 °C to rt. Even this tactic gave suboptimal yields, and in these cases the addition of 1 equiv of zinc chloride at rt improved the outcome. The reaction of 2a with morpholine led to an unreactive precipitate being formed when zinc chloride was added, but we found that heating this reaction to 55 °C without the Lewis acid was a viable set of conditions.
Primary amines could also be used in the aromatization sequence; screening showed that the best conditions in these cases involved heating at 55 °C, with the addition of zinc chloride having a small but detrimental effect on the yield. The formation of compounds 3l, 3m, 3n, and 3o was then accomplished using the optimal conditions. Unfortunately, less nucleophilic sources of nitrogen, such as anilines, amides, and bulky amines were poor in the cyclization. The deprotection of 3m to form 3-phenyl aniline was then carried out using standard conditions (H2, Pd–C, MeOH, 92% yield) to show the potential of this method to form primary aromatic amines.
It was also desirable to examine the reaction of a relatively precious amine in order to determine whether acceptable yields could be obtained with less than 5 equiv. Therefore, we tested (+)-dehydroabietylamine and its participation in the cyclization to form 3q. In all cases we found that zinc chloride was beneficial to the reaction and that the slow addition (syringe pump) of the diketone to the reaction vessel allowed 1.5 equiv of the amine to be employed in a reaction that yielded a respectable 54% of compound 3q.
The cyclization also proved to be successful with an N,N-disubstituted hydrazine, 1-aminopiperidine, but only at 85 °C. Interestingly, the reaction of 2a with ammonia (excess) in CH2Cl2 with or without zinc chloride only gave pyridine 5 as the product, with no evidence for formation of the aniline.
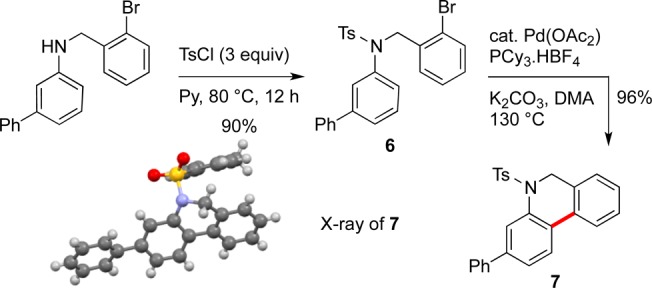
To illustrate the utility of the products of this reaction, aryl amine 3o was subjected to palladium cyclization conditions developed by Fagnou.11 Tosyl protection of aryl amine 3o gave 6 which was then submitted to cyclization conditions to furnish tricycle 7 in 92% yield (Scheme 2) (the structure of 7 was determined by X-ray crystallography12).
Scheme 2. Cyclization of a Bromo-Substituted Substrate after Aniline Formation.
Clearly, the mechanism of this cyclization is of interest. Following the reaction of 2a and pyrrolidine by NMR spectroscopy showed rapid formation of intermediate C, which slowly disappeared as either aryl amine 3a or side product enamine 4 emerged, depending upon the number of equivalents of amine used in the reaction and the temperature (Scheme 3, with higher equivalents of amine and a lower temperature favoring formation of the aryl amine product). Enamine 4 is an unwanted side product and was unreactive when resubjected to the reaction conditions.
Scheme 3. A Preliminary Mechanistic Interpretation.
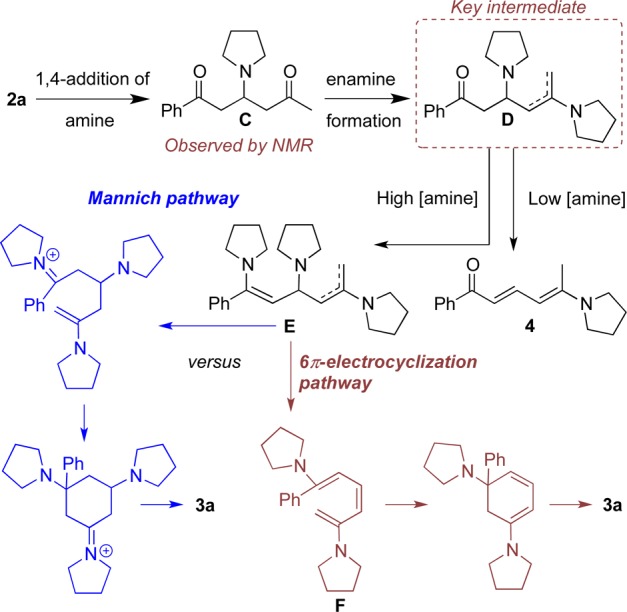
At this point we suggest the following mechanism: proceeding from C with a second equivalent of amine, enamine formation may take place on the more electrophilic carbonyl of intermediate C, resulting in the formation of another intermediate D. When lower equivalents of amine are used in the reaction, D undergoes elimination to form the side product 4. However, when a larger excess of amine is used, a second enamine formation takes place resulting in formation of bis-enamine E. We can conceive of two pathways by which this bis-enamine can form the aryl amine product 3a, originating from (i) Mannich type cyclization, followed by elimination of two molecules of pyrrolidine, or (ii) elimination of pyrrolidine and 6π-electrocyclization, followed by another elimination. We are not presently able to distinguish between these two pathways, but note that the substituted intermediate F bears a strong similarity to systems in which a facile 6π-electrocyclization is supported by calculation13 and other experimental observations.14 However, if the electrocyclization pathway is followed, then it is not clear what drives the formation of the cis-alkene that must be present in F for cyclization to occur.
In summary, a useful de novo synthesis of substituted anilines from unsaturated 1,5-dicarbonyls has been discovered. This methodology provides access to substituted anilines in good yields, and with a wide array of substituents and substitution patterns being possible. We have also performed some preliminary mechanistic experiments and suggested two distinct possibilities for aryl amine formation. Future work will be focused on further exploration of the reaction mechanism and the application of this methodology to synthetic targets.
Acknowledgments
We thank the EPSRC and AstraZeneca for support and A. L. Thompson (University of Oxford) for help with crystallographic analysis.
Supporting Information Available
Experimental procedures, characterization data for all new compounds; copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Guram A. S.; Buchwald S. L. J. Am. Chem. Soc. 1994, 116, 7901. [Google Scholar]; b Paul F.; Patt J.; Hartwig J. J. Am. Chem. Soc. 1994, 116, 5969. [Google Scholar]; For recent reviews, see:; c Surrya D. S.; Buchwald S. L. Chem. Sci. 2011, 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Surry D.; Buchwald S. L. Angew. Chem., Int. Ed. 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chan D. M. T.; Monaco K. L.; Wang R. P.; Winters M. P. Tetrahedron Lett. 1998, 39, 2933. [Google Scholar]; b Lam P. Y. S.; Clark C. G.; Saubern S.; Adams J.; Winters M. P.; Chan D. M. T.; Combs A. Tetrahedron Lett. 1998, 39, 2941. [Google Scholar]; c Monnier F.; Tailefer M. Angew. Chem., Int. Ed. 2009, 48, 6954. [DOI] [PubMed] [Google Scholar]; For a recent review, see:; d Qiao J. X.; Lam P. Y. S.. Recent Advances in Chan–Lam Coupling Reaction: Copper-Promoted C–Heteroatom Bond Cross-Coupling Reactions with Boronic Acids and Derivatives, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, 2nd ed.; Hall D. G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Vols. 1 & 2, Chapter 6. [Google Scholar]
- a Ullmann F.; Bielecki J. Ber. 1901, 34, 2174. [Google Scholar]; b Ullmann F. Liebigs Ann. 1904, 332, 38. [Google Scholar]; For recent reviews, see:; c Evano G.; Blanchard N.; Toumi M. Chem. Rev. 2008, 108, 3054. [DOI] [PubMed] [Google Scholar]; d Thomas A. W.; Ley S. V. Angew. Chem., Int. Ed. 2003, 42, 5400. [DOI] [PubMed] [Google Scholar]
- a Thirunavukkarasu V. S.; Kozhushkov S. I.; Ackermann L. Chem. Commun. 2014, 50, 29. [DOI] [PubMed] [Google Scholar]; b Collet F.; Dodd R. H.; Dauban P. Chem. Commun. 2009, 34, 5061. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Hong W. P.; Iosub A. V.; Stahl S. J. Am. Chem. Soc. 2013, 135, 13664. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cossy J.; Belotti D. Org. Lett. 2002, 4, 2557. [DOI] [PubMed] [Google Scholar]; c Neumann H.; Jocobi von Wangelin A.; Klaus S.; Stubing D.; Gordes D.; Beller M. Angew. Chem., Int. Ed. 2003, 42, 4503. [DOI] [PubMed] [Google Scholar]
- a Li L.; Zhao M.; Ren Z.; Li J.; Guan Z. Org. Lett. 2012, 14, 3506. [DOI] [PubMed] [Google Scholar]; b Sibgatulin D. A.; Volochnyuk D. A.; Kostyuk A. N. Tetrahedron Lett. 2007, 48, 2775. [Google Scholar]; c Kiren S.; Padwa A. J. Org. Chem. 2009, 74, 7781. [DOI] [PubMed] [Google Scholar]; d Barluenga J.; López L. A.; Martınez S.; Tomas M. J. Org. Chem. 1998, 63, 22. [Google Scholar]; For recent reviews on de novo synthesis of arene rings, see:; e Serra S.; Fuganti C.; Brenna E. Chem.—Eur. J. 2007, 13, 6783. [DOI] [PubMed] [Google Scholar]; f Wessig P.; Muller G. Chem. Rev. 2008, 108, 2051. [DOI] [PubMed] [Google Scholar]
- a Donohoe T. J.; Orr A. J.; Bingham M. Angew. Chem., Int. Ed. 2006, 45, 2664. [DOI] [PubMed] [Google Scholar]; b Donohoe T. J.; Bower J. F.; Basutto J. A.; Fishlock L. P.; Panayiotis A. P.; Callens C. K. Tetrahedron 2009, 8969. [Google Scholar]; c Donohoe T. J.; Fishlock L. P.; Basutto J. A.; Bower J. F.; Procopiou P. A.; Thompson A. L. Chem. Commun. 2009, 21, 3008. [DOI] [PubMed] [Google Scholar]; d Donohoe T. J.; Fishlock L. P.; Procopiou P. A. Chem.—Eur. J. 2008, 14, 5716. [DOI] [PubMed] [Google Scholar]
- a Donohoe T. J.; Bower J. F.; Chan L. K. Org. Biomol. Chem. 2012, 10, 1322. [DOI] [PubMed] [Google Scholar]; b Donohoe T. J.; Bower J. F. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3373. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Donohoe T. J.; Race N. J.; Bower J. F.; Callens C. K. Org. Lett. 2010, 12, 4094. [DOI] [PubMed] [Google Scholar]; d Donohoe T. J.; Basutto J. A.; Rathi A. H.; Bower J. F. Org. Lett. 2011, 13, 1036. [DOI] [PubMed] [Google Scholar]; e Donohoe T. J.; Bower J. F.; Baker D. B.; Basutto J. A.; Chan L. K.; Gallagher P. Chem. Commun. 2011, 47, 10611. [DOI] [PubMed] [Google Scholar]
- Zhan Z.-Y. J. U.S. Patent, 2007, 20070043180; this catalyst is commerically available. It was necessary to use 5 equiv of the enone component to ensure maximum yields from the CM reaction.
- Dess D. B.; Martin J. C. J. Org. Chem. 1983, 48, 4155. [Google Scholar]
- Campeau L.; Parisien M.; Jean J.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 681. [DOI] [PubMed] [Google Scholar]
- Single crystal diffraction data for 7 were collected on Oxford Diffraction SuperNova diffractometer (λ(Mo Kα) = 0.71070 Å) at 120 and 300 K. Cell parameters and intensity data for both data sets were determined using CrysAlisPro, and the structure was solved by charge-flipping using ‘Superflip’:; a Palatinus L.; Chapuis G. J. Appl. Crystallogr. 2007, 40, 786. [Google Scholar]; Both structures were refined by full-matrix least squares on F2 using the CRYSTALS suite:; b Betteridge P. W.; Carruthers J. R.; Cooper R. I.; Prout K.; Watkin D. J. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar]; c Cooper R. I.; Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2010, 43, 1100–1107. [Google Scholar]; d Cooper R. I.; Gould R. O.; Parsons S.; Watkin D. J. J. Appl. Crystallogr. 2002, 35, 168–174. [Google Scholar]; Full refinement details are given in the Supporting Information (CIF); crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre (CCDC 986297) and can be obtained via www. ccdc.cam.ac.uk/data_request/cif.
- Yu T.; Fu Y.; Liu L.; Guo Q. J. Org. Chem. 2006, 71, 6157. [DOI] [PubMed] [Google Scholar]
- For examples of 6π-electrocyclization based routes to aryl amines:; a Davies I. W.; Marcoux J.; Kuethe J. T.; Lankshear M. D.; Taylor J. D.; Tsou N.; Dormer P. G.; Hughes D. L.; Houk K. N.; Guner V. J. Org. Chem. 2004, 69, 1298. [DOI] [PubMed] [Google Scholar]; b Guner V. A.; Houk K. N.; Davies I. W. J. Org. Chem. 2004, 69, 8024. [DOI] [PubMed] [Google Scholar]; c Bianchi L.; Dell’Erba C.; Maccagno M.; Petrillo G.; Rizzato E.; Sancassan F.; Severi E.; Tavani C. J. Org. Chem. 2005, 70, 8734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.