

Abstract
The electronic and steric effects in the stoichiometric dehydrocoupling of secondary and primary phosphine–boranes H3B·PR2H [R = 3,5-(CF3)2C6H3; p-(CF3)C6H4; p-(OMe)C6H4; adamantyl, Ad] and H3B·PCyH2 to form the metal-bound linear diboraphosphines H3B·PR2BH2·PR2H and H3B·PRHBH2·PRH2, respectively, are reported. Reaction of [Rh(L)(η6-FC6H5)][BArF4] [L = Ph2P(CH2)3PPh2, ArF = 3,5-(CF3)2C6H3] with 2 equiv of H3B·PR2H affords [Rh(L)(H)(σ,η-PR2BH3)(η1-H3B·PR2H)][BArF4]. These complexes undergo dehydrocoupling to give the diboraphosphine complexes [Rh(L)(H)(σ,η2-PR2·BH2PR2·BH3)][BArF4]. With electron-withdrawing groups on the phosphine–borane there is the parallel formation of the products of B–P cleavage, [Rh(L)(PR2H)2][BArF4], while with electron-donating groups no parallel product is formed. For the bulky, electron rich, H3B·P(Ad)2H no dehydrocoupling is observed, but an intermediate Rh(I) σ phosphine–borane complex is formed, [Rh(L){η2-H3B·P(Ad)2H}][BArF4], that undergoes B–P bond cleavage to give [Rh(L){η1-H3B·P(Ad)2H}{P(Ad)2H}][BArF4]. The relative rates of dehydrocoupling of H3B·PR2H (R = aryl) show that increasingly electron-withdrawing substituents result in faster dehydrocoupling, but also suffer from the formation of the parallel product resulting from P–B bond cleavage. H3B·PCyH2 undergoes a similar dehydrocoupling process, and gives a mixture of stereoisomers of the resulting metal-bound diboraphosphine that arise from activation of the prochiral P–H bonds, with one stereoisomer favored. This diastereomeric mixture may also be biased by use of a chiral phosphine ligand. The selectivity and efficiencies of resulting catalytic dehydrocoupling processes are also briefly discussed.
Short abstract
The effect of changing the steric and electronic profile of the phosphine of primary and secondary phosphine−boranes H3B·PR2H (R = H, aryl, adamantyl) on the Rh-catalyzed dehydrocoupling to form metal-bound linear diboraphosphines is reported.
Introduction
The development of efficient catalytic methods for the formation of bonds between main group elements is of considerable interest for the continued development of main group chemistry. Such processes enable new discoveries to be made in the promising application areas that main group species are now occupying, such as high performance polymers, emissive materials, etch resists for lithography, and precursors to ceramic thin films or devices.1−6 However, the development of this field lags substantially behind the advances made in catalytic C–C and C–X bond formation, for which there are now a myriad of efficient ways to promote such unions that are important for the construction of new molecules. Catalytic dehydrocoupling5,7,8 of amine– and phosphine–boranes is one method that has emerged for the formation of B–N and B–P bonds, and development in the area has been spurred on by the potential for ammonia–borane to act as a hydrogen carrying vector.9−11 In addition, polymeric materials that can arise from dehydropolymerization of primary analogues are also of significant interest as they are valence isoelectronic with technologically ubiquitous polyolefins. Although the metal catalyzed formation of polyaminoboranes has attracted recent attention,12−18 catalytic routes to polyphosphinoboranes have also been known since 1999.19 Perhaps the best example is that of the [Rh(COD)2][OTf] catalyzed dehydrocoupling of secondary, H3B·PR2H, and primary, H3B·PRH2, phosphine–boranes to give oligomeric and polymeric materials (Scheme 1).19−21
Scheme 1. Phosphine–Borane Dehydrocoupling.

In contrast to amine–borane dehydrocoupling,8,10,15,22−24 the mechanism of catalytic dehydrocoupling of phosphine–boranes has received less attention. Although initial reports demonstrated that catalysis using [Rh(COD)2][OTf] was a homogeneous process (i.e., not colloidal),25 there has been only sporadic further work on elucidating the mechanistic details.26−29 Progress has no doubt been slowed due to the fact that the reaction conditions reported for phosphine–borane dehydrocoupling often require melt conditions, thus making interrogation of the catalytic cycle problematic. Recently, we have reported that the Rh(I) complexes [Rh(PtBu2H)2(η6-FC6H5)][BArF4],30 and [Rh(L)(η6-FC6H5)][BArF4],31 [L = Ph2P(CH2)3PPh2] are particularly well-suited to the study of the dehydrocoupling mechanism of secondary phosphine–boranes in solvents such as fluorobenzene; and on the basis of the observation of intermediates, kinetic studies, and H/D exchange experiments we have proposed a catalytic cycle for the dehydrocoupling of H3B·PR2H (R= Ph, tBu; Scheme 2). For this cycle, intermediate species were isolated, but their structures could not be confirmed by X-ray crystallography. In particular for R = Ph, a β-B-agostic σ complex B, and the product of dehydrocoupling F, that is proposed to sit off cycle, could be isolated and spectroscopically characterized. Under stoichiometric conditions the observation that B transforms into F on gentle heating allowed for kinetic parameters to be determined that suggested that the rate-determining step(s) for dehydrocoupling were located within the transformations B to D. In solution phase the turnover limiting step for catalysis is proposed to be the displacement of the linear diboraphosphine product (i.e., F to A), although under the melt conditions used for efficient catalysis this may well be different. Further insight comes from the observations that for R = tBu the barrier to dehydrocoupling is higher (70 °C versus 25 °C for reaction), P–H activation appears also to be a higher energy process, different intermediates (A and E) are observed, and the turnover limiting process in catalysis is now suggested to be the P–H activation/dehydrocoupling steps. Prior work has demonstrated a similar difference in relative rates of dehydrocoupling of secondary H3B·PR2H [R = p-(CF3)C6H4, Ph, tBu, iBu] and primary H3B·PRH2 [R = Ph, tBu, iBu] phosphine–boranes using the [Rh(COD)2][OTf] catalyst, and this was suggested to be due to a combination of steric and electronic (relative P–H bond strengths) factors,21,32,33 although the mechanism of dehydrocoupling of phosphine–boranes using this catalyst is currently not known.20,25,30 Interestingly, the related dehydrogenation of aryl amine–boranes shows that the activity of the N–H bond is such that spontaneous dehydrocoupling occurs in the absence of catalyst, with electron-withdrawing aryl groups [p-(CF3)C6H4] undergoing faster reaction than electron-donating [p-(OMe)C6H4].34 Very recent work has shown that paramagnetic Ti(III) centers might also be involved in dehydrocoupling of phosphine– and amine–boranes when using Cp2Ti-based catalysts,35 while oligomerization of base-stabilized phosphino–boranes at Cp2Ti centers has been described.29 Likely decomposition routes in Rh-systems for phosphine–borane dehydrocoupling to form bis(phosphine)boronium salts have also recently been discussed.36
Scheme 2. Proposed Catalytic Cycle for the Dehydrocoupling of H3B·PR2H To Give H3B·PR2BH2·PR2H.

[BArF4]− anions are not shown.
In this Article, we report an extension of our investigations into the mechanism of phosphine–borane dehydrocoupling using the {Rh(Ph2P(CH2)3PPh2)}+ fragment, by varying the electronic and steric profile of the secondary phosphine–boranes H3B·PR2H [R = 3,5-(CF3)2C6H3; p-(CF3)C6H4; p-(OMe)C6H4; adamantyl], as well as investigations with the primary phosphine–borane H3B·PCyH2. Dehydrocoupling forms the corresponding metal–bound linear diboraphosphines H3B·PR2BH2·PR2H and H3B·PRHBH2·PRH2, respectively. These studies provide insight into the determining role of the electronics and sterics of the phosphine–borane in the dehydrocoupling process, as well as providing as yet unreported examples of the solid-state structures of the intermediates related to the catalytic cycle. We also report for the first time the partial control of diastereoselectivity in dehydrocoupling of primary phosphine–boranes, that can additionally be biased by use of a chiral chelating phosphine on the rhodium center.
Results and Discussion
Phosphine–Borane and Diboraphosphine Starting Materials
A range of secondary phosphine–boranes with differing electronic and steric properties have been used in this study (1, 2, 3, and 4, Figure 1), which also provide comparison with the previously reported Ph, 6, and tBu, 7, analogues.31 The primary phosphine–borane 5 has also been used.37 Compounds 2(33) and 3(38) are known adducts and offer electron-withdrawing and donating aryl groups, respectively. Bis-CF3-substituted 1 is a new complex and offers an alternative to 2. The synthesis of adamantyl-substituted phosphine, 4, an analogue of 7, has been reported in the patent literature.39 Compared with the tButyl group, adamantyl has a greater steric bulk due to its larger volume and rigid structure.40,41 The new linear diboraphosphines, 10–13, have also been synthesized to aid in the identification of final dehydrocoupling products. Complexes 10–12 are synthesized by a Rh-catalyzed process from the corresponding phosphine–boranes, while primary phosphine containing 13 has been synthesized in good isolated yield (85%) by addition of [NBu4][BH4] to the bis(phopshine)boronium [(CyH2P)2BH2]Br.36
Figure 1.

Phosphine–boranes 1–7 and diboraphosphines 8–13.
Stoichiometric Dehydrocoupling of Secondary Phosphine–Boranes
Addition of 2 equiv of 1 to [Rh(L)(η6-FC6H5)][BArF4] [L = Ph2P(CH2)3PPh2] in 1,2-F2C6H4 solution at 25 °C rapidly (on time of mixing) resulted in the formation of [Rh(L)(H)(σ,η-PR2BH3)(η1-H3B·PR2H)][BArF4], 14 [R = 3,5-(CF3)2(C6H3), Scheme 3], which was characterized by NMR spectroscopy, ESI-MS (electrospray ionization mass spectrometry), and single crystal X-ray diffraction. Likewise, the use of 2 equiv of phosphine–borane 2 or 3 results in the formation of the analogous complexes 15 [R = p-(CF3)C6H4] and 16 [R = p-(OMe)C6H4], respectively, which were fully characterized using solution techniques. All these complexes proceed to dehydrocouple (vide infra), and only for 14 was an analytically pure crystalline solid obtained. Even so, dissolution of crystalline material of 14 resulted in the observation of small amounts (approximately 5–10%) of the associated dehydrocoupling product in the solution NMR spectra after short periods of time. Complexes 15 and 16 could only be isolated as oils, but their characterization by NMR spectroscopy and ESI-MS was fully consistent with their formulation.
Scheme 3. Synthesis of Complexes 14, 15, and 16.
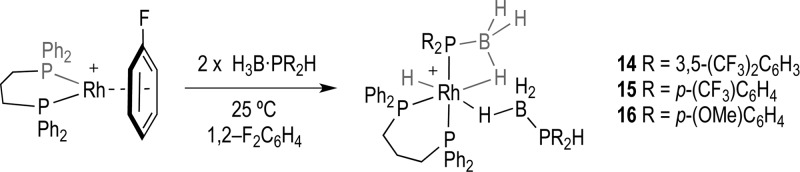
[BArF4]− anions are not shown.
The solution NMR spectra for 14, 15, and 16 are very similar to those previously reported for [Rh(L)(H)(σ,η-PPh2BH3)(η1-H3B·PPh2H)][BArF4] (i.e., B, Scheme 2(31)), and data for 14 is discussed in detail. The 31P{1H} NMR spectrum of 14 shows four different phosphorus environments. Two of the resonances are broadened significantly compared to the other two, suggesting these phosphorus atoms are bound to a quadrupolar boron center. One of these shows both a large trans PP coupling [J(PP) 244 Hz] and coupling to 103Rh [J(RhP) 75 Hz], while the other is a broad singlet. The other two signals are sharper and are assigned to the two 31P environments of the Ph2P(CH2)3PPh2 ligand. One of these sharper resonances [δ 29.5, ddd, J(RhP) 130, J(PP) 35, J(PP) 21 Hz] is assigned to the phosphorus atom trans to the weakly bound β-B-agostic interaction on the basis of the larger 103Rh coupling constant, while the other signal [δ 11.3, ddd, J(RhP) 103, J(PP) 244, J(PP) 35 Hz] is assigned to the phosphorus atom trans to the coordinated phosphido ligand. In the 1H NMR spectrum of 14 one broad, relative integral 3H, signal is observed at δ −0.78, indicative of a Rh···H3B σ interaction in which the B–H bonds are undergoing rapid site exchange on the NMR spectroscopic time scale between terminal and bridging sites.42 A broad, relative integral 1H, resonance at δ −6.12 is assigned to a static β-B-agostic B–H interaction. Cooling of the solution to 0 °C led to the resolution of this signal as doublet [J(PH) = 65 Hz], fully consistent with its trans disposition to a phosphine. The remaining BH(terminal) signals are not observed, and it is likely they are coincident with the {CH2}3 signals. A sharper signal at δ −16.21, relative integral 1H, is assigned to a metal–hydride resonance, in which the coupling to both 103Rh and 31P is clearly small and unresolved. The PH group is observed at δ 5.81 that collapses into a singlet in the 1H{31P} NMR spectrum. The 11B NMR spectrum shows a broad signal centered at δ −39.8, which is not shifted significantly from that of free phosphine–borane 1 (δ −42.0). This is assigned to a coincidence of the η1 β-B–H···Rh agostic and σ Rh···H3B signals, as has been noted previously.31,43 Complexes 15 and 16 have similar 1H, 11B, and 31P NMR spectra, and thus we assign very similar structures.
Crystals of complex 14 of suitable quality for analysis by X-ray diffraction were obtained by layering of a 1,2-F2C6H4 solution with pentane at −26 °C. The structure of 14 in the solid-state (Figure 2) is fully consistent with the structure deduced from the solution NMR spectroscopic data. The formally Rh(III) center adopts a pseudo-octahedral geometry, with the chelating phosphine ligand and the hydride located on one of the faces of the octahedron. Two of the three remaining coordination sites are occupied by a phosphine–borane unit that has undergone P–H activation, and is bound to the metal via a phosphido bond [Rh1–P3, 2.3045(10) Å] and a β-B-agostic bond [Rh1–B1, 2.515(4) Å]. The other phosphine–borane unit occupies the last coordination site via a σ η1-Rh···H–B interaction.42 All the hydrides (B–H and Rh–H) were located in the final difference map. The structure is in full accord with the solution NMR spectroscopic data, confirming the spectroscopic assignments that have been made previously.31 β-B-agostic interactions are known,35,44,45 and we have recently reported [Rh(κ1,η-PPh2BH2·PPh3)(PPh3)2][BArF4] in which a base-stabilized phosphine–borane adopts a β-B-agostic interaction with the Rh-center.36 σ phosphine–boranes are also known,42,46,47 and bimetallic complexes showing both B-agostic and σ borane coordination modes have been reported.48 Compared to a Rh(I) complex that shows a bidentate η2-coordination mode for the σ borane, [Rh(PtBu2H)2(η2-H3B·PtBu2H)][BArF4],30 the Rh···B distance for the η1-interaction in 14 is considerably longer [2.188(3) Å versus 2.740(4) Å, respectively], consistent with this different binding motif. Similar changes in M···B distance have been noted on moving between η1 and η2 coordination modes in chelating phosphine–boranes.43
Figure 2.

Molecular structure of the cation of 14. Displacement ellipsoids are drawn at 50% probability level. Some hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Rh1–P1, 2.778(10); Rh1–P2, 2.3163(9); Rh1–P3, 2.3045(10); P3–B1, 1.913(4); P4–B2, 1.918(4); Rh1···B1, 2.515(4); Rh1···B2, 2.740(4); Rh1–P3–B1, 72.54(14); Rh1–B2–P4, 121.3(2).
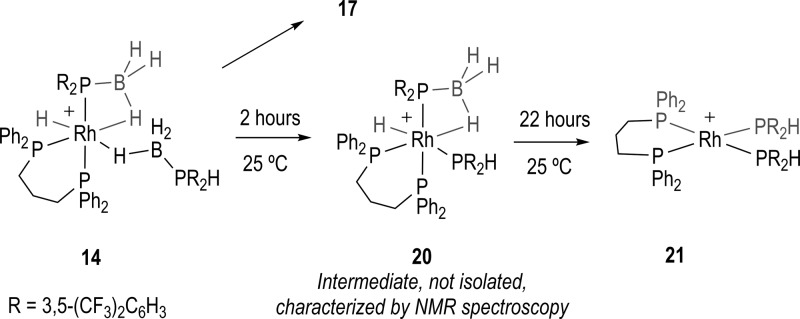
Complexes 14–16 undergo spontaneous dehydrocoupling (25 °C) to form products of the general formula [Rh(L)H(σ,η2-PR2·BH2PR2·BH3)][BArF4]: 17, R = 3,5-(CF3)2C6H3; 18, R = p-(CF3)C6H4; 19, R = p-(OMe)C6H4 (Scheme 4). This process also results in the liberation of H2 (observed, 1H NMR spectroscopy). For 17 and 18 there are additional products formed, assigned as [Rh(L)(PR2H)2][BArF4], 21 and 22, respectively, on the basis of NMR spectroscopic data. These complexes are formed in parallel to 17 and 18, as preformed 17 (vide infra) does not proceed to form 21. Complex 21 has been independently prepared by addition of two equivalents of HP((CF3)2C6H3)2 to [Rh(L)(η6-FC6H5)][BArF4].
Scheme 4. Dehydrocoupling of Complexes 14–16.
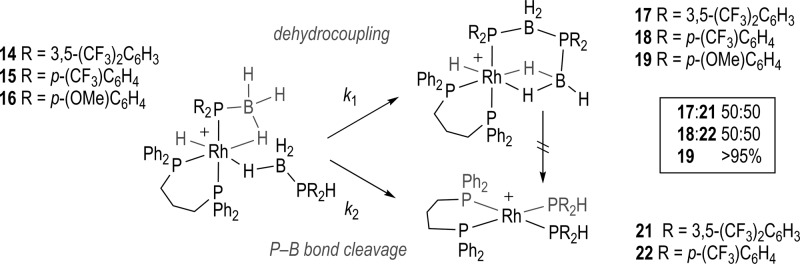
[BArF4]− anions are not shown. Time = 6 h 17/21, 18/22 (25 °C); 8 h 16/19 (35 °C).
This mixture of products observed for the electron-withdrawing phosphine substituents (i.e., 1 and 2) contrasts with that found for when R = Ph31 and p-(OMe)C6H4, which yield the dehydrocoupled (e.g., 19 and F, Scheme 2) product in essentially quantitative form (∼95% by 31P{1H} NMR spectroscopy). Complex 17 has been synthesized cleanly from direct addition of the preformed dehydrocoupled diboraphosphine product, 10, to [Rh(L)(η6-FC6H5)][BArF4], Scheme 5. It was from this reaction that material of 17 suitable for single crystal X-ray diffraction was obtained.
Scheme 5. Synthesis of 17 by Direct Addition of the Linear Diboraphosphine 10.
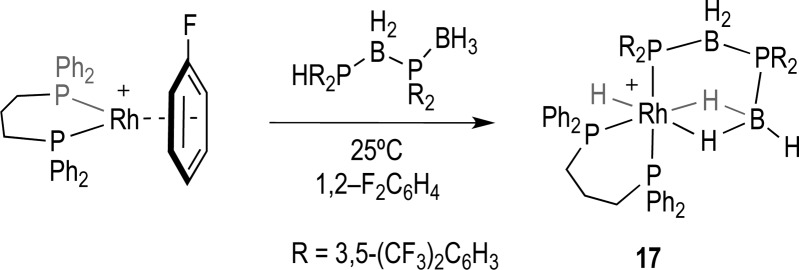
[BArF4]− anions are not shown.
Figure 3 shows the solid-state structure of 17, in which the diboraphosphine acts as a chelate to the Rh(III) center, via a phosphido group and two B-agostic interactions: [Rh(L)H(σ,η2-PR2·BH2PR2·BH3)][BArF4] [R = 3,5-(CF3)2C6H3]. All the hydride ligands (B–H and Rh–H) were located in the final difference map. The Rh(III) center has pseudo-octahedral geometry, in which the oligomeric phosphine–borane is bound tridentate to the metal through η2-BH2···Rh [B2–Rh1, 2.280(5) Å] and phosphido [P3–Rh1, 2.3925(10) Å] interactions. The hydride ligand is positioned trans to one of the B–H···Rh interactions. The Rh···B distance is considerably shorter than those observed in 14, consistent with the η2-bidentate binding mode of the borane. This distance is similar to others reported for chelating phosphine–borane complexes with Rh.49−52
Figure 3.

Molecular structure of the cation of 17. Displacement ellipsoids are drawn at 50% probability level. Some hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Rh1–P1, 2.3241(11); Rh1–P2, 2.2650(11); Rh1–P3, 2.3925(10); Rh1···B2, 2.280(5); Rh1–P3–B1, 110.88(15); B1–P4–B2, 107.5(2).
The NMR spectroscopic data for 17 are fully consistent with the solid-state structure being retained in solution and are also very similar to that reported for the analogous complex formed from the deydrocoupling of 6 (R = Ph).31 The 31P{1H} NMR spectrum shows four different phosphorus environments. Two of these signals are well-resolved and show coupling to 103Rh, δ 46.6 [J(RhP) 111 Hz] and δ 12.8 [J(RhP) 91 Hz], and are attributed to the chelating phosphine ligand. One of these signals (δ 12.8) also shows large 31P–31P coupling [J(PP) 260 Hz] suggesting a trans position relative to the phosphido center. The other two environments are broad, typical of those observed when coupling to a quadrupolar boron center. For one of these trans J(PP) coupling is also observed. The 1H NMR spectrum shows three different broad, relative integral 1H, environments assigned to the BH3 moiety [δ −4.54, −1.20, and 4.37]. This indicates that the BH3 unit is not undergoing exchange on the NMR spectroscopic time scale, as noted previously for similar η2-M···H3B systems.31,43,50,52 The Rh–H signal is observed at δ −13.98 as a sharper signal, although this also shows unresolved coupling. The 11B NMR spectrum shows two different environments [δ −27.1 and 0.21] for the two boron atoms present in the diboraphosphine, with the latter assigned to the η2-H3B unit on the basis of the large downfield shift from free ligand (Δδ = +36.8).43 Spectroscopic data for complexes 18 and 19, that are produced by the direct dehydrocoupling route are similar, although for 18 this is also formed as a mixture with 22.
The dehydrocoupling reaction (i.e., 14 to 17) shows a dependence on the substituents on the phosphine. For electron-withdrawing aryl groups (e.g., p-CF3), it is faster when compared with electron rich groups (i.e., p-OMe). Following these processes in situ using NMR spectroscopy demonstrated that these dehydrocoupling reactions follow a first order rate profile for the consumption of the starting material over at least three half-lives (see Supporting Information): 1 3 h (25 °C); 2 3 h (25 °C); 6 14 h (25 °C);313 8 h (35 °C), ∼120 h (25 °C). That the parallel products 21 and 22 are formed in approximately equal ratio to the dehydrocoupled product (17, 18, respectively)) suggests that k1 ≈ k2 (Scheme 4). In addition to this parallel process, direct comparison of the rate constants is further complicated by the fact that 16 → 19 required heating to 35 °C to make the reaction run over a convenient time scale for analysis by NMR spectroscopy. Nevertheless these relative rates reflect previous observations on the rate of catalytic dehydrocoupling when the electronics of a system are changed, in as much as electron-withdrawing groups promote the reaction.21 Interestingly, for all the aryl complexes initial P–H activation to form a phosphido hydride complex (i.e., 14) is very rapid, occurring on time of mixing. This suggests that for aryl-substituted phosphine–boranes it is not initial P–H activation that is rate-determining for the dehydrocoupling event, as we have commented on for R = Ph.31 In this study we suggested that B–H activation/reorganization in intermediates such as B (Scheme 2) prior to P–B bond formation might be the rate limiting process.31 This might well be promoted by a weaker B–H bond, and calculations on analogous H3B·L (L = Lewis base) systems show that the B–H bond is considerably weaker when there are electron-withdrawing groups on the Lewis base.53 However, we cannot rule out that the relative P–H bond strengths in intermediates such as 14 also might play a role, or that there is a change in the rate determining step on changing the phosphine–borane ligand, as the intimate details of the mechanism leading to P–B formation still remain to be resolved. The observation that for an electron-withdrawing phosphine there is a significant proportion of parallel product formed that results from P–B bond cleavage is consistent with the weakening of the P–B bond with increasingly electron-withdrawing aryl substiutents.8,54 P–B bond cleavage has been noted previously in σ phosphine–borane complexes to give either simple adducts47 or further reaction to yield bis(phosphine)boronium salts.30
Prior to the formation of the parallel product 21 (R = 3,5-(CF3)2C6H3) an intermediate is observed that has been characterized by 1H and 31P{1H} NMR spectroscopy as [Rh(L)H(σ,η-PR2·BH3)(PR2H)][BArF4] 20, i.e., a complex that sits directly between 14 and 21 by loss of one “BH3” fragment (Scheme 6). Complex 20 results from P–B bond cleavage, formally of the σ-H3B·PR2H ligand, to afford a complex with a β-B-agostic interaction from a phosphide borane ligand (as for 14) and a simple PR2H ligand trans to a hydride. Complex 20 was not isolated in pure form, being observed alongside 14 and the final products 17/21. However, after 2 h reaction a significant proportion of 20 is present (∼20% by 31P NMR spectroscopy), allowing for its identification aided by comparison with the NMR spectroscopic data for 14 (Supporting Information). In particular four environments are observed in the 31P NMR spectrum, with only one of these broadened significantly by coupling to quadrupolar boron. This signal also shows a large, mutual, trans J(PP) coupling with another phosphine environment. In the high-field region of the 1H NMR spectrum a broad doublet is observed at δ −7.06 [J(HP) = 76 Hz] which is assigned to the β-B-agostic interaction, while there is a relatively sharper one at δ −9.61 [J(HP) = 165 Hz] assigned to Rh–H, and again 103Rh coupling is not resolved. These assignments were confirmed by 1H{31P}, 1H{11B}, and 1H/31P correlation experiments.
Scheme 6. Formation of the Parallel Products 17 and 21 from 14.
[BArF4]− anions are not shown.
Addition of 2 equiv of the bulky and electron rich phosphine–borane H3B·P(adamantyl)2H, 4, to [Rh(L)(η6-FC6H5)][BArF4] in 1,2-F2C6H4 solution at 25 °C rapidly results in a color change from orange to purple and the formation of the new σ bound Rh(I) phosphine–borane complex [Rh(L)(η2-H3B·P(adamantyl)2H)][BArF4], 23, which was characterized in situ by NMR spectroscopy. This complex could not be isolated as it undergoes further reaction, by P–B bond cleavage at room temperature, to form 24 (Scheme 7). Addition of 1 equiv of 4 resulted in a final mixture of 24 and [Rh(L)(η6-FC6H5)][BArF4].
Scheme 7. Synthesis of Complex 24 by Direct and Indirect Routes.

[BArF4]− anions are not shown.
The 1H NMR spectrum of complex 23 immediately after preparation shows a broad, relative integral 3H, signal at δ −1.36 characteristic of a σ-bound phosphine–borane that is undergoing site exchange between the coordinated and uncoordinated B–H environments.42 Two signals are observed in the 31P{1H} NMR spectrum, in a 2:1 ratio at δ 35.1 [J(RhP) 167 Hz] and δ 30.1 (br). Over time (1 h), complex 23 disappears to be replaced by a new complex that has been characterized by NMR spectroscopy and a solid-state X-ray diffraction experiment as [Rh(L)(PHR2)(η1–H3B·PHR2)][BArF4] (24, R = adamantyl). Figure 4 shows the structure of the cation present in 24 in the solid-state. A Rh(I) center is in a pseudo-square-planar geometry with a chelating ligand, and the other two coordination sites are occupied by P(adamantyl)2H and a η1-H3B·P(adamantyl)2H [Rh···B, 2.457(7) Å] ligands, respectively. The BH and PH hydrogen atoms were located in the final difference map. The solution NMR spectroscopic data for 24 are fully consistent with the solid-state structure, and in particular the trans disposition of P1 and P3, and the η1-H3B·PR2H ligand.
Figure 4.

Molecular structure of the cation of 24. Displacement ellipsoids are drawn at 50% probability level. Some hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Rh1–P1, 2.2262(16); Rh1–P2, 2.2861(16); Rh1–P3, 2.3568(15); Rh1···B1, 2.457(7); B1–P4, 1.936(7).
A significant amount of P–B bond cleavage product is thus observed for both electron poor aryl phosphine–boranes (e.g., 14) and very bulky electron rich phosphine–boranes (e.g., 24), but not the electron rich aryl phosphine 3 or H3B·PPh2H (6).31 Interestingly we have recently reported that for H3B·PtBu2H P–B bond cleavage is also observed during dehydrocoupling catalysis being accompanied by a further dehydrocoupling step, through which bis(phosphine)boronium salts are ultimately formed.30,36 Similar complexes can be prepared on rhodium using H3B·PPh2H and PPh3 under stoichiometeric conditions.36 One suggested mechanism for this process is the reaction of a short-lived phosphino–borane (or its masked equivalent) with coordinated phosphine, not dissimilar to the mechanism suggested for the formation of diaminoboranes from amine–boranes and amines catalyzed by alkaline earth catalysts.55 Complexes 20 and 24 serve as models for intermediates in this process [Rh(III) and Rh(I), respectively], although we do not observe the formation of corresponding bis(phosphine)boronium salts in this case.
Stoichiometric Dehydrocoupling of Primary Phosphine–Boranes
The dehydrocoupling of primary phosphine–boranes can yield polyphosphinoboranes, rather than the simple oligomers observed with secondary phosphine–boranes (Scheme 1). With an appreciation of the intermediate metal complexes formed with secondary phosphine–boranes from this and previous work,30,31,36 it was of interest to explore whether the proposed dehydrocoupling mechanism for secondary phosphine–boranes using [Rh(L)(η6-FC6H5)][BArF4] could be applied to primary analogues. Such insight into the mechanism of dehydropolymerization of phosphine–boranes is important, as these processes currently remain unresolved due to the melt conditions employed that make following intermediates or kinetics problematic.20,28,33
In situ investigations using stoichiometric quantities of primary phosphine–boranes H3B·PPhH2 resulted in immediate reaction when combined with [Rh(L)(η6-FC6H5)][BArF4], but a number of products were formed which we have not been able to convincingly characterize. This mixture of species observed is in contrast with H3B·PPh2H where single products are formed analogous to 14–16.31 However, reaction of [Rh(L)(η6-FC6H5)][BArF4] with a slight excess of H3B·PCyH2 (5) in 1,2-F2C6H4 solution at 25 °C led to the instantaneous formation of only two complexes in a 1:1 ratio, 25a and 25b, [Rh(L)H(σ,η-PCyH·BH3)(η1-H3B·PCyH2)][BArF4], as a proposed diastereomeric pair (Scheme 8). This stereoisomerism comes from P–H activation at the prochiral primary phosphine. These new products are directly analogous to those formed with secondary phosphine–boranes (i.e., 14), and the NMR spectroscopic data match closely. The 31P{1H} NMR spectrum from this reaction shows 8 resonances, in addition to a broad peak at δ −35.5 due to excess phosphine–borane, as each diastereomer contains four distinct phosphorus environments. Signals centered at δ 31.7 and 30.5 are assigned to one of the chelating phosphine ligand 31P environments in each diastereoisomer, and show characteristic J(RhP) coupling constants consistent with a Rh(III) center. Complex overlapping multiplets at δ 11.8 [2 × ddd] represent the resonances for both diastereomers of the second chelated phosphorus center, which is trans to the phosphide position, displaying a large trans PP coupling constant [J(PP) ∼ 200 Hz] in addition to coupling to 103Rh and cis-31P. The remaining 4 signals are broad indicating the phosphorus centers are bound to a quadrupolar 11B nucleus. Of these, peaks at δ −11.0 and −32.1 are assigned to the phosphide centers of each diastereomer trans to the chelating phosphine [J(PP) ∼200 Hz], and resonances at δ −39.8 and δ −44.2 as assigned to phosphorus centers in the σ-bound phosphine–borane unit. These large differences in chemical shift of the phosphido signal (Δδ 21.2) might reflect significant local difference in steric pressure between 25a and 25b at this group. Interestingly, a much smaller difference is observed with the dehydrocoupled products (26a/b, Δδ 3.5) in which the phosphide group is part of a chelate ring. The 1H NMR spectrum does not have the necessary resolution to separate out the diastereomers in the hydride region, with broad resonances observed at δ −2.3 (3 H, BH3), δ −7.9 (1 H, Rh–H–B), δ −17.5 (Rh–H).
Scheme 8. Synthesis of 25a, 25b, and the Dehydrocoupled Products 26a and 26b.

[BArF4]− anions are not shown.
Complexes 25a/b cannot be isolated in pure form, and characterization by NMR spectroscopy is best performed on freshly prepared samples, as after 1 h (25 °C) they have undergone dehydrocoupling to give a mixture of two resolvable diastereomers 26a and 26b, with one of the diastereomers present in a significantly larger amount ∼6:1 (Scheme 8), indicating that the dehydrocoupling step occurs with some stereocontrol.56 The decomposition product [Rh(L)(PH2Cy)2]+, analogous to 21/22, was also observed. NMR spectroscopic and ESI-MS analysis suggests that the dehydrocoupling products formed are direct analogues of 17. This mixture of diastereomers can also be synthesized cleanly by direct reaction of [Rh(L)(η6-FC6H5)][BArF4] with the preformed diaboraphosphine H3B·PCyHBH2·PCyH2 (13) in 1,2-F2C6H4 solution at 25 °C (Figure 5 for the solid-state structure). Immediate measurement of the 31P{1H} NMR spectrum after mixing showed clean conversion to complexes 26a and 26b in an approximate 1:1 ratio, interestingly different from the 1:6 ratio observed from dehydrocoupling.
Figure 5.

Molecular structure of 13. Displacement ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (deg): P1–B1 1.9267(18), B1–P2 1.9381(18), P2–B2 1.926(2); P1–B1–P2 108.34(9), B1–P2–B2 113.32(9).
Resonances in the 31P{1H} NMR spectrum of 26 can, again, be assigned aided by reference to those of structurally characterized 17. Peaks centered at δ 37.9 and 34.5 result from the chelated phosphorus trans to the B-agostic site, while the signals for the phosphorus trans to the phosphido group overlap at δ 10.7, and display characteristic J(PP) trans coupling [255 Hz]. The broad resonances of the diboraphosphine are observed at δ 19.8 and 16.2 for the phosphido center [J(PP) 255] and δ −14.9 and −16.6 ppm for the remaining site. The high-field region of the 1H NMR spectrum of 26a/26b shows a slight downfield shift of the Rh–H hydride resonance to δ −16.1, when compared to 25a/25b, while the η2-BH2···Rh units are observed as two broadened resonances at δ −2.98 (1H) and δ −5.98 (1H). For these hydride signals the separate signals are not resolved for each diasteroisomer, although each resonance is rather asymmetric suggesting two overlapping environments.
A 31P{1H} NMR spectrum taken of this mixture after 18 h at 25 °C showed a significant change in the ratios of the diastereomers 26a/26b (Scheme 9). The peaks for one isomer at [δ 34.5, 16.2, 10.7, and −14.9] have reduced relative area, giving an approximate ratio of 6:1 for the two diastereoisomers. This ratio is similar to that found from direct dehydrocoupling in 25a/25b after 1 h (vide supra), underscoring the stereocontrol occurring in the P–B bond forming process. Leaving this solution for one week resulted in no significant change to this ratio, suggesting equilibrium had been reached. We suggest that the mechanism for equilibration involves reductive elimination of the phosphido and hydride ligands to form a Rh(I) σ phosphine–borane complex,30 similar to E in Scheme 2, which then undergoes rapid oxidative addition of the other P–H bond. This must be a reversible process, leading to a thermodynamic ratio of the diastereoisomers and the resulting selectivity. Unfortunately we were unable to deduce the stereochemistry of the preferred isomer using ROESY experiments or a solid-state structure. However, inspection of models leads us to propose that the thermodynamic product is likely to have the cyclohexyl group pointing away from the chelating phosphine ligand’s phenyl groups, i.e., 26b. That these diastereoisomers are a result of the metal activation of the prochiral terminal P–H bonds in 13 is shown by addition of an excess of dppe to 26a/b.56 This affords [Rh(dppe)(L)][BArF4]31 with the concomitant formation of free 13 (Scheme 9).
Scheme 9. Change in Diastereoisomeric Ratio and Release of the Diboraphosphine.

[BArF4]− anions are not shown.
We have briefly explored the use of a chiral metal/ligand fragment in dehydrocoupling, [Rh(BDPP)]+ [S,S-BDPP = (2S,4S)-2,4-bis(diphenylphosphino)pentane]. This chiral ligand was chosen as electronically and sterically (i.e., bite angle) it is similar to Ph2P(CH2)3PPh2. Addition of H3B·PCyH2, 5, to [Rh(BDPP)(η6-FC6H5)][BArF4] results in the immediate formation of two diastereoisomers of [Rh(BDPP)H(σ,η-PCyHBH3)(η1-H3B·PCyH2)][BArF4], 27, in a 3:1 ratio (Scheme 10). Although we are unable to comment on the absolute configuration of these isomers, it is interesting to note that this is biased away from the 1:1 ratio observed in the achiral system. Compounds 27a/b proceed on to dehydrocouple to form diastereoisomers of [Rh(BDPP)H(σ,η2-PRH·BH2PRH·BH3)][BArF4], 28, 1:5:3:0 ratio. The same mixture of diasteroisomers can be formed by direct addition of 13 to [Rh(BDPP)(η6-FC6H5)][BArF4]. Initially a 2:1:2:1 ratio of 4 isomers is observed, that changes to a 5:1:3:0 ratio after 18 h. We are unable to comment in more detail on the conformation of these isomers, although the observation of stereocontrol in the direct dehydrocoupling is similar to that observed for the achiral system. Addition of excess dppe to this mixture forms a product identified by ESI-MS as [Rh(BDPP)(dppe)]+ and free 13 (by 31P and 11B NMR spectroscopy). We have not explored whether there is enantiocontrol at the central {PCyH unit} arising from this PB coupling event on release from the metal.
Scheme 10. Use of a Chiral Ligand in Dehydrocoupling.

[BArF4]− anions are not shown.
For these experiments with H3B·PCyH2 it is interesting to note that P–H activation is rapid and reversible with the Rh(I) precursor. This is in contrast to results obtained with secondary phosphine–boranes H3B·PtBu2H and H3B·PtBu2BH2·PtBu2H, which on addition to [Rh(L)(η6-FC6H5)][BArF4] gave the corresponding Rh(I) σ-phosphine–borane complexes with no P–H activation.31 Such selectivity for primary over secondary phosphines in P–H activation at a metal center has been described previously for both phosphine57 and phosphine–borane ligands.27 In particular it has been shown that addition of H3B·PPhH2 to Pt(PEt3)2H(PPh2·BH3) results in exchange of the metal bound phosphide complex to give the primary phosphido–borane complex.26 Here it was suggested that the greater thermodynamic driving force for formation of the primary phosphido–borane complex comes from steric effects, as M–P bonds to smaller primary phosphido ligands are likely to be stronger.
Catalytic Dehydrocoupling of Secondary Phosphine–Boranes
Under the standard catalytic melt conditions (90 °C, 5 mol %),20 [Rh(L)(η6-FC6H5)][BArF4] will dehydrocouple the secondary aryl phosphine–boranes used in this study to form the corresponding linear diboraphosphines 10–12, although we have not explored in detail the temporal evolution of these systems due to the problems associated with directly interrogating the melt. However, trends can be observed. For electron-withdrawing groups (1 and 2), complete consumption of starting material occurs in 4 h (Table 1). The reaction at this temperature is not selective, and although the main product is the linear diboraphosphine, there are products that we tentatively identify as the cyclic oligomers (BH2PR2)n (n = 3, 4).20,33 Our results are broadly in line with the previously reported catalyzed dehydrocoupling of 2 using [Rh(COD)Cl]2, which, at a slightly lower temperature (60 °C, 16 h, melt), affords the linear diboraphosphine product in 69% isolated yield, while at 100 °C only the cyclic oligomers are isolated. The mechanism of formation of the higher cyclic oligomers, (BH2PR2)n, remains to be resolved.20 For electron-donating 3 the reaction is slower using the [Rh(L)(η6-FC6H5)][BArF4] catalyst (8 h) but overall is more selective. For R = Ph we have previously shown that [Rh(L)(η6-FC6H5)][BArF4] catalyzes dehydocoupling to give the corresponding linear diboraphosphine in greater than 95% conversion after 4 h.31 For secondary phosphine–boranes, H3B·PPh2H thus offers balance between overall rate and selectivity.
Table 1. Conversion of H3B·PR2H with Timea.
| H3B·PR2H | time/h | H3B·PR2H/% | H3B·PR2BH2PR2H/% | (BH2PR2)n/% |
|---|---|---|---|---|
| 1 | 1 | 10 | 55 | <5 |
| 4 | <5 | 45 | 10 | |
| 8 | <5 | 35 | 50 | |
| 2 | 1 | 10 | 70 | 10 |
| 4 | <5 | 70 | 15 | |
| 8 | <5 | 70 | 15 | |
| 3 | 1 | 50 | 30 | <5 |
| 4 | 30 | 45 | 5 | |
| 8 | 20 | 60 | 5 |
R = aryl, see Figure 1. [Cat.] = [Rh(L)(η6-FC6H5)][BArF4], L = Ph2P(CH2)3PPh2. Conversions calculated from 31P{1H} NMR spectra. Conditions: [Rh(L)(η6-FC6H5)][BArF4], 5 mol %, 90°C, melt.
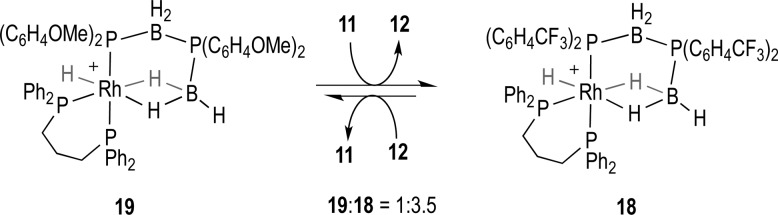
Given the product distributions and likely decomposition pathways in the melt it is inappropriate to comment in detail on the nature of the rate-determining steps during catalysis under these conditions. However, on the basis of the solution studies, P–B bond formation, (dehydrocoupling) is faster with electron-withdrawing groups. The temporal differences in observed product conversion in the melt could reflect a difference in the rate of the P–B bond forming event, or alternatively, they could reflect the ease at which the bound product is substituted on the metal center, i.e., a turnover limiting step. To probe this latter scenario, reaction between 19 (aryl-OMe) and diboraphosphine 11 (aryl-CF3) to form 18 and free 12 demonstrates that an equilibrium is established slightly in favor of 18 (Scheme 11). This suggests that there is not a strong inherent difference in binding strengths between the two products, with the implication being that the observed rate differences in the melt arise from the dehydrocoupling step. Although this is different from what is observed in solution at room temperature, in which release of the product is likely the turnover limiting step, it is consistent with the high local concentration of H3B·PR2H that being under melt conditions (90 °C) would promote such a substitution.
Scheme 11. Competition Experiments between Linear Diboraphosphines.
[BArF4]− anions are not shown.
Catalytic Dehydrocoupling of Primary Phosphine–Boranes
[Rh(L)(η6-FC6H5)][BArF4] also acts as a catalyst for the dehydrocoupling of primary phosphine–boranes. Under melt conditions (90 °C, 5 mol %, 4 h) H3B·PPhH2 is dehydrocoupled to give a major product which is identified by 31P NMR spectroscopy as being polymeric (BH2PPhH)n by comparison with previously reported19,20 data for purified material coming from the [Rh(COD)2][OTf] catalyzed process [δ −49.3, d, J(PH) ∼350 Hz, 1,2–F2C6H4; lit.: δ −48.9, δ, J(PH) 360 Hz, CDCl3]. There were also other species observed ∼δ −55, which could be reduced in relative concentration (to ∼10%) by precipitation into hexanes. Such species have been previously suggested to be short-chain oligomers.20 Interestingly, these proposed shorter chain oligomers are present in a greater proportion at shorter reaction times, which might suggest that polycondensation is occurring to give higher molecular weight polymer. Under non-melt conditions20 (toluene heated to reflux, 0.5 mol %, 16 h) these shorter oligomers are by far the dominant species (Supporting Information). It thus appears that a high local concentration of phosphine–borane is necessary for productive dehydropolymerization. Positive mode ESI-MS (electrospray mass spectrometry) of the melt reaction product demonstrated polymerization, showing repeat units of [H(PPhHBH2)nPPhH2]+ up to n = 10 (Supporting Information). Similar analyses have been reported for amine–borane dehydropolymerization.12,14,58 That these polymers are terminated by {PPhH2} groups rather than {BH3} is confirmed by inspection of the corresponding isotopomer patterns. This formulation also argues against cyclic oligomers being observed by ESI-MS, and presumably the additional phosphine arises from P–B bond cleavage. Use of H3B·PCyH2 under these conditions afforded significantly more complex mixtures that we were unable to resolve.
Conclusions
The solid-state structures of the intermediates in the dehydrocoupling of secondary phosphine–boranes using the {Rh(Ph2PCH2CH2CH2PPh2)}+ fragment have been determined. This demonstrates that the complex that precedes dehydrocoupling to form a linear diboraphosphine has σ bound and P–H activated phosphine–borane ligands, while the product has a linear diboraphosphine bound to the metal center. For aryl phosphine–boranes, electron-withdrawing groups (CF3) promote stoichiometric dehydrocoupling faster than for more electron-donating (OMe) groups. This increase in rate is accompanied by a significant degree of parallel and competitive P–B bond cleavage to afford metal complexes with two monodentate phosphine ligands, which we suggest is due to a weakening of the P–B bond with electron-withdrawing aryl groups. These systems also turnover catalytically under melt conditions, with the overall rate of conversion broadly following the relative dehydrocoupling rates observed in the stoichiometric studies, suggesting that the dehydrocoupling step under melt conditions might also be the turnover limiting step. P–B bond cleavage also occurs for very bulky electron rich adamantyl phosphine–boranes, to such an extent that stoichiometric dehydrocoupling is not observed. For this phosphine–borane we suggest that sterics play a role in this process.
A significant observation is that, for primary phosphine–boranes, which are precursors to polyphosphinoboranes, use of the {Rh(Ph2PCH2CH2CH2PPh2)}+ fragment results in some apparent diastereoselectivity in the dehydrocoupling step, at least in the stoichiometric reactions that produce metal-bound diboraphosphines. Such selectivity could well have implications in the control of the stereochemistry of polymer that would result from further insertion events. A significant future challenge is to harness any inherent bias in each dehydrocoupling insertion step productively while also developing the necessary spectroscopic and physical characterization markers to interrogate the oligomer and polymer stereochemistry.
Experimental Section
All manipulations, unless otherwise stated, were performed under an atmosphere of argon, using standard Schlenk and glovebox techniques. Glassware was oven-dried at 130 °C overnight and flamed under vacuum prior to use. Hexane and pentane were dried using a Grubbs type solvent purification system (MBraun SPS-800) and degassed by successive freeze–pump–thaw cycles.59 CD2Cl2, C6H5F, and 1,2-F2C6H4 were distilled under vacuum from CaH2 and stored over 3 Å molecular sieves, 1,2-F2C6H4 was stirred over alumina for 2 h prior to drying. Bis-1,3-(diphenylphosphino)propane (dpp3) and (2S,4S)-2,4-bis(diphenylphosphino)pentane (BDPP) were purchased from Aldrich. [Rh(nbd)Cl]260 and [Rh(nbd)(dpp3)][BArF4]16 were prepared as previously described. (4-Methoxyphenyl)2HP·BH3 (3), (adamantyl)2HP·BH3 (4), and CyH2P·BH3 (5) were prepared by the same method as Me3P·BH361 but with the phosphine changed. (4-Trifluoromethylphenyl)2PH·BH3 (2) and (3,5-bis(trifluoromethyl)phenyl)2PCl were prepared according to literature procedures reported by Clark et al.33 NMR spectra were recorded on a Bruker AVD 500 MHz spectrometer at room temperature unless otherwise stated. In 1,2-C6H4F2, 1H NMR spectra were referenced to the center of the downfield solvent multiplet (δ 7.07), and 31P and 11B NMR spectra were referenced against 85% H3PO4 (external) and BF3·OEt2 (external), respectively. The spectrometer was prelocked and preshimmed using a C6D6 (0.1 mL) and 1,2-C6H4F2 (0.3 mL) sample. Chemical shifts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on a Bruker micrOTOF instrument.62 In all ESI-MS spectra there was a good fit to both the principal molecular ion and the overall isotopic distribution. Signals in the 31P{1H} NMR spectra were integrated relative to those in similar environments (i.e., Rh–P or B–P) to obtain the relative ratios of products, and data was acquired with a pulse repetition time of 1 s. This avoids potential problems with different relaxation times for different phosphorus environments. Nevertheless, the quoted relative ratios based upon this data should be treated as qualitative rather than quantitative.
Synthesis and Characterization of New Complexes
Synthesis of H3B·PR2H [R = 3,5-Bis(trifluoromethyl)phenyl] (1)
A solution of (3,5-bis(trifluoromethyl)phenyl)2PCl (1.48 g, 3.0 mmol) in diethyl ether (5 mL) was added dropwise to a diethyl ether (20 mL) suspension of LiBH4 (0.070 g, 3.21 mmol) cooled to 5 °C with an ice bath. The mixture became cloudy immediately and was allowed to stir for 30 min. The diethyl ether was removed in vacuo, and the residue was dissolved in hexanes (30 mL) and filtered through Celite. The hexanes were reduced in vacuo to ∼10 mL, and the solution was placed in the freezer (−20 °C) overnight yielding colorless crystals. Excess hexanes were decanted, and crystals were dried to afford a fine white powder which was subsequently washed with 2 × 3 mL of cold hexanes. Removal of all volatiles under reduced pressure yielded 630 mg of fine white powder (1).
1H NMR (300 MHz, CDCl3): δ 8.14 (br s, 1 H, p-Ar-H), 8.09 (br s, 2 H, o-Ar-H), 6.58 (dm, 1JHP = 388 Hz, 1 H, PH), 0.3–2.0 (br m, 3 H, BH). 31P{1H} NMR (121 MHz, CDCl3): δ 4.7 (br s, PH). 11B{1H} NMR (160 MHz, CDCl3): δ −41.7 (br s, BH3). 19F NMR (282 MHz, CDCl3): δ −62.9 (s, CF3). EI-MS (70 eV) m/z (%): 458 (62) [M+ – BH3]. Anal. Found: C 40.71%, H 2.02%. Calcd for C16H10BF12P: C 40.68%, H 2.14%.
Synthesis of (Adamantyl)2PH·BH3 (4)
(Adamantyl)2PH·BH3 was prepared under the same conditions as Me3P·BH361 but with (adamantyl)2PH instead of PMe3.
1H NMR (300 MHz, CDCl3): δ 3.61 (dm, 1 H, 1JHP = 379 Hz, PH), 2.11 to 1.83 (30 H, adamantyl-H), 0.41 to −0.15 (br m, 3 H, BH). 31P{1H} NMR (121 MHz, CDCl3): δ 40.1 (br m, PH). 11B{1H} NMR (160 MHz, CDCl3): δ −44.8 (br d, BH3). Anal. Found: C 75.78%, H 10.71%. Calcd for C20H34BP: C 75.89%, H 10.84%.
Synthesis of H3B·PR2BH2·PR2H [R = 3,5-Bis(trifluoromethyl)phenyl (10); 4-Trifluoromethylphenyl (11); 4-Methoxyphenyl (12)]
A Youngs flask charged with 0.25 mmol of R2PH·BH3 (118 mg of 1, 84 mg of 2, 65 mg of 3) and 5 mol % of [Rh(dpp3)(C6H5F)][BArF4] (18.4 mg, 0.0125 mmol) was heated to 90 °C for 4 h (10 and 11) or 8 h (12) in melt conditions. The resulting solids were washed with n-hexane and recrystallized from a mixture of diethyl ether and hexane at −18 °C (10 30 mg, 25%; 11 22 mg, 26%; 12 32 mg, 49%).
Details follow for 10. 1H NMR (300 MHz, CDCl3): δ 8.09 to 7.89 (12 H, Ar–H), 7.32 (dm, 1JHP = 412 Hz, 1 H, PH), 2.45 (br m, 2 H, BH2), 1.11 (br m, 3 H, BH3). 31P{1H} NMR (121 MHz, CDCl3): δ −1.7 (br s, PHR2), −14.0 (br s, PR2). 11B{1H} NMR (160 MHz, CDCl3): δ −33.2 (br s, BH2), −37.7 (br s, BH3). Anal. Found: C 40.90%, H 1.83%. Calcd for C32H18B2F24P2: C 40.76%, H 1.93%.
Details follow for 11. 1H NMR (500 MHz, CD2Cl2): δ 7.77 to 7.52 (16 H, Ar–H), 7.04 (dt, 1JHP = 426 Hz, 3JHH = 7.8 Hz, 1 H, PH), 2.37 (br m, 2 H, BH2), 1.02 (br m, 3 H, BH3). 31P{1H} NMR (202 MHz, CD2Cl2): δ −3.5 (br s, PHR2), −15.4 (br s, PR2). 11B{1H} NMR (160 MHz, CD2Cl2): δ −33.7 (br s, BH2), −37.6 (br s, BH3).
Details follow for 12. 1H NMR (500 MHz, CD2Cl2): δ 7.64 to 6.77 (16 H, Ar–H), 6.69 (dt, 1JHP = 415 Hz, 3JHH = 6.6 Hz, 1 H, PH), 3.84 (s, 6 H, CH3), 3.80 (s, 6 H, CH3), 2.14 (br m, 2 H, BH2), 0.96 (br m, 3 H, BH3). 31P{1H} NMR (202 MHz, CD2Cl2): δ −7.6 (br s, PHR2), −22.1 (br s, PR2). 11B{1H} NMR (160 MHz, CD2Cl2): δ −33.3 (br s, BH2), −37.1 (br s, BH3).
Synthesis of [Rh(dpp3)H(PR2·BH3)(H3B·PHR2)][BArF4] [R = 3,5-Bis(trifluoromethyl)phenyl (14); 4-Trifluoromethylphenyl (15); 4-Methoxyphenyl (16)]
To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 2 equiv of H3B·PR2H (32 mg of 1, 23 mg of 2, 18 mg of 3 0.068 mmol) was added 1,2-F2C6H4 (5 mL). The solution was stirred at room temperature 10 min, and a change in color from pale orange to bright yellow was observed. Complexes 15 and 16 were isolated as yellow oils, and characterized in situ by NMR spectroscopy and ESI-MS. Complex 14 could be crystallized at −24 °C in the freezer inside the glovebox (yield 29.6 mg, 37%). Complexes 15 and 16 could not be isolated cleanly, and attempts to do so led to intractable mixtures of 15 and 16 with 18 and 19, respectively.
Details follow for 14. Slow diffusion of pentane (10 mL) over a solution of 14 in 1,2-F2C6H4 at −24 °C afforded yellow crystals (one of which was employed for an X-ray diffraction study).
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 5.81 (d, 1JHP = 435 Hz, 1 H, PH), 3.12–0.81 (8 H, 3CH2 dpp3 + BH2), −0.78 (br, 3 H, BH3), −6.12 (br, 1 H, BH–Rh), −16.21 (s, 1 H, Rh–H). Signals from the aromatics were not observed due to being overlapped by signals from 1,2-F2C6H4.
1H NMR (500 MHz, 1,2-F2C6H4, selected data at 0 °C): δ −6.12 (d, 1JHP = 65 Hz, 1 H, BH–Rh). 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 29.5 (ddd, JPRh = 130 Hz, JPP(cis)= 35 Hz, JPP(cis) = 21 Hz, Ph2P(CH2)3PPh2), 11.3 (ddd, JPP(trans) = 244 Hz, JPRh = 103 Hz, JPP(cis) = 35 Hz, Ph2P(CH2)3PPh2), −0.7 (dd, JPP(trans) = 248 Hz, JPRh = 75 Hz, Rh-PR2BH3), −2.62 (br s, PHR2BH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ −6.2 (BArF4), −39.8 (br, 2 × BH3). ESI-MS (1,2-C6H4F2, 60 °C) positive ion: m/z = 1431.07 (calcd 1431.07, M+ – 2BH3). Anal. Found: C 46.82%, H 2.39%. Calcd for C91H58B3F48P4Rh: C 47.02%, H 2.52%.
Details follow for 15. 1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 4.90 (d, JHP = 414 Hz, PH), 3.01–1.10 (8 H, 3CH2 dpp3 + BH2), −1.18 (br, 3H, BH3), −6.95 (d, JHP = 78 Hz, 1 H, BH–Rh), −16.51 (s, 1 H, Rh–H). Signals from the aromatics were not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 27.3 (ddd, JPRh = 127 Hz, JPP(cis) = 37 Hz, JPP(cis) = 16 Hz, Ph2P(CH2)3PPh2), 11.8 (ddd, JPP(trans) = 231 Hz, JPRh = 100 Hz, JPP(cis) = 37 Hz, Ph2P(CH2)3PPh2), −0.3 (dd, JPP(trans) = 231 Hz, JPRh = 70 Hz, Rh-PR2BH3), −5.96 (br s, PHR2BH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ −6.2 (BArF4) −39.9 (br, 2 × BH3). ESI-MS (1,2-C6H4F2, 60 °C) positive ion: m/z = 1159.13 (calcd 1159.13, M+ – 2BH3).
Details follow for 16. 1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 4.93 (d, JHP = 409 Hz, 1 H, PH), 3.77 (s, 6 H, −OCH3), 3.69 (s, 6 H, −OCH3), 3.16–0.58 (8 H, 3CH2 dpp3 + BH2), −1.11 (br, 3 H, BH3), −6.53 (d, JHP = 73 Hz, 1 H, BH–Rh), −16.49 (s, 1 H, Rh–H). Signals from the aromatics were not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 27.3 (ddd, JPRh = 132 Hz, JPP(cis) = 37 Hz, JPP(cis) = 12 Hz, Ph2P(CH2)3PPh2), 9.5 (ddd, JPP(trans) = 227 Hz, JPRh = 98 Hz, JPP(cis) = 37 Hz, Ph2P(CH2)3PPh2), 0.7 (br dd, JPP(trans) = 227 Hz, JPRh = 65 Hz, Rh-PR2BH3), −11.2 (br s, PHR2BH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ −6.2 (BArF4) −38.7 to −49.8 (br, 2 × BH3). ESI-MS (1,2-F2C6H4, 60 °C) positive ion: m/z = 1021.25 (calcd 1021.25, M+ – BH3), 914.19 (calcd 914.20, M+ – BH3 – C6H4OMe), 900.17 (calcd 900.17, M+ – 2BH3, −C6H4OMe), 775.17 (calcd 775.17, M+ – (MeOC6H4)2HP·BH3).
Synthesis of [Rh(dpp3)H(PR2·BH2PR2·BH3)][BArF4] [R = 3,5-Bis(trifluoromethyl)phenyl (17); 4-Trifluoromethylphenyl (18); 4-Methoxyphenyl (19)]
Method A follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 2 equiv of H3B·PR2H (32 mg of 1, 23 mg of 2, 18 mg of 3 0.068 mmol) was added 1,2-F2C6H4 (5 mL). The solution was stirred at room temperature for 24 h. The formation of H2 gas is also observed. Complex 19 was isolated as yellow oil (37 mg, 61%). Complexes 17 and 18 could not be isolated cleanly; they were observed with 22 and 23, respectively.
Method B follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 1 equiv of 10 (32 mg, 0.068 mmol) was added 1,2-F2C6H4 (4 mL). Complex 17 was isolated as yellow solid (65 mg, 82%).
Details follow for 17. Slow diffusion of pentane (10 mL) over a solution of 17 in 1,2-F2C6H4 at −24 °C afforded yellow crystals (one of which was employed for an X-ray diffraction study).
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8H, BArF4), 7.69 (s, 4H, BArF4), 4.40 (vbr, 1H, BH) 3.10–2.12 (8H, 3CH2 dpp3 + BH2), −1.20 (vbr, 1H, BH), −4.54 (vbr, 1H, BH), −13.98 (s, 1H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 46.6 (dd, JRh–P= 111, JP2–P1(cis)= 36, Ph2P(CH2)3PPh2), 29.5 (m, JP–P(trans) = 260, Rh-PR2BH3PR2HBH3), 12.8 (ddd,, JP–P(trans) = 260, JRh–P= 91, JP–P(cis) = 33, Ph2P(CH2)3PPh2), −2.7 (s, Rh-PR2BH3PR2HBH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ 0.21 (br), −6.2 (s, BArF4), −27.1 (br). ESI-MS (1,2-F2C6H4, 60 °C) positive ion: m/z = 1457.09 (calcd 1457.12, M+). Anal. Found: C 47.15%, H 2.34%. Calcd for C91H56B3F48P4Rh: C 47.07%, H 2.43%.
Details follow for 18. 1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 4.24 (v br, 1 H, BH), 2.61–1.72 (8 H, 3CH2 dpp3 + BH2), −1.29 (v br, 1 H, BH), −4.65 (v br, 1 H, BH), −14.90 (s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 42.5 (dd, JPRh = 106 Hz, JPP(cis) = 34 Hz, Ph2P(CH2)3PPh2), 28.6 (m, JPP(trans) = 272 Hz, Rh-PR2BH3PR2HBH3), 15.3 (ddd, JPP(trans) = 272 Hz, JPRh = 101 Hz, JPP(cis) = 33 Hz, Ph2P(CH2)3PPh2), −6.4 (s, Rh-PR2BH3PR2HBH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ 0.4 (br), −6.2 (s, BArF4), −25.9 (br). ESI-MS (1,2-F2C6H4) positive ion: m/z = 1185.17 (calcd 1185.18, M+).
Details follow for 19. 1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 3.77 (s, 3 H, −OCH3), 3.76 (s, 3 H, −OCH3), 3.69 (s, 6 H, −OCH3), 2.85–1.72 (8 H, 3CH2 dpp3 + BH2), −1.03 (v br, 1 H, BH), −4.00 (v br, 1 H, BH), −14.55 (s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 42.7 (ddd, JPRh = 109 Hz, JPP(cis) = 35 Hz, JPP(cis) = 12 Hz Ph2P(CH2)3PPh2), 28.1 (br m, JPP(trans) = 279 Hz, Rh-PR2BH3PR2HBH3), 12.5 (ddd, JPP(trans) = 279 Hz, JPRh = 88 Hz, JPP(cis) = 12 Hz, Ph2P(CH2)3PPh2), −11.7 (br s, Rh-PR2BH3PR2HBH3). 11B{1H} NMR (160 MHz, C6H4F2): δ 3.42 (br), −6.2 (s, BArF4), −27.7 (br). ESI-MS (1,2-F2C6H4) positive ion: m/z = 1033.27 (calcd 1033.27, M+).
Synthesis of [Rh(dpp3)(PHR2)2][BArF4] [R = 3,5-Bis(trifluoromethylphenyl) (21)]
Method A follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 2 equiv of H3B·PR2H (32 mg of 1) was added 1,2-F2C6H4 (5 mL). The solution was stirred at room temperature for 24 h. The formation of H2 (gas) is also observed. Complex 21 could not be isolated cleanly as 17 was also observed.
Method B follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (20 mg, 0.034 mmol) and 2 equiv of PHR2 (31 mg, 0.068 mmol, R = 3,5-bis(trifluoromethyl)phenyl) was added 1,2-F2C6H4 (1 mL). After stirring for 10 min the solution was evaporated to dryness and the solid washed with pentane (2 mL). Complex 21 was isolated as yellow solid (yield 17.8 mg, 57%).
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.33 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 6.41 (dm, JHP = 375 Hz, 2 H, PH), 2.62 (br, 4 H, 2 CH2 dpp3), 2.17 (m, 2 H CH2, dpp3). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 9.6 (m, 2P, AA′BB′M), 5.5 (m, 2P, AA′BB′M). ESI-MS (1,2-F2C6H4) positive ion: m/z = 1431.04 (calcd 1431.07, M+). Anal. Found: C 47.71%, H 2.21%. Calcd for C91H52BF48P4Rh: C 47.60%, H 2.28%.
[Rh(dpp3)(PHR2)2][BArF4] [R = 4-Trifluoromethylphenyl (22)]
Complex 22 was characterized by in situ NMR spectroscopy. 1H NMR (500 MHz, 1,2-F2C6H4): δ 8.33 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 5.91 (dm, JHP = 359 Hz, 2 H, PH), 2.57 (br, 4 H, 2 × CH2 dpp3), 2.06 (m, 2 H × CH2, dpp3). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 12.2 (m, 2P, AA′BB′M), 5.2 (m, 2P, AA′BB′M).
Synthesis of [Rh(dpp3)(η2-H3B·PR2H)][BArF4] [R = Adamantyl (23)]
To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and H3B·PR2H (4) (11 mg, 0.034 mmol) was added 1,2-F2C6H4 (5 mL). The solution was stirred at room temperature for 1 min, and a change in color from pale orange to purple was observed. Complex 23 could not be isolated because further reaction to form 24 occurred.
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.68 (s, 4 H, BArF4), 3.45 (d, 1 H, 1JHP = 380 Hz, B-PH), 2.52–1.12 (36 H, adamantyl-H + dpp3 CH2), −1.36 (br, 3 H, BH3). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 35.1 (d, JPRh = 167 Hz, dpp3), 30.5 (br, B–P). 11B NMR (160 MHz, 1,2-F2C6H4): δ −0.8 (br), −6.0 (s, BArF4). ESI-MS (1,2-F2C6H4) positive ion: m/z = 629.09 (calcd 629.08, [Rh(dpp3)(C6H4F2)]+). The weakly bound σ-complex could not be observed.
Synthesis of [Rh(dpp3)(PR2H)(H3B·PR2H)][BArF4] [R = Adamantyl (24)]
Method A follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 2 equiv of H3B·PR2H (4) (22 mg, 0.068 mmol) was added 1,2-F2C6H4 (5 mL). The solution was stirred at room temperature for 24 h. Complex 24 was isolated as orange solid.
Method B follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 1 equiv of PHR2 (10 mg, 0.034 mmol) was added 1,2-F2C6H4 (5 mL). The solution was stirred for 5 min, and then 1 equiv of H3B·PR2H (4) (11 mg, 0.034 mmol) was added. The solution was stirred for 5 min, and complex 24 was isolated as orange solid (yield 48 mg, 71%). Slow diffusion of pentane (10 mL) into a solution of 24 in 1,2-F2C6H4 at −24 °C afforded yellow crystals (one of which was employed for X-ray diffraction studies).
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.33 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 3.30 (d, 1JHP = 362 Hz, 1 H, B-PH), 2.87 (d, 1JHP = 412 Hz, 1 H, PH), 2.45–1.56 (66 H, dpp3 CH2 and adamantyl-H), −0.24 (br, 3 H, BH3). 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 60.4 (ddd, JPP(trans) = 266 Hz, JPRh = 142 Hz, JPP(cis) = 30 Hz, Ph2P(CH2)3PPh2), 30.5 (s, PHR2BH3), 22.8 (dd, JPRh = 163 Hz, JPP(cis) = 30 Hz, PPh2(CH2)3PPh2), 6.6 (ddd, JPP(trans) = 270 Hz, JPRh = 144 Hz, JPP(cis) = 30 Hz, Rh-PR2BH3). 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ −6.0 (s, BArF4), −42.2 (br, BH3). ESI-MS (1,2-F2C6H4) positive ion: m/z = 956.30 (unidentified fragment). Anal. Found: C 59.38%, H 4.99%. Calcd for C99H103B2F24P4Rh: C 59.50%, H 5.20%.
Synthesis of [Rh(dpp3)H(PCyH·BH3)(H3B·PCyH2)][BArF4] (25a and 25b)
To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) was added 1,2-F2C6H4 (5 mL). A 2 equiv portion of H3B·PH2Cy (5) (0.68 mL, 0.1 M solution in 1,2-F2C6H4, 0.068 mmol) was then added. The solution was stirred at room temperature for 1 min, and a change in color from orange to pale yellow was observed. Complexes 25a and 25b were observed as an approximate 1:1 ratio of isomers and were characterized in situ by NMR spectroscopy. Complexes 25a and 25b could not be isolated as they reacted quickly to form complexes 26a and 26b. The 31P{1H} NMR spectrum of this reaction mixture indicates that 2 diastereomers are present; while we were able to identify the 2 sets of 4 resonances each (labeled † and §, based on coupling constants and approximate integrations) it was not possible to determine which set of signals belonged to which diastereomer. See Figure S4, Supporting Information, for more detail.
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 2.73–0.32 (32 H, 3 CH2 dpp3 + BH2, CyH, PH), −2.29 (v br, 3 H, BH), −7.92 (br d, 1 H, BH-Rh), −17.51 (s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 31.7 (dm, JPRh = 134 Hz, Ph2P1†(CH2)3PPh2), 30.5 (dm, JPRh = 129 Hz, Ph2P1§(CH2)3PPh2), 11.8 (overlapping ddd, JPP(trans) = approximately 200 Hz, JPRh = approximately 104 Hz, JPP(cis) = approximately 25 Hz, Ph2P(CH2)3P2†§Ph2), −11.0 (br d, JPP(trans) = approximately 200 Hz, Rh-P3§HCy-B), −32.1 (br d, JPP(trans) = approximately 200 Hz, Rh-P3†HCy-B), −39.8 (br s, Rh–H3BP4†H2Cy), −44.2 (br s, Rh–H3BP4§H2Cy). ESI-MS (1,2-F2C6H4, 60 °C) positive ion: m/z = 747.20 (calcd 747.21, M+ – 2BH3).
Synthesis of [(CyH2P)2BH2]Br
To a stirred solution of PCyH2 (3.40 mL, 10% wt in hexane, 2.0 mmol) in dichloromethane (20 mL) was added BrH2B·SMe2 (1.0 mL, 1.0 M in CH2Cl2, 1.0 mmol) and the solution stirred at room temperature for 2 h. The resulting colorless solution was concentrated in vacuo to approximately 3 mL, and diethyl ether (30 mL) was added to precipitate a white solid. The solvent was decanted off and the solid washed with a further 10 mL of diethyl ether. The solid was redissolved in dichloromethane, filtered, and recrystallized from a mixture of dichloromethane and diethyl ether at −18 °C to yield white crystals (first crop 0.190 g, second crop 0.041 g, overall yield 71%). At room temperature in CD2Cl2 [(CyH2P)2BH2]Br undergoes a degenerate exchange reaction; [(CyH2P)2BH2]Br is in equilibrium with CyH2PBH2Br + PH2Cy. This exchange process does not occur at −60 °C on the NMR time scale, and each of these species can be observed. In the solid-state the complex exists as [(CyH2P)2BH2]Br.
1H NMR (500 MHz, −60 °C, CD2Cl2): δ 5.46 (dm, 1JHP = 429 Hz, [(CyH2P)2BH2]+), 4.75 (dm, 1JHP = 388 Hz, CyH2PBH2Br), 2.52 (dm, 1JHP = 196 Hz, CyH2P), 2.37–1.16 (CyH and BH). 31P{1H} NMR (202 MHz, −60 °C, CD2Cl2): δ −35.5 (br s, [(CyH2P)2BH2]+), −38.2 (br s, CyH2PBH2Br), −107.5 (s, CyH2P). Anal. Found: C 44.35%, H 8.74%. Calcd for C12H28BBrP2: C 44.30%, H 8.68%.
Synthesis of CyH2P·BH2PCyH·BH3 (13)
[(CyH2P)2BH2]Br (0.150 g, 0.461 mmol) (prepared as above) and [NnBu4][BH4] (0.119 g, 0.461 mmol) were added to a Schlenk tube. Dichloromethane (10 mL) was added, and effervescence was observed; the solution was stirred at room temperature for 1 h. The solvent was removed in vacuo, and n-hexane (20 mL) was added to the white solid. The solution was filtered to remove [NnBu4]Br and concentrated to approximately 2 mL. Storage of this solution for 16 h at −18 °C yielded colorless crystals of 13 (0.102 g, 86%).
1H NMR (500 MHz, 25 °C, CD2Cl2): δ 4.68 (dm, 1JHP = 386 Hz, CyH2P·BH2PHCy·BH3), 3.50 (dm, 1JHP = 326 Hz, CyH2P·BH2PHCy·BH3), 2.17–0.02 (CyH and BH). 31P{1H} NMR (202 MHz, 25 °C, CD2Cl2): δ −36.6 (br, CyH2P·BH2PHCy·BH3), −45.1 (br, CyH2P·BH2PHCy·BH3). Anal. Found: C 55.79%, H 11.82%. Calcd for C12H30B2P2: C 55.77%, H 11.71%.
Synthesis of [Rh(dpp3)H(PHCy·BH2PHCy·BH3)][BArF4] (26a and 26b)
Method A follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) was added 1,2-F2C6H4 (5 mL) followed by 2 equiv of H3B·PCyH2 (5) (0.68 mL, 0.1 M solution in 1,2-F2C6H4, 0.068 mmol). The solution was stirred at room temperature for 12 h. The formation of H2 is also observed. Complexes 26a and 26b were characterized as a mixture in solution by NMR spectroscopy and ESI-MS. The 31P{1H} NMR spectrum of this reaction mixture indicates that 2 diastereomers are present; we were able to identify the 2 sets of 4 resonances each (labeled 26a and 26b, based on coupling constants and approximate integrations) and tentatively assigned the individual diastereomers (Scheme S5, Supporting Information) by inspection of a model.
Method B follows. To a Youngs flask charged with [Rh(dpp3)(C6H5F)][BArF4] (50 mg, 0.034 mmol) and 1 equiv of CyH2P·BH2PCyH·BH3 (13) (8.8 mg, 0.034 mmol) was added 1,2-F2C6H4 (4 mL). Complexes 26a and 26b were characterized as a mixture in solution by NMR spectroscopy and ESI-MS.
1H NMR (500 MHz, 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 4.35–3.01 (PH and BH), 2.92–0.92 (30 H, 3CH2 dpp3, BH2, CyH), −2.98 (br, 1 H, BH-Rh), −5.98 (br, 1 H, BH-Rh), −16.08 (s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from 1,2-F2C6H4. 31P{1H} NMR (202 MHz, 1,2-F2C6H4): δ 37.9 (dm, JPRh = 102 Hz, Ph2P1b(CH2)3PPh2), 34.5 (dm, JPRh = 102 Hz, Ph2P1a(CH2)3PPh2), 19.8 (br d, JPP(trans) = approximately 255 Hz, Rh-P3bHCyB), 16.2 (br d, JPP(trans) = approximately 255 Hz, Rh-P3aHCy-B), 10.7 (overlapping dm, JPP(trans) = 255 Hz, JPRh = 88 Hz, JPP(cis) = approximately 25 Hz, Ph2P(CH2)3P2abPh2), −14.9 (br s, Rh-PHCyBH2P4aHCyBH3), −16.6 (br s, Rh-PHCyBH2P4bHCyBH3). ESI-MS (1,2-F2C6H4, 60 °C) positive ion: m/z = 773.26 (calcd 773.24, M+).
Synthesis of [Rh(BDPP)(nbd)][BArF4]
[Rh(nbd)2][BArF4] was synthesized by an adaptation of the preparation of [Rh(cod)2][BArF4].63 [Rh(nbd)2][BArF4] (0.100 g, 0.0869 mmol) was dissolved in CH2Cl2 (10 mL) to produce a deep red solution. (2S,4S)-2,4-Bis(diphenylphosphino)pentane (BDPP) (0.0383 g, 0.0869 mmol) was added, and a color change to red was observed. The solution was stirred for 1 h at room temperature before the solvent was removed in vacuo. n-Pentane (20 mL) was added, and the solution was sonicated to produce an orange powder. The solvent was decanted and the solid washed with pentane (2 × 20 mL). The product was dried in vacuo and isolated (yield 0.102 g, 78%).
1H NMR (300 MHz, CD2Cl2): δ 7.72 (s, 8 H, BArF4), 7.56 (s, 4 H, BArF4), 7.70–7.36 (m, P-Ph), 4.86 (m, 2 H, =C—H), 4.30 (m, 2 H, =C—H), 3.90 (m, 2 H, nbd C—H), 2.77 (m, 2 H, P—C—H), 1.84 (tt, 3JHH = 6.45 Hz, 3JHP = 19.95 Hz, 2 H, CH2), 1.58 (m, 2 H, nbd CH2), 1.13 (m, 6 H, CH3). 31P{1H} NMR (121.6 MHz, CD2Cl2): δ 27.4 (d, JPRh = 149 Hz). Anal. Found: C 54.32%, H 3.18%. Calcd for C68H50BF24P2Rh: C 54.47%, H 3.36%.
Synthesis of [Rh(BDPP)(C6H5F)][BArF4]
[Rh(BDPP)(nbd)][BArF4] prepared as above (0.020 g, 0.0133 mmol) was added to a high-pressure NMR tube and dissolved in fluorobenzene (0.5 mL). The sample was degassed by the freeze–pump–thaw method and hydrogen gas (4 atm) introduced. The sample was mixed for 30 min and then degassed by the freeze–pump–thaw method and placed under argon.
1H NMR (500 MHz, C6H5F): δ 8.32 (s, 8 H, BArF4), 7.61 (s, 4 H, BArF4), 5.63 (m, 2 H, Ar–H), 5.43 (m, 2 H, Ar–H), 5.10 (m, 1 H, Ar–H), 2.38 (m, 2 H, P-CH), 1.40 (m, 2 H, CH2), 0.78 (m, 6 H, CH3). Signals from P-Ph not observed due to being overlapped by signals from C6H5F. Signals from norbornane (from hydrogenation of norbornadiene) can also be observed at δ 2.12 (m, 2 H, nba C–H), 1.40 (m, 2 H, nba CH2, overlapped), 1.11 (m, 2 H, nba CH2). 31P{1H} NMR (202 MHz, C6H5F): δ 39.9 (d, JPRh = 194 Hz). ESI-MS (C6H5F, 60 °C) positive ion: m/z = 639.13 (calcd 639.12, M+).
Synthesis of [Rh(BDPP)H(PCyH·BH3)(H3B·PCyH2)][BArF4] (27a and 27b)
[Rh(BDPP)(C6H5F)][BArF4] (26) was prepared in a high-pressure NMR tube as above. To this was added 2 equiv of H3B·PH2Cy (5) (0.27 mL, 0.1 M solution in 1,2-F2C6H4, 0.027 mmol) and the solution mixed for 1 min. Complexes 27a and 27b were observed as ratio of isomers and were characterized in situ by NMR spectroscopy. Complexes 27a and 27b could not be isolated as they reacted quickly to form complexes of 28; signals for 28 can be observed in both 1H and 31P{1H} NMR spectra of 27a and 27b which, along with the presence of H2, show that the complexes rapidly undergo dehydrocoupling to form complexes of 28. The 31P{1H} NMR spectrum of this reaction mixture indicates that 2 diastereomers are present; while we were able to identify the 2 sets of 4 resonances (labeled † and §, based on coupling constants and approximate integrations), it was not possible to determine which set of signals belonged to which diastereomer. See Figure S6, Supporting Information.
1H NMR (500 MHz, C6H5F + 1,2-F2C6H4): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 4.19 to 0.32 (CH3, CH2, CH, BDPP, BH2, CyH, PH), −1.80 to −3.31 (v br, 3 H, BH3), −7.54 to −8.86 (br d, 1 H, BH-Rh), −17.73 (br s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from C6H5F and 1,2-F2C6H4. H2 can be observed in the NMR spectrum as a sharp singlet at δ 4.52 ppm suggesting that some dehydrocoupling to complex 28 has occurred. This is further evidenced by the small Rh–H hydride signal at δ −16.07 ppm which is observed for complex 28. 31P{1H} NMR (202 MHz, C6H5F + 1,2-F2C6H4): δ 42.5 (dm, JPRh = 129 Hz, Ph2P1†(CH2)3PPh2), 32.6 (dm, JPRh = 119 Hz, Ph2P1§(CH2)3PPh2), 28.3 (overlapping ddd, JPP(trans) = approximately 208 Hz, JPRh = approximately 98 Hz, JPP(cis) = approximately 32 Hz, Ph2P(CH2)3P2†§Ph2), 3.6 (br d, JPP(trans) = approximately 208 Hz, Rh-P3§HCy-B), −12.1 (br d, JPP(trans) = approximately 208 Hz, Rh-P3†HCy-B), −42.1 (br s, Rh–H3BP4§H2Cy), −44.2 (br s, Rh–H3BP4†H2Cy).
Synthesis of [Rh(BDPP)H(PCyH·BH2PCyH·BH3)][BArF4] (28a, 28b, 28c, and 28d)
Method A follows. [Rh(BDPP)(C6H5F)][BArF4] was prepared on an NMR scale from [Rh(BDPP)(nbd)][BArF4] (0.020 g, 0.0133 mmol) as above. This solution was transferred by cannula to an NMR tube containing CyH2P·BH2PHCy·BH3 (13) (3.4 mg, 0.0133 mmol), and the solution was mixed briefly to yield a pale yellow solution.
Method B follows. [Rh(BDPP)(C6H5F)][BArF4] was prepared on an NMR scale from [Rh(BDPP)(nbd)][BArF4] (0.020 g, 0.0133 mmol) as above. To this was added 2 equiv of H3B·PH2Cy (5) (0.27 mL, 0.1 M solution in 1,2-C6H4F2, 0.027 mmol) and the solution mixed for 18 h.
The 31P{1H} NMR spectrum arising from method A indicates that 4 diastereomers are present; while we were able to identify some of the signals from the 4 sets of 4 resonances (labeled †, §, $, and & based on coupling constants and approximate integrations), it was not possible to determine which set of signals belonged to which of the diastereomers. See Figure S7, Supporting Information. Method B affords, essentially, only one diastereoisomer.
1H NMR (500 MHz, C6H5F): δ 8.32 (s, 8 H, BArF4), 7.69 (s, 4 H, BArF4), 5.47–2.46 (PH), 2.11–0.63 (CH, CH2, and CH3 BDPP, BH2, CyH), −3.21 (br, 1 H, BHRh), −5.65 to −6.85 (br, 1 H, BHRh), −16.09, −16.20, and −17.04 (s, 1 H, Rh–H). Signals from aromatics not observed due to being overlapped by signals from C6H5F and 1,2-C6H4F2. 31P{1H} NMR (202 MHz, C6H5F): δ 58.9 (ddd, JPRh = 107 Hz, JPP(cis) = 30 Hz, JPP(cis) = 12 Hz, Ph2P1§(CH2)3PPh2), 58.0 (dm, JPRh = 102 Hz, Ph2P1†(CH2)3PPh2), 46.6 (dm, JPRh = 100 Hz, Ph2P1$ (CH2)3PPh2), 43.6 (dm, JPRh = 102 Hz, Ph2P1&(CH2)3PPh2), 28.7 (ddd, JPP(trans) = 254 Hz, JPRh = 90 Hz, JPP(cis) = 32 Hz, Ph2P(CH2)3P2$Ph2), 27.5 (ddd, JPP(trans) = 250 Hz, JPRh = 92 Hz, JPP(cis) = 32 Hz, Ph2P(CH2)3P2&Ph2), 24.8–17.7 (overlapping m, JPP(trans) = approximately 254 Hz, JPRh = approximately 87 Hz, JPP(cis) = approximately 26 Hz, Ph2P(CH2)3P2†§Ph2 and Rh-P3HCy-B), −13.5 to −20.6 (br s of 4 isomers, Rh-PHCyBH2P4HCyBH3). ESI-MS (1,2-C6H4F2, 60 °C) positive ion: m/z = 801.29 (calcd 801.29, M+).
In order to establish that dehydrocoupling had occurred when method B was used to form complexes 28, excess (10 equiv) of 1,2-bis(diphenylphosphino)ethane was added to the reaction mixture to release the dehydrocoupled product, CyH2P·BH2PHCy·BH3, from the metal center. The 31P{1H} NMR spectrum showed two broad signals at δ −37.9 and −43.9 which are in agreement with those found for compound 13.
Attempted Polymerization of PhH2P·BH3 in Solution
In a procedure similar to that reported by Manners et al.,20 PhH2P·BH3 (0.248 g, 2.0 mmol) was dissolved in toluene (10 mL) either in the presence of [Rh(dpp3)(C6H5F)][BArF4] (14.7 mg, 0.01 mmol) or with no catalyst present. The solution was heated to reflux for 16 h before cooling to room temperature. The solution was concentrated in vacuo and added to stirred hexane (100 mL) to produce a white precipitate. The solvent was decanted and the solid washed with hexane (2 × 50 mL). The solid was dried in vacuo and isolated in air (yield 0.110 g Rh catalyzed, 0.101 g uncatalyzed). The 31P{1H} NMR spectrum of the isolated solids produced by the different methods are very similar with several very broad peaks from δ −45 to −57 and very broad peaks of lower intensity from δ −72 to −87 . See Figure S8, Supporting Information. This is in agreement with the results obtained by Manners et al. for uncatalyzed polymerization of PhH2P·BH3.20
Melt Polymerization of PhH2P·BH3
A Youngs flask charged with PhH2P·BH3 (31 mg, 0.25 mmol) and 5 mol % of [Rh(dpp3)(C6H5F)][BArF4] (18.4 mg, 0.0125 mmol) was heated to 90 °C for 4 h in melt conditions. The resulting solid was dissolved in 1,2-difluorobenzene and analyzed by NMR spectroscopy. The 31P{1H} NMR spectrum shows a peak at δ −49.3 ppm in agreement with that observed by Manners et al.7 for polymeric material and a lower intensity resonance at δ −55.0 ppm. See Supporting Information Figure S9. In the 31P{1H} NMR spectrum the peak at δ −49.3 ppm split into a broad doublet with a JPH coupling constant of approximately 350 Hz. Analysis by ESI-MS of the reaction mixture showed a repeating pattern corresponding to the polymeric repeat unit −[PhHP·BH2]–, see Figure S10, Supporting Information.
Acknowledgments
The authors acknowledge European Union (FP7, Marie Curie Action “Dehydrocouple”), and the EPSRC (EP/J02127X/1,EP/J020826/1).
Supporting Information Available
Further experimental and characterization details, including selected NMR data and X-ray crystallographic data (including data in CIF format). This material is available free of charge via the Internet at http://pubs.acs.org. Crystallographic data have been deposited with the Cambridge Crystallographic Data Center (CCDC) and can be obtained via www.ccdc.cam.ac.uk/data_request/cif.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Chivers T.; Manners I.. Inorganic Rings and Polymers of the p-Block Elements; RSC: Cambridge, U.K., 2009. [Google Scholar]
- Gauvin F.; Harrod J. F.; Woo H. G. In Advances in Organometallic Chemistry; Stone F. G. A., Robert W., Eds.; Academic Press: New York, 1998; Vol. 42. [Google Scholar]
- Jaska C. A.; Bartole-Scott A.; Manners I. Dalton Trans. 2003, 4015–4021. [Google Scholar]
- Reichl J. A.; Berry D. H. In Advances in Organometallic Chemistry; Robert W., Anthony F. H., Eds.; Academic Press: New York, 1998; Vol. 43. [Google Scholar]
- Clark T. J.; Lee K.; Manners I. Chem.—Eur. J. 2006, 12, 8634–8648. [DOI] [PubMed] [Google Scholar]
- Leitao E. M.; Jurca T.; Manners I. Nat. Chem. 2013, 5, 817–829. [DOI] [PubMed] [Google Scholar]
- Waterman R. Chem. Soc. Rev. 2013, 42, 5629–5641. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Robertson A. P. M.; Sloan M. E.; Manners I. Chem. Rev. 2010, 110, 4023–4078. [DOI] [PubMed] [Google Scholar]
- Hamilton C. W.; Baker R. T.; Staubitz A.; Manners I. Chem. Soc. Rev. 2009, 38, 279–293. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Robertson A. P. M.; Manners I. Chem. Rev. 2010, 110, 4079–4124. [DOI] [PubMed] [Google Scholar]
- Wright W. R. H.; Berkeley E. R.; Alden L. R.; Baker R. T.; Sneddon L. G. Chem. Commun. 2011, 47, 3177–3179. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Sloan M. E.; Robertson A. P. M.; Friedrich A.; Schneider S.; Gates P. J.; Guànne J. S.; Manners I. J. Am. Chem. Soc. 2010, 132, 13332–13345. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Presa Soto A.; Manners I. Angew. Chem., Int. Ed. 2008, 47, 6212–6215. [DOI] [PubMed] [Google Scholar]
- Dietrich B. L.; Goldberg K. I.; Heinekey D. M.; Autrey T.; Linehan J. C. Inorg. Chem. 2008, 47, 8583–8585. [DOI] [PubMed] [Google Scholar]
- Johnson H. C.; Robertson A. P. M.; Chaplin A. B.; Sewell L. J.; Thompson A. L.; Haddow M. F.; Manners I.; Weller A. S. J. Am. Chem. Soc. 2011, 133, 11076–11079. [DOI] [PubMed] [Google Scholar]
- Dallanegra R.; Robertson A. P. M.; Chaplin A. B.; Manners I.; Weller A. S. Chem. Commun. 2011, 47, 3763–3765. [DOI] [PubMed] [Google Scholar]
- Baker R. T.; Gordon J. C.; Hamilton C. W.; Henson N. J.; Lin P.-H.; Maguire S.; Murugesu M.; Scott B. L.; Smythe N. C. J. Am. Chem. Soc. 2012, 134, 5598–5609. [DOI] [PubMed] [Google Scholar]
- Marziale A. N.; Friedrich A.; Klopsch I.; Drees M.; Celinski V. R.; Schmedt auf der Günne J.; Schneider S. J. Am. Chem. Soc. 2013, 135, 13342–13355. [DOI] [PubMed] [Google Scholar]
- Dorn H.; Singh R. A.; Massey J. A.; Lough A. J.; Manners I. Angew. Chem., Int. Ed. 1999, 38, 3321–3323. [DOI] [PubMed] [Google Scholar]
- Dorn H.; Singh R. A.; Massey J. A.; Nelson J. M.; Jaska C. A.; Lough A. J.; Manners I. J. Am. Chem. Soc. 2000, 122, 6669–6678. [Google Scholar]
- Dorn H.; Vejzovic E.; Lough A. J.; Manners I. Inorg. Chem. 2001, 40, 4327–4331. [DOI] [PubMed] [Google Scholar]
- Friedrich A.; Drees M.; Schneider S. Chem.—Eur. J. 2009, 15, 10339–10342. [DOI] [PubMed] [Google Scholar]
- Vance J. R.; Robertson A. P. M.; Lee K.; Manners I. Chem.—Eur. J. 2011, 17, 4099–4103. [DOI] [PubMed] [Google Scholar]
- Sewell L. J.; Lloyd-Jones G. C.; Weller A. S. J. Am. Chem. Soc. 2012, 134, 3598–3610. [DOI] [PubMed] [Google Scholar]
- Jaska C. A.; Manners I. J. Am. Chem. Soc. 2004, 126, 9776–9785. [DOI] [PubMed] [Google Scholar]
- Jaska C. A.; Dorn H.; Lough A. J.; Manners I. Chem.—Eur. J. 2003, 9, 271–281. [DOI] [PubMed] [Google Scholar]
- Jaska C. A.; Lough A. J.; Manners I. Dalton Trans. 2005, 326–331. [DOI] [PubMed] [Google Scholar]
- Lee K.; Clark T. J.; Lough A. J.; Manners I. Dalton Trans. 2008, 2732–2740. [DOI] [PubMed] [Google Scholar]
- Thoms C.; Marquardt C.; Timoshkin A. Y.; Bodensteiner M.; Scheer M. Angew. Chem., Int. Ed. 2013, 52, 5150–5154. [DOI] [PubMed] [Google Scholar]
- Huertos M. A.; Weller A. S. Chem. Commun. 2012, 48, 7185–7187. [DOI] [PubMed] [Google Scholar]
- Huertos M. A.; Weller A. S. Chem. Sci. 2013, 4, 1881–1888. [Google Scholar]
- Dorn H.; Rodezno J. M.; Brunnhöfer B.; Rivard E.; Massey J. A.; Manners I. Macromolecules 2003, 36, 291–297. [Google Scholar]
- Clark T. J.; Rodezno J. M.; Clendenning S. B.; Aouba S.; Brodersen P. M.; Lough A. J.; Ruda H. E.; Manners I. Chem.—Eur. J. 2005, 11, 4526–4534. [DOI] [PubMed] [Google Scholar]
- Helten H.; Robertson A. P. M.; Staubitz A.; Vance J. R.; Haddow M. F.; Manners I. Chem.—Eur. J. 2012, 18, 4665–4680. [DOI] [PubMed] [Google Scholar]
- Helten H.; Dutta B.; Vance J. R.; Sloan M. E.; Haddow M. F.; Sproules S.; Collison D.; Whittell G. R.; Lloyd-Jones G. C.; Manners I. Angew. Chem., Int. Ed. 2013, 52, 437–440. [DOI] [PubMed] [Google Scholar]
- Shuttleworth T. A.; Huertos M. A.; Pernik I.; Young R. D.; Weller A. S. Dalton Trans. 2013, 42, 12917–12925. [DOI] [PubMed] [Google Scholar]
- McNulty J.; Zhou Y. Tetrahedron Lett. 2004, 45, 407–409. [Google Scholar]
- Van Overschelde M.; Vervecken E.; Modha S. G.; Cogen S.; Van der Eycken E.; Van der Eycken J. Tetrahedron 2009, 65, 6410–6415. [Google Scholar]
- Eastham G., Tindale N.Catalyst System for Carbonylating Ethylenically Unsaturated Compounds. WO2005079981A1, 2005.
- Zapf A.; Beller M. Chem. Commun. 2005, 431–440. [DOI] [PubMed] [Google Scholar]
- Fleckenstein C. A.; Plenio H. Chem. Soc. Rev. 2010, 39, 694–711. [DOI] [PubMed] [Google Scholar]
- Shimoi M.; Nagai S.; Ichikawa M.; Kawano Y.; Katoh K.; Uruichi M.; Ogino H. J. Am. Chem. Soc. 1999, 121, 11704–11712. [Google Scholar]
- Merle N.; Koicok-Kohn G.; Mahon M. F.; Frost C. G.; Ruggerio G. D.; Weller A. S.; Willis M. C. Dalton Trans. 2004, 3883–3892. [DOI] [PubMed] [Google Scholar]
- Forster T. D.; Tuononen H. M.; Parvez M.; Roesler R. J. Am. Chem. Soc. 2009, 131, 6689–6691. [DOI] [PubMed] [Google Scholar]
- Spielmann J.; Piesik D. F. J.; Harder S. Chem.—Eur. J. 2010, 16, 8307–8318. [DOI] [PubMed] [Google Scholar]
- Kawano Y.; Yamaguchi K.; Miyake S.-y.; Kakizawa T.; Shimoi M. Chem.—Eur. J. 2007, 13, 6920–6931. [DOI] [PubMed] [Google Scholar]
- Ledger A. E. W.; Ellul C. E.; Mahon M. F.; Williams J. M. J.; Whittlesey M. K. Chem.—Eur. J. 2011, 17, 8704–8713. [DOI] [PubMed] [Google Scholar]
- Chaplin A. B.; Weller A. S. Angew. Chem., Int. Ed. 2010, 49, 581–584. [DOI] [PubMed] [Google Scholar]
- Macías R.; Rath N. P.; Barton L. Angew. Chem., Int. Ed. 1999, 38, 162–164. [Google Scholar]
- Ingleson M.; Patmore N. J.; Ruggiero G. D.; Frost C. G.; Mahon M. F.; Willis M. C.; Weller A. S. Organometallics 2001, 20, 4434–4436. [Google Scholar]
- Wagler J.; Hill A. F. Organometallics 2008, 27, 2350–2353. [Google Scholar]
- Nguyen D. H.; Lauréano H.; Jugé S.; Kalck P.; Daran J.-C.; Coppel Y.; Urrutigoity M.; Gouygou M. Organometallics 2009, 28, 6288–6292. [Google Scholar]
- Rablen P. R. J. Am. Chem. Soc. 1997, 119, 8350–8360. [Google Scholar]
- Méndez M.; Cedillo A. Comput. Theor. Chem. 2013, 1011, 44–56. [Google Scholar]
- Bellham P.; Hill M. S.; Kociok-Kohn G.; Liptrot D. J. Chem. Commun. 2013, 49, 1960–1962. [DOI] [PubMed] [Google Scholar]
- In principle there should be four diastereoisomers for 25 as both P-centers are stereogenic. However, we assume that the second center (which is next to the BH3) is far enough removed from the metal center so as not to influence the 31P or 1H chemical shifts significantly.
- Wicht D. K.; Paisner S. N.; Lew B. M.; Glueck D. S.; Yap G. P. A.; Liable-Sands L. M.; Rheingold A. L.; Haar C. M.; Nolan S. P. Organometallics 1998, 17, 652–660. [Google Scholar]
- Robertson A. P. M.; Leitao E. M.; Jurca T.; Haddow M. F.; Helten H.; Lloyd-Jones G. C.; Manners I. J. Am. Chem. Soc. 2013, 135, 12670–12683. [DOI] [PubMed] [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Organometallics 1996, 15, 1518–1520. [Google Scholar]
- Neumann E.; Pfaltz A. Organometallics 2005, 24, 2008–2011. [Google Scholar]
- Schmidbaur H.; Weiss E.; Müller G. Synth. React. Inorg. Met.-Org. Chem. 1985, 15, 401–413. [Google Scholar]
- Lubben A. T.; McIndoe J. S.; Weller A. S. Organometallics 2008, 27, 3303–3306. [Google Scholar]
- Guzel B.; Omary M. A.; Fackler J. P. Jr; Akgerman A. Inorg. Chim. Acta 2001, 325, 45–50. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.