

Abstract
Wnt and Rho GTPase signaling play critical roles in governing numerous aspects of cell physiology, and have been shown to be involved in endochondral ossification and osteoarthritis (OA) development. In this review, current studies of canonical Wnt signaling in OA development, together with the differential roles of Rho GTPases in chondrocyte maturation and OA pathology are critically summarized. Based on the current scientific literature together with our preliminary results, the strategy of targeting Wnt and Rho GTPase for OA prognosis and therapy is suggested, which is instructive for clinical treatment of the disease.
Introduction
The disability burden and prevalence of osteoarthritis (OA) in both developed and developing countries is increasing due to an aging population. OA is a degenerative joint disease that is characterized by cartilage degradation and osteophyte formation. It involves multiple components of the joint, including the synovial joint lining, peri-articular bone and adjacent supporting connective tissue elements [1]. Current OA treatment modalities mainly function as intermittent symptom relief without long-term improvement in disease prognosis due to our current limited understanding of OA pathophysiology. Better understanding of the underlying mechanisms of OA initiation and progression might therefore facilitate identification of appropriate therapeutic targets for OA treatment [2].
The mechanism of OA is currently not well defined, as multiple factors can in more than one way lead to articular cartilage destruction and loss of joint function. Recently, increasing numbers of studies have implicated chondrocyte terminal differentiation (hypertrophy-like changes) in the pathogenesis of OA. This is similar to the chondrocyte differentiation process during endochondral ossification (EO). The close resemblance between terminal differentiation in OA cartilage and EO suggests that new OA therapeutic targets can potentially be identified from EO biology. Normal articular chondrocytes located at the ends of long bones do not develop into the hypertrophic state, thus avoiding terminal differentiation. However, OA chondrocytes lose their stable phenotype and undergo hypertrophy, which is characterized by cell enlargement as well as expression of various chondrocyte maturation and osteogenesis markers such as COLX [3], matrix metalloproteinase (MMP)13 (also known as collagenase 3) [3-5], a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-5 [6-8], osteopontin, osteocalcin, Indian Hedgehog [9], Runx2 [10], vascular endothelial growth factor (VEGF) [11], and transglutaminase-2 (TG-2) [12].
The developmental biology of EO is of key importance in understanding the process of OA, and there is much scientific evidence indicating that signaling pathways modulating joint formation and homeostasis are of central importance in the pathogenesis of OA. The Wnt signaling pathway is well established to be a key regulator in EO [13,14], a process through which bone and articular cartilage are formed. At the same time, most studies support the notion that activation of Wnt/β-catenin signaling is associated with articular chondrocyte matrix catabolism and stable phenotype loss [15]. Recent years have also seen a number of studies indicating that Rho GTPases play central roles in both chondrocyte differentiation and articular chondrocyte physiology, which will be discussed below.
Wnt and Rho GTPase signaling and their interaction
In the canonical Wnt signaling pathway, most β-catenin in the cytoplasm is sequestered within an oligomeric complex of casein kinase, axin, the adenomatous polyposis coli tumor suppressor protein (APC) and glycogen synthase kinase 3β (GSK3β) [16]. However, when Wnt ligands bind to cell membrane receptors, signaling through the frizzled receptors inhibits this degradation process, thereby increasing the levels of free cytoplasmic β-catenin. Accumulation of cytoplasmic β-catenin results in its translocation to the nucleus, where it binds to transcription factors such as lymphoid enhancing factor (LEF)/T cell factor (TCF) to generate a transcriptionally active complex that targets genes such as those encoding MYC, cyclin D1, MMP3 and CD44 [17]. In addition, there are some natural extracellular inhibitory factors that regulate canonical wnt signaling, including members of the secreted frizzled receptor protein (sFRP) family, Dickkopf (Dkk) proteins [18], Wnt inhibitory factor [19], cerberus [20] and sclerostin [21] (Figure 1).
Figure 1.
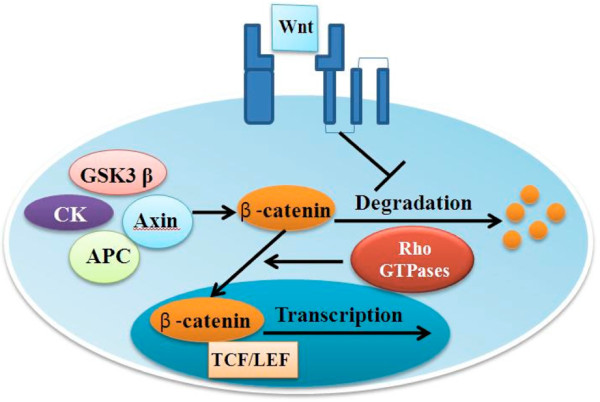
Schematic representation of the canonical Wnt signaling pathway. In canonical Wnt signaling, most β-catenin in the cytoplasm is sequestered in an oligomeric complex of glycogen synthase kinase 3β (GSK3β), casein kinase (CK), axin and adenomatous polyposis coli tumor suppressor protein (APC). When Wnt ligands bind to their cognate cell membrane receptors, signals are released to inhibit this degradation process, resulting in β-catenin accumulation and nuclear translocation regulated by Rac1, DKK1 and FRZB, which are all antagonists of canonical Wnt signaling. LEF, lymphoid enhancing factor; TCF, T cell factor.
The Rho family of GTPases includes 20 members, which are 'Ras-like' proteins. Amongst these, Cdc42, Rac1, and RhoA have been intensively studied. Guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs) and guanine nucleotide dissociation inhibitors (GDIs) are all regulators of the switch between the active and inactive forms of Rho-GTP. Rho GTPases have also been referred to as 'molecular switches' for transducing signals from the chondrocyte extracellular matrix to affect cytoskeletal actin dynamics and cellular morphology, which in turn regulate cell proliferation, apoptosis and gene expression [22].
Meanwhile, a new study indicates that Rho GTPases play a role in nuclear transportation of cytoplasmic β-catenin. Constitutive activation of Rac1 in colon cancer cells significantly enhances TOPFlash promoter activity and nuclear β-catenin accumulation. This effect is inhibited by dominant-negative Rac1 [23]. Mutation of RacGap50C, a negative regulator of Rac1, in Drosophila embryos stimulated canonical Wnt signaling [24]. Similarly, Rac1-specific activator Tiam1 was demonstrated to transcriptionally activate β-catenin/TCF complexes in response to Wnt3a [25]. In another study, Wu and colleagues [26] reported that Rac1 acted cooperatively with JNK2 activation during β-catenin phosphorylation and nuclear localization. This was further supported by phenotype similarity between Rac1 and β-catenin ablation in mouse limb bud ectoderm. Although neither stabilization nor nuclear localization of β-catenin requires RhoA activation, Wnt3a induction of osteogenic differentiation of stem cells requires both RhoA and β-catenin activation [27] (Figure 1).
By contrast, much less attention has been paid to non-canonical Wnt signaling, which is characterized as being β-catenin/TCF independent. One example of non-canonical Wnt signaling is the planar cell polarity pathway, which promotes cell organization in a particular orientation [28,29], through the action of Rho GTPases on assembly of the actin cytoskeleton [30,31].
Roles of Wnt and Rho GTPases in regulating chondrocyte hypertrophy and maturation
Canonical Wnt signaling is known to induce chondrocyte hypertrophy and final maturation. During skeletal development and growth, chondrocyte hypertrophy, calcification, and expression of MMPs, ADAMTS and VEGF in limb buds or growth plates require activation of canonical Wnt signaling [32,33]. Forced expression of the constitutively active form of LEF in chick chondrocytes stimulates ectopic EO [34]. Additionally, mis-expression of Frzd-1, a Wnt antagonist, led to delayed chondrocyte maturation, metalloprotease expression and marrow/bone formation [35], thus suggesting a positive role of Wnt signaling in promoting chondrocyte maturation. These data confirmed the pivotal role of Wnt-β-catenin in chondrocyte maturation and hypertrophy during EO.
Recent studies also suggest that GTPases play a significant role in both chondrocyte development and maturation. Rac1 and Cdc42 are co-expressed in both articular and growth plate chondrocytes, and they function to accelerate the rate of chondrocyte differentiation by increasing COLX promoter activity [36]. Kerr and colleagues [37] found that levels of active Rho GTPases increased with chick chondrocyte maturation. The activated Rac1 expression induced chondrocyte enlargement and MMP13 upregulation, suggesting a positive role of Rac1 in chondrocyte maturation. Additionally, Rac1 and Cdc42 are required for chondrocyte condensation mediated by N-cadherin and act as positive regulators of chondrogenesis [38]. The regulatory effect on chondrocyte differentiation was verified by gene mutation studies in mice. In vivo, Rac1-deficient growth plates displayed delayed ossification, reduced chondrocyte proliferation and increased apoptosis [39], partly due to reduced mitogenic activity through Rac1-inducible nitric oxide synthase-nitric oxide signaling in EO [40]. Similar results were observed in limb bud development. One study reported that the specific deletion of Rac1 (Msx-2 cre) caused severe truncations of limb buds due to impaired nuclear transport of β-catenin [26]. Studies by Kamijo and colleagues reported that both Rac1 [41] and Cdc42 [42] are essential for interdigital programmed cell death through regulation of Bmp, Msx1, and Msx2 gene expression.
A study by Beier and colleagues [43] demonstrated an antagonistic effect of RhoA/ROCK signaling on chondrocyte differentiation, in contrast to Rac1/Cdc42 signaling [44]. Over-expression of RhoA in ATDC5 cells resulted in delayed hypertrophic differentiation with reduced COLX and MMP13 expression. However, pharmacological inhibition of RhoA/ROCK by Y27632 increases Sox9, COLII and aggrecan mRNA levels during chondro-genesis in monolayer culture systems. The observed effects of RhoA/ROCK signaling appeared to be antagonistic in a three-dimensional micromass culture system [45]. Similarly, the study by Lassar and colleagues also reported that RhoA/ROCK signaling regulated Sox9 transcriptional activity through actin polymerization mediated by protein kinase A phosphorylation of Sox9 [46]. By contrast, studies of D'Lima and colleagues [47] demonstrated that ROCK, a downstream effector of RhoA, directly phosphorylates Sox9, which in turn regulates chondrogenesis. This suggests that RhoA functions through signaling pathways other than ROCK in modulating chondrogenesis [48]. Recently, Sox9 has been demonstrated to correlate with Mef2c in modulating chondrocyte terminal differentiation [49], suggesting that Rho GTPases may function upstream of Sox9 during chondrocyte differentiation.
In summary, Rac1/Cdc42 and RhoA/ROCK signaling pathways are all expressed during chondrogenesis and have adverse effects on chondrocyte terminal differentiation (hypertrophy-like change). The Rac1/Cdc42 signaling pathway accelerates chondrocyte hypertrophy while the RhoA/ROCK signaling pathway delays chondrocyte maturation through regulation of Sox9, as illustrated in Figure 2, but the underlying mechanisms are still poorly understood.
Figure 2.
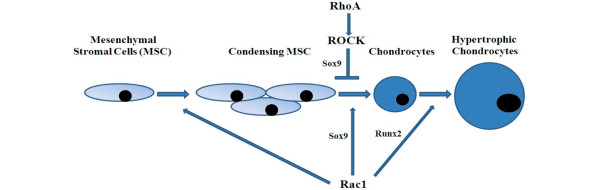
Rho GTPase signaling in chondrocyte development and maturation. Rac1/Cdc42 and RhoA/ROCK signaling pathways are all required for chondrogenesis, and have antagonistic effects on chondrocyte terminal differentiation, which is probably mediated by interaction with Sox9 and Runx2.
Canonical Wnt signaling and pathological changes in osteoarthritis
Wnt-β-catenin signaling is activated in both human and mice OA cartilage. In fact, many animal model studies utilizing a genetic approach have strengthened this view. Mechanical injury, a major cause of OA, leads to down-regulation of Wnt antagonist FRZB and up-regulation of ligand Wnt16 and target genes encoding β-catenin, Axin-2, C-JUN and LEF-1 [50]. Furthermore, transcrip-tome analysis demonstrated that expression of Wnt1-inducible signaling protein 1 (WISP-1) is increased twofold in cartilage lesions compared to healthy intact cartilage [51]. These findings indicate that Wnt signaling may function as an OA initiation factor upon mechanical injury. Corr and colleagues [52,53] first reported that Arg200Trp and Arg324Gly Frzb variants, encoding sFRP3, an extracellular inhibitor of Wnt-β-catenin signaling, contributed to genetic susceptibility of women to hip OA. However, the same conclusions were not reached by another two groups that investigated other populations [54,55]. Although Min and colleagues [56] thought that these two variants are also associated with other generalized OA at multiple sites, there is still no direct evidence implicating Frzb variants in knee OA. Frzb knockout mice display increased cartilage damage and thicker cortical bone formation [57]. Given the close relationship between bone shape and OA development, Baker-Lepain and colleagues [58] believed that SNPs in Frzb are associated with the shape of proximal femur and further contribute to hip OA development. However, some pertinent questions remain: do these two variants increase wnt ligand binding with the Frizzled protein to activate Wnt-β-catenin signaling; and does mis-function of Frzb in chondrocytes directly lead to OA or Frzb modulation of bone shape, disrupting mechanical loading on cartilage and consequently leading to OA? The inhibition of Dickkopf-1 (Dkk1), a negative regulator of Wnt-β-catenin signaling, has been reported to be able to reverse the bone-destructive characteristics of rheumatoid arthritis to the bone-forming characteristics of OA [59]. Another study on the mouse OA model also demonstrated that control of Dkk1 expression prevents joint cartilage deterioration in osteoarthritic knees through attenuating the apoptosis regulator Bax, MMP3 and RANKL (receptor activator of nuclear factor kappa-B ligand) [60]. Additionally, Blom and colleagues [61] showed that stimulation of Wnt-induced signaling protein 1 (WISP1) in chondrocytes resulted in IL1-dependent induction of MMPs and aggrecanase, suggesting induction of chondrocyte maturation. LRP5 is located in chromosome 11q12-13, which is thought to be an OA susceptibility locus [62]. Lrp5-/- mice displayed increased cartilage degradation, probably due to low bone mass density [63]. These studies thus provide indirect evidence for Wnt-β-catenin participation in OA progression. Zhu and colleagues [64] provided direct evidence for the first time that β-catenin is implicated in the development of OA. The conditional activation of β-catenin in articular chondrocytes of adult mice caused OA-like cartilage degradation and osteophyte formation, and this was associated with accelerated chondrocyte maturation and MMP13 expression. Later, the authors reported a somewhat contradictory finding that selective suppression of β-catenin signaling in articular chondrocytes also causes OA-like cartilage degradation in Col2a1-ICAT (inhibitor of β-catenin and TCF) transgenic mice [65]. This led Kawaguchi [66] to hypothesize that β-catenin induces chondrocyte maturation similarly to Runx2, whereas it suppresses chondrocyte apoptosis similarly to osteoprotegerin (Table 1).
Table 1.
Overview of the roles of various elements of the Wnt signaling pathway in osteoarthritis development, as demonstrated by human genetic studies or animal models
| Effect on | ||||
|---|---|---|---|---|
| Element treatment/SNPs | Wnt signaling | Results | Conclusions | |
| Receptor | ||||
| LRP5 | Haplotype (C-G-C-C-A) in LRP5 | Inhibition | This haplotype predisposes to increased risk of OA | LRP5 variant may predispose patients to OA [56] |
| Lrp5 knockout in mice | Inhibition | Increased cartilage degradation, decreased bone mineral density | Loss of function of Lrp5 leads to OA [57] | |
| Wnt ligands | ||||
| Wnts (up-regulated in OA) | Mechanical injury | Activation | Up-regulation of Wnt16/WISP-1, down-regulation of FRZB, up- regulation of β-catenin, axin-2, C-JUN and LEF-1 | Mechanical injury activates Wnt signaling [43] |
| Wnt antagonists | ||||
| Frzb (up-regulated in OA) | Arg200Trp/Arg324Gly Frzb variants | Activation | These two variants are associated with female hip OA from an epidemiological viewpoint | These two variants confer genetic susceptibility to female hip OA [46,47] |
| Arg200Trp/Arg324Gly Frzb variants | Activation | These two variants are associated with other generalized OA by epidemiological analysis | These two variants contribute to female hip OA [50] | |
| Frzb knockout mice | Activation | Increased cartilage damage, thicker cortical bone formation | Loss of function of Frzb contributes to the development of OA [51] | |
| DKK1 (up-regulated in OA) | Inhibition of DKK1 by antibody | Activation | Blocks bone erosion, promotes bone formation, reverses RA to OA | Wnt signaling is a key regulator of joint remodeling [53] |
| OA rat knees were treated with end-capped phosphorothioate Dkk-1 antisense oligonucleotide (Dkk-1-AS) | Inhibition | Alleviated Mankin score, cartilage fibrillation, and serum cartilage degradation markers | Dkk1 expression prevents OA cartilage destruction and subchondral bone damage [54] | |
| Transcription factor | ||||
| β-Catenin (up- regulated in OA) | Activation of β-catenin in articular chondrocytes | Activation | OA-like cartilage degradation, osteophyte formation, accelerated chondrocyte maturation and MMP13 expression | Wnt/β-catenin activation promotes OA development by accelerating chondrocyte maturation [58] |
| Suppression of β-catenin in articular chondrocytes | Inhibition | OA-like cartilage degradation, increased chondrocyte apoptosis | Wnt/β-catenin inhibition promotes OA development by increasing chondrocyte apoptosis [59] |
LEF, lymphoid enhancing factor; MMP, matrix metalloproteinase; OA, osteoarthritis; RA, rheumatoid arthritis.
Although most current studies in the scientific literature demonstrate the involvement of canonical Wnt-β-catenin signaling in OA development, the role of this signaling pathway in OA pathophysiology is actually dependent on patient characteristics. For instance, two SNPs in FRZB were initially thought to be associated with an increased risk of primary hip OA among female patients [52,53]. However, conflicting data were reported by different studies [54,55]. The relationship between FRZB SNPs and human OA development may be dependent on the characteristics of the patient population, that is, sex and age-related differences. Excessive or insufficient β-catenin signaling in mice chondrocytes has been shown to increase susceptibility to OA phenotype [64,65], thus suggesting that balanced β-catenin levels are essential for maintaining homeostasis of articular chondrocytes. Factors impairing this balance could lead to pathological changes in chondrocytes by promoting either terminal differentiation or apoptosis.
Moreover, because OA is a systemic joint disease affecting overall joint tissues, including cartilage, sub-chondral bone and synovium, imbalance of β-catenin signaling in tissues other than cartilage could also initiate or promote OA development. For example, because canonical Wnt signaling has direct roles in osteogenesis, excessive Wnt signaling can also lead to increased bone formation, which might be associated with osteophyte formation. Two Wnt antagonists, sFRP1, which binds to RANKL [67], and DKK1, which promotes osteoprotegerin secretion [58], can alter the balance between osteoclast and osteoblast development. Additionally, up-regulated DKK1 levels in synovial fibroblasts contribute to synovial hypervascularity in OA [68], which would imply that modulating DKK1 expression in synovial fibroblasts may be a potential therapeutic strategy for OA-induced synovitis and joint degradation.
Rho GTPases and pathological changes in osteoarthritis chondrocytes
With increasing recognition of the role of Rho GTPase activities in chondrocyte hypertrophy-like changes, their effects on OA have attracted much attention and have been investigated using both human genetic studies and animal models. Epidemiological studies from different groups reported a relationship between SNPs in RhoB and OA susceptibility in some populations [69,70]. Meanwhile, rodent OA models treated with the Rho kinase inhibitor AS1892802 displayed alleviation of cartilage damage [71]. RhoB is downregulated in OA articular chondrocytes and is thought to be responsible for significant DNA damage observed in the pre-apoptotic phenotype of OA chondrocytes [72]. RhoA-ROCK signaling is thought to be involved in early phase response to abnormal mechanical stimuli, which is accepted as a contributory factor to OA initiation and progression [73]. In addition, RhoA-ROCK signaling has also been demonstrated to interact with other pathological factors associated with OA such as transforming growth factor-epidermal growth factor receptor signaling factors [74], IL1a, insulin-like growth factor-1 (IGF-1) [75] and leptin [76], suggesting a global role of RhoA-ROCK in OA progression. With regards to the Rac1/Cdc42 signaling pathway in OA progression, Cdc42-GTP content decreases [77] while Rac1-GTP increases with chondrocyte aging. This provides new insights into age-related OA development. Additionally, Rac1 regulates CTGF/CCN2 gene expression [78], which is upregulated in OA, and has been shown to be beneficial for articular cartilage regeneration in a mono-iodoacetate (MIA)-induced OA model and articular cartilage defect model [79]. A recent study by Long and colleagues [80] showed that Rac1 is involved in Fnf-induced MMP13 production, thus suggesting a metabolic role of Rac1 activation in cartilage (Figure 3).
Figure 3.
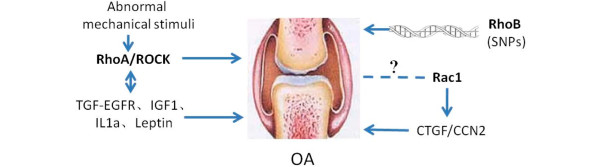
Rho GTPases play direct or indirect roles in osteoarthritis (OA). SNPs in RhoB have been found in OA populations; Rac1 regulates CTGF/CCN2 expression, which in turn play a pathological role in OA; RhoA/ROCK interact with OA pathological factors such as transforming growth factor-epidermal growth factor receptor (TGF-EGFR), insulin-like growth factor-1 (IGF-1), IL1a, and leptin in regulating OA progression; RhoA-ROCK signaling is suggested to be involved in OA early phase response to abnormal mechanical stimuli.
The role of Rho GTPases in OA progression may not only be limited to cartilage, but may also involve synovium and osteochondral bone. Rac and its regulators - GEFs and GAPs - have been proven to play vital roles in STAT signaling transduction [81-85], which is essential for the inflammatory response, thus suggesting the important role of Rac GTPases in OA joint inflammation [86]. Our preliminary results also showed that intra-articular administration of the Rac1 inhibitor NSC2376 efficaciously decreases mRNA transcript levels of pro-inflammatory factors in joint tissue (unpublished data). Moreover, Rho GTPases also have important roles in mature osteoclasts by regulating the formation of actin rings and resorption lacunae [87] and are required for osteoclast differentiation [88]. The definitive role of Rho GTPase expression in osteochondral bone that contributes to OA progression needs to be further studied.
Our preliminary study investigating human OA cartilage shows that Rac1 is activated in OA chondrocytes and the level of Rac1-GTP is greatly upregulated by IL1b in a chondrocyte monolayer culture system (unpublished data), suggesting the important role of Rac1 in pro-inflammatory factor-induced OA progression. Furthermore, primary chondrocytes from OA calcified cartilage (one phenotype of OA) is significantly inhibited by the Rac1 specific inhibitor NSC23766, as demonstrated by Alizarin Red staining (unpublished data). Constitutive over-expression of Rac1 resulted in up-regulation of COLX, Runx2 and ADAMTS-5 and intra-articular injection of NSC23766 delayed mice OA development (unpublished data). Due to the high level of expression of Rac1 in human and mouse articular chondrocytes (Figure 4), further studies are focusing on the role of Rac1 in OA development in vivo, and its underlying mechanism. Additionally, the defined role of Rho GTPase in OA progression should be further investigated with animal models utilizing both genetic and pharmacological tools.
Figure 4.
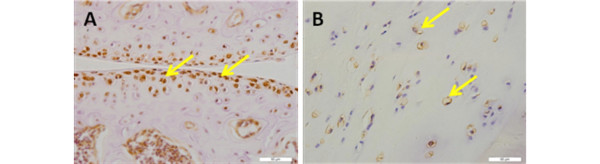
Rac1 is expressed in both mouse and human cartilage. (A) Robust expression of Rac1 was detected at the surface and middle zones of mouse cartilage but chondrocytes in calcified zone displayed reduced expression. (B) Rac1 is distributed ubiquitously in human articular cartilage. Arrows in both panels indicate representative Rac1-positive chondrocytes. Scale bars = 50 μm.
As mentioned earlier, Wnt/β-catenin signaling activation leads to elevated articular chondrocyte catabolism, hypertrophy-like changes and cartilage degradation, which are all key features of OA [66]. Rho GTPases have recently been discovered to function as key mediators of β-catenin nuclear translocation and the available data demonstrated significant roles of GTPases in chondrocyte hypertrophy, maturation and OA development [69-80]. Interaction between canonical Wnt signaling and GTPases independent of actin cytoskeletal changes in OA development has not yet been addressed. The preliminary results from our study indicate that Rho GTPase modulation of OA may partially function through control of β-catenin nuclear translocation in canonical Wnt signaling.
Wnt signaling and Rho GTPases as targets for OA treatment
Current treatment modalities of OA, including pharmacological and surgical procedures, are mainly focused on promoting partial regeneration and relieving pain. For example, acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDS) and cyclooxygense 2 (COX-2) [89] are all utilized to relieve arthritic pain and can achieve good short-term results. Surgical treatment, including lavage, abrasion arthroplasty and microfracture, has long been considered as a palliative therapy for pain, possibly due to removal of inflammatory factors and bone marrow mesenchymal stem cell-mediated fibrous cartilage regeneration on the subchondral bone [90]. Concerns about later re-emergence of pain and durability of the newly formed fibrous cartilage by micro-fracture makes it imperative to develop more effective OA treatment modalities.
Recently, tissue engineering for cartilage regeneration has achieved much progress. Autologous chondrocyte implantation has often been used to treat simple cartilage defects [91,92]. However, chondrocytes in the newly formed cartilage by these procedures are likely to undergo calcification and hypertrophy-like changes, thereby affecting cartilage function [93]. Therefore, to improve therapeutic efficacy and maintain the functional status of regenerated cartilage, OA treatment should be focused on removing the causes or risk factors of OA. Small molecules targeted to OA-specific molecular pathophysiology may be a good strategy.
Available evidence suggests a critical role of Wnt signaling in EO as well as OA development. Excessive levels of some Wnt ligands and β-catenin have been observed in degenerating cartilage. However, this seems to be a paradox because several Wnt signaling antagonists, including DKK1, FRP1, FRP2, and FRP4, are strongly expresssed in OA synovium and cartilage. This may possibly be explained by the conjecture that both gain or loss of function of Wnt/β-catenin signaling would disrupt cartilage homeostasis and lead to pathological changes associated with OA. Aberrant expression of Wnt ligands and Wnt antagonists in synovium may function as an early signal to initiate OA, which in turn can be utilized as an easily accessible OA prognostic marker.
Both genetic and experimental studies have highlighted the great potential of locally modulating the Wnt signaling pathway to alter OA prognosis. Rho GTPases have been recently discovered to modulate β-catenin nuclear translocation and control β-catenin/TCF transcription activity. An altered level of Rho GTPases in articular chondrocytes might therefore be recognized as a new marker for OA development. Hence, Rho GTPases may be good targeting candidates to develop small molecule drugs for OA therapy. In fact, many ROCK inhibitors have recently emerged and have been reported in the patent literature. Some of these are utilized for inflammatory disorders such as multiple sclerosis and asthma. In particular, fasudil hydrochloride, a potent ROCK inhibitor, has been clinically used to treat cerebral vaso-spasm [94] and pulmonary hypertension [95].
Although blocking the activity of some members of the Rho GTPases family is able to prevent chondrocytes from undergoing hypertrophy and ossification, there are several pertinent problems to be solved before this strategy can be utilized as a means of OA therapy. Theoretically, Rho GTPases interact with the Sox9 and Runx2 pathways in maintaining a fine balance between chondrogenesis and chondrocyte terminal differentiation. The underlying mechanism needs further investigation to identify more specific intervening signal molecules implicated in chondrocyte hypertrophy-like changes. Alternatively, Rho GTPase effectors could be more promising drug targets, because each of these effectors mediates specific roles of Rho GTPases. To date, modulating Rho GTPases to prevent chondrocytes from undergoing hypertrophy-like change has been evaluated in several animal studies and have demonstrated significant efficacy in OA therapy [71]. However, many scientific questions about the application of Rho GTPases for OA treatment still remain to be answered.
Last but not least, since Wnt and Rho GTPases have important signaling roles in numerous cell types, systemic administration of modulators of these pathways could be dangerous. Localized drug delivery may be a solution. Some biomaterials, such as chitosan and alginate microspheres, may serve as delivery vehicles for controlled drug release in designated tissues. Because Wnt and Rho GTPase signaling pathways modulate both early chondrogenesis (which should be promoted for cartilage repair) and hypertrophic differentiation (which should be suppressed), there should ideally be programmed drug administration for initial activation of these signaling pathways to promote chondrogenesis, followed by inhibition at a later time point to prevent chondrocyte terminal differentiation. Unpublished results from our lab showed that mesenchymal stem cells seeded on biomaterials incorporated with cytokines promoted cartilage repair. Thereafter, intra-articular injection of Rho GTPase inhibitors at a later time point could block terminal differentiation of the newly formed chondrocytes.
Conclusion
OA articular chondrocytes undergo hypertrophy-like changes, which is a similar process to EO. Wnt/β-catenin and Rho GTPases, mainly RhoA, Rac1 and Cdc42, are well recognized as crucial regulators or mediators of chondrocyte development and chondrocyte hypertrophy during EO. It is now well established that Wnt/β-catenin and Rho GTPases have similar roles in OA progression and local modulation of the Wnt signaling pathway delays OA development. Preliminary studies have illustrated that Rac1 inhibition suppressed OA articular chondrocytes from undergoing hypertrophy-like changes both in vivo and in vitro. Moreover, Rac1 inhibitors may also be promising drugs for preventing chondrocyte ossification in cartilage tissue engineering. Other members of the Rho GTPase family may also possess similar potential as molecular targets for OA therapy. It was only in the last few years that the roles of Rho GTPases in modulating chondrocyte development and OA were intensively studied. Their regulatory effects on chondro-cyte hypertrophy-like change warrants the use of Rho GTPase activators or inhibitors for OA prevention and cartilage tissue engineering. However, several concerns need to be addressed before Rho GTPase modulation is utilized as a means of OA therapy: the dosage and timing of intervention should be carefully investigated; appropriate controlled release systems may potentiate sustained function of Rho GTPases in OA joints; and drugs targeting specific effectors of Rho GTPases should be further developed to avoid side effects.
Abbreviations
ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; EO: endochondral ossification; FRP: frizzled receptor protein; GAP: GTPase-activating protein; GEF: guanine nucleotide exchange factor; IL: interleukin; LEF: lymphoid enhancing factor; MMP: matrix metalloproteinase; OA: osteoarthritis; RANKL: receptor activator of nuclear factor kappa-B ligand; sFRP: secreted frizzled receptor protein; SNP: single-nucleotide polymorphism; TCF: T cell factor; VEGF: vascular endothelial growth factor.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Shouan Zhu, Email: anhuiange2007@yahoo.cn.
Huanhuan Liu, Email: hhliu012@yahoo.com.cn.
Yan Wu, Email: wuyanzju@126.com.
Boon Chin Heng, Email: alexis.heng@bsse.ethz.ch.
Pengfei Chen, Email: 790048914@qq.com.
Hua Liu, Email: hua94126@yahoo.com.cn.
Hong Wei Ouyang, Email: hwoy@zju.edu.cn.
Acknowledgements
This work was supported by National key scientific research projects (2012CB966604), the National Natural Science Fund (81125014, 81101356, 81201395, J1103603), Zhejiang province public welfare fund (2012C3112), New century talent fund (NCET-08-0487), The National High Technology Research and Development Program of China (2012AA020503) and Sponsored by Regenerative Medicine in Innovative Medical Subjects of Zhejiang Province.
References
- Brandt KD, Radin EL, Dieppe PA, de Putte L. Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis. 2006;15:1261–1264. doi: 10.1136/ard.2006.058347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;15:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, Min BH, Chun JS. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;15:687–693. doi: 10.1038/nm.2153. [DOI] [PubMed] [Google Scholar]
- Shlopov BV, Gumanovskaya ML, Hasty KA. Autocrine regulation of collagenase 3 (matrix metalloproteinase 13) during osteoarthritis. Arthritis Rheum. 2000;15:195–205. doi: 10.1002/1529-0131(200001)43:1<195::AID-ANR24>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;15:585–594. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Majumdar MK, Askew R, Schelling S, Stedman N, Blanchet T, Hopkins B, Morris EA, Glasson SS. Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 2007;15:3670–3674. doi: 10.1002/art.23027. [DOI] [PubMed] [Google Scholar]
- Malfait AM, Liu RQ, Ijiri K, Komiya S, Tortorella MD. Inhibition of ADAM-TS4 and ADAM-TS5 prevents aggrecan degradation in osteoarthritic cartilage. J Biol Chem. 2002;15:22201–22208. doi: 10.1074/jbc.M200431200. [DOI] [PubMed] [Google Scholar]
- Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, Fourie AM, Fosang AJ. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;15:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- Wei F, Zhou J, Wei X, Zhang J, Fleming BC, Terek R, Pei M, Chen Q, Liu T, Wei L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthritis Cartilage. 2012;15:755–763. doi: 10.1016/j.joca.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G, Nakamura K, Chung UI, Kawaguchi H. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;15:2462–2470. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- Jansen H, Meffert RH, Birkenfeld F, Petersen W, Pufe T. Detection of vascular endothelial growth factor (VEGF) in moderate osteoarthritis in a rabbit model. Ann Anat. 2012;15:452–456. doi: 10.1016/j.aanat.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Huebner JL, Johnson KA, Kraus VB, Terkeltaub RA. Transglutaminase 2 is a marker of chondrocyte hypertrophy and osteoarthritis severity in the Hartley guinea pig model of knee OA. Osteoarthritis Cartilage. 2009;15:1056–1064. doi: 10.1016/j.joca.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;15:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;15:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Otani T, Koike T, Iwamoto M, Enomoto-Iwamoto M. Wnt/betacatenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: its possible role in joint degeneration. Lab Invest. 2008;15:264–274. doi: 10.1038/labinvest.3700747. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;15:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Bae S, Reid CD, Kessler DS. Siamois and Twin are redundant and essential in formation of the Spemann organizer. Dev Biol. 2011;15:367–381. doi: 10.1016/j.ydbio.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;15:357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;15:431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Agius E, Leyns L, Bhattacharyya S, Grunz H, Bouwmeester T, De Robertis EM. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature. 1999;15:707–710. doi: 10.1038/17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;15:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Beier F. Regulation of chondrocyte differentiation by the actin cytoskeleton and adhesive interactions. J Cell Physiol. 2007;15:1–8. doi: 10.1002/jcp.21110. [DOI] [PubMed] [Google Scholar]
- Esufali S, Bapat B. Cross-talk between Rac1 GTPase and dysregulated Wnt signaling pathway leads to cellular redistribution of beta-catenin and TCF/ LEF-mediated transcriptional activation. Oncogene. 2004;15:8260–8271. doi: 10.1038/sj.onc.1208007. [DOI] [PubMed] [Google Scholar]
- Jones WM, Bejsovec A. RacGap50C negatively regulates wingless pathway activity during Drosophila embryonic development. Genetics. 2005;15:2075–2086. doi: 10.1534/genetics.104.039735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buongiorno P, Pethe VV, Charames GS, Esufali S, Bapat B. Rac1 GTPase and the Rac1 exchange factor Tiam1 associate with Wnt-responsive promoters to enhance beta-catenin/TCF-dependent transcription in colorectal cancer cells. Mol Cancer. 2008;15:73. doi: 10.1186/1476-4598-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;15:340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossol-Allison J, Stemmle LN, Swenson-Fields KI, Kelly P, Fields PE, McCall SJ, Casey PJ, Fields TA. Rho GTPase activity modulates Wnt3a/beta-catenin signaling. Cell Signal. 2009;15:1559–1568. doi: 10.1016/j.cellsig.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutt DI. The asymmetric subcellular localisation of components of the planar polarity pathway. Semin Cell Dev Biol. 2002;15:225–231. doi: 10.1016/S1084-9521(02)00041-1. [DOI] [PubMed] [Google Scholar]
- Klein TJ, Mlodzik M. Planar cell polarization: an emerging model points in the right direction. Annu Rev Cell Dev Biol. 2005;15:155–176. doi: 10.1146/annurev.cellbio.21.012704.132806. [DOI] [PubMed] [Google Scholar]
- Fanto M, Weber U, Strutt DI, Mlodzik M. Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr Biol. 2000;15:979–988. doi: 10.1016/S0960-9822(00)00645-X. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;15:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;15:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, Yamada Y, Costantini F, Wakisaka S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005;15:19185–19195. doi: 10.1074/jbc.M414275200. [DOI] [PubMed] [Google Scholar]
- Kitagaki J, Iwamoto M, Liu JG, Tamamura Y, Pacifci M, Enomoto-Iwamoto M. Activation of beta-catenin-LEF/TCF signal pathway in chondrocytes stimulates ectopic endochondral ossification. Osteoarthritis Cartilage. 2003;15:36–43. doi: 10.1053/joca.2002.0863. [DOI] [PubMed] [Google Scholar]
- Enomoto-Iwamoto M, Kitagaki J, Koyama E, Tamamura Y, Wu C, Kanatani N, Koike T, Okada H, Komori T, Yoneda T, Church V, Francis-West PH, Kurisu K, Nohno T, Pacifici M, Iwamoto M. The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev Biol. 2002;15:142–156. doi: 10.1006/dbio.2002.0802. [DOI] [PubMed] [Google Scholar]
- Wang G, Beier F. Rac1/Cdc42 and RhoA GTPases antagonistically regulate chondrocyte proliferation, hypertrophy, and apoptosis. J Bone Miner Res. 2005;15:1022–1031. doi: 10.1359/JBMR.050113. [DOI] [PubMed] [Google Scholar]
- Kerr BA, Otani T, Koyama E, Freeman TA, Enomoto-Iwamoto M. Small GTPase protein Rac-1 is activated with maturation and regulates cell morphology and function in chondrocytes. Exp Cell Res. 2008;15:1301–1312. doi: 10.1016/j.yexcr.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Wang G, Dupuis H, Shao Z, Beier F. Rac1 signaling stimulates N-cadherin expression, mesenchymal condensation, and chondrogenesis. J Biol Chem. 2007;15:23500–23508. doi: 10.1074/jbc.M700680200. [DOI] [PubMed] [Google Scholar]
- Wang G, Woods A, Agoston H, Ulici V, Glogauer M, Beier F. Genetic ablation of Rac1 in cartilage results in chondrodysplasia. Dev Biol. 2007;15:612–623. doi: 10.1016/j.ydbio.2007.03.520. [DOI] [PubMed] [Google Scholar]
- Wang G, Yan Q, Woods A, Aubrey LA, Feng Q, Beier F. Inducible nitric oxide synthase-nitric oxide signaling mediates the mitogenic activity of Rac1 during endochondral bone growth. J Cell Sci. 2011;15:3405–3413. doi: 10.1242/jcs.076026. [DOI] [PubMed] [Google Scholar]
- Suzuki D, Yamada A, Amano T, Yasuhara R, Kimura A, Sakahara M, Tsumaki N, Takeda S, Tamura M, Nakamura M, Wada N, Nohno T, Shiroishi T, Aiba A, Kamijo R. Essential mesenchymal role of small GTPase Rac1 in interdigital programmed cell death during limb development. Dev Biol. 2009;15:396–406. doi: 10.1016/j.ydbio.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Aizawa R, Yamada A, Suzuki D, Iimura T, Kassai H, Harada T, Tsukasaki M, Yamamoto G, Tachikawa T, Nakao K, Yamamoto M, Yamaguchi A, Aiba A, Kamijo R. Cdc42 is required for chondrogenesis and interdigital programmed cell death during limb development. Mech Dev. 2012;15:38–50. doi: 10.1016/j.mod.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Beier F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem. 2005;15:11626–11634. doi: 10.1074/jbc.M409158200. [DOI] [PubMed] [Google Scholar]
- Wang G, Woods A, Sabari S, Pagnotta L, Stanton LA, Beier F. RhoA/ROCK signaling suppresses hypertrophic chondrocyte differentiation. J Biol Chem. 2004;15:13205–13214. doi: 10.1074/jbc.M311427200. [DOI] [PubMed] [Google Scholar]
- Woods A, Beier F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J Biol Chem. 2006;15:13134–13140. doi: 10.1074/jbc.M509433200. [DOI] [PubMed] [Google Scholar]
- Kumar D, Lassar AB. The transcriptional activity of Sox9 in chondrocytes is regulated by RhoA signaling and actin polymerization. Mol Cell Biol. 2009;15:4262–4273. doi: 10.1128/MCB.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudenschild DR, Chen J, Pang N, Lotz MK, D'Lima DD. Rho kinasedependent activation of SOX9 in chondrocytes. Arthritis Rheum. 2010;15:191–200. doi: 10.1002/art.25051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Kim S, Kim Y, Jin EJ, Sonn JK. Inhibition of RhoA but not ROCK induces chondrogenesis of chick limb mesenchymal cells. Biochem Biophys Res Commun. 2012;15:500–505. doi: 10.1016/j.bbrc.2012.01.053. [DOI] [PubMed] [Google Scholar]
- Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, Lefebvre V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. 2012;15:597–609. doi: 10.1016/j.devcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;15:1410–1421. doi: 10.1002/art.23444. [DOI] [PubMed] [Google Scholar]
- Blom AB, Brockbank SM, van Lent PL, van Beuningen HM, Geurts J, Takahashi N, van der Kraan PM, van de Loo FA, Schreurs BW, Clements K, Newham P, van den Berg WB. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009;15:501–512. doi: 10.1002/art.24247. [DOI] [PubMed] [Google Scholar]
- Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, Ferreira A, Ciesielski C, Carson DA, Corr M. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci USA. 2004;15:9757–9762. doi: 10.1073/pnas.0403456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane NE, Lian K, Nevitt MC, Zmuda JM, Lui L, Li J, Wang J, Fontecha M, Umblas N, Rosenbach M, de Leon P, Corr M. Frizzled-related protein variants are risk factors for hip osteoarthritis. Arthritis Rheum. 2006;15:1246–1254. doi: 10.1002/art.21673. [DOI] [PubMed] [Google Scholar]
- Lories RJ, Boonen S, Peeters J, de Vlam K, Luyten FP. Evidence for a differential association of the Arg200Trp single-nucleotide polymorphism in FRZB with hip osteoarthritis and osteoporosis. Rheumatology (Oxford) 2006;15:113–114. doi: 10.1093/rheumatology/kei148. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lopez J, Pombo-Suarez M, Liz M, Gomez-Reino JJ, Gonzalez A. Further evidence of the role of frizzled-related protein gene polymorphisms in osteoarthritis. Ann Rheum Dis. 2007;15:1052–1055. doi: 10.1136/ard.2006.065938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JL, Meulenbelt I, Riyazi N, Kloppenburg M, Houwing-Duistermaat JJ, Seymour AB, Pols HA, van Duijn CM, Slagboom PE. Association of the Frizzled-related protein gene with symptomatic osteoarthritis at multiple sites. Arthritis Rheum. 2005;15:1077–1080. doi: 10.1002/art.20993. [DOI] [PubMed] [Google Scholar]
- Lories RJ, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, Thomas JT, Luyten FP. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007;15:4095–4103. doi: 10.1002/art.23137. [DOI] [PubMed] [Google Scholar]
- Baker-Lepain JC, Lynch JA, Parimi N, McCulloch CE, Nevitt MC, Corr M, Lane NE. Variant alleles of the Wnt antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012;15:1457–1465. doi: 10.1002/art.34526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;15:156–163. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]
- Weng LH, Wang CJ, Ko JY, Sun YC, Wang FS. Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum. 2010;15:1393–1402. doi: 10.1002/art.27357. [DOI] [PubMed] [Google Scholar]
- Blom AB, Brockbank SM, van Lent PL, van Beuningen HM, Geurts J, Takahashi N, van der Kraan PM, van de Loo FA, Schreurs BW, Clements K, Newham P, van den Berg WB. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009;15:501–512. doi: 10.1002/art.24247. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Gidley J, Sandy JR, Perry MJ, Elson CJ, Kirwan JR, Spector TD, Doherty M, Bidwell JL, Mansell JP. Haplotypes of the low-density lipoprotein receptor-related protein 5 (LRP5) gene: are they a risk factor in osteoarthritis. Osteoarthritis Cartilage. 2005;15:608–613. doi: 10.1016/j.joca.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Lodewyckx L, Luyten FP, Lories RJ. Genetic deletion of low-density lipoprotein receptor-related protein 5 increases cartilage degradation in instability-induced osteoarthritis. Rheumatology (Oxford) 2012;15:1973–1978. doi: 10.1093/rheumatology/kes178. [DOI] [PubMed] [Google Scholar]
- Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier RN, O'Keefe RJ, Zuscik M, Chen D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009;15:12–21. doi: 10.1359/jbmr.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O'Keefe RJ, Chen D. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008;15:2053–2064. doi: 10.1002/art.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi H. Regulation of osteoarthritis development by Wnt-betacatenin signaling through the endochondral ossification process. J Bone Miner Res. 2009;15:8–11. doi: 10.1359/jbmr.081115. [DOI] [PubMed] [Google Scholar]
- Häusler KD, Horwood NJ, Chuman Y, Fisher JL, Ellis J, Martin TJ, Rubin JS, Gillespie MT. Secreted frizzled-related protein-1 inhibits RANKL-dependent osteoclast formation. J Bone Miner Res. 2004;15:1873–1881. doi: 10.1359/JBMR.040807. [DOI] [PubMed] [Google Scholar]
- Weng LH, Ko JY, Wang CJ, Sun YC, Wang FS. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012;15:3267–3277. doi: 10.1002/art.34602. [DOI] [PubMed] [Google Scholar]
- Shi D, Nakamura T, Nakajima M, Dai J, Qin J, Ni H, Xu Y, Yao C, Wei J, Liu B, Ikegawa S, Jiang Q. Association of single-nucleotide polymorphisms in RHOB and TXNDC3 with knee osteoarthritis susceptibility: two casecontrol studies in East Asian populations and a meta-analysis. Arthritis Res Ther. 2008;15:R54. doi: 10.1186/ar2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin J, Meulenbelt I, Min J, Mustafa Z, Sinsheimer JS, Carr A, Slagboom PE. Genetic association analysis of RHOB and TXNDC3 in osteoarthritis. Am J Hum Genet. 2007;15:383–386. doi: 10.1086/511443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita N, Yoshimi E, Hatori C, Kumakura F, Seki N, Shimizu Y. Alleviating effects of AS1892802, a Rho kinase inhibitor, on osteoarthritic disorders in rodents. J Pharmacol Sci. 2011;15:481–489. doi: 10.1254/jphs.10319FP. [DOI] [PubMed] [Google Scholar]
- Gebhard PM, Soder S, Bau B, Aigner T. Down-regulation of the GTPase RhoB might be involved in the pre-apoptotic phenotype of osteoarthritic chondrocytes. Front Biosci. 2004;15:827–833. doi: 10.2741/1254. [DOI] [PubMed] [Google Scholar]
- Haudenschild DR, Nguyen B, Chen J, D'Lima DD, Lotz MK. Rho kinasedependent CCL20 induced by dynamic compression of human chondrocytes. Arthritis Rheum. 2008;15:2735–2742. doi: 10.1002/art.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton CT, Usmani SE, Mort JS, Beier F. Rho/ROCK and MEK/ERK activation by transforming growth factor-alpha induces articular cartilage degradation. Lab Invest. 2010;15:20–30. doi: 10.1038/labinvest.2009.111. [DOI] [PubMed] [Google Scholar]
- Novakofski K, Boehm A, Fortier L. The small GTPase Rho mediates articular chondrocyte phenotype and morphology in response to interleukin-1alpha and insulin-like growth factor-I. J Orthop Res. 2009;15:58–64. doi: 10.1002/jor.20717. [DOI] [PubMed] [Google Scholar]
- Liang J, Feng J, Wu WK, Xiao J, Wu Z, Han D, Zhu Y, Qiu G. Leptin-mediated cytoskeletal remodeling in chondrocytes occurs via the RhoA/ROCK pathway. J Orthop Res. 2011;15:369–374. doi: 10.1002/jor.21257. [DOI] [PubMed] [Google Scholar]
- Fortier LA, Miller BJ. Signaling through the small G-protein Cdc42 is involved in insulin-like growth factor-I resistance in aging articular chondrocytes. J Orthop Res. 2006;15:1765–1772. doi: 10.1002/jor.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Pala D, Kennedy L, McLean S, Rockel JS, Wang G, Leask A, Beier F. Rac1 signaling regulates CTGF/CCN2 gene expression via TGFbeta/Smad signaling in chondrocytes. Osteoarthritis Cartilage. 2009;15:406–413. doi: 10.1016/j.joca.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Nishida T, Kubota S, Kojima S, Kuboki T, Nakao K, Kushibiki T, Tabata Y, Takigawa M. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor) J Bone Miner Res. 2004;15:1308–1319. doi: 10.1359/JBMR.040322. [DOI] [PubMed] [Google Scholar]
- Long DL, Willey JS, Loeser RF. Rac1 is required for matrix metalloproteinase-13 production by chondrocytes in response to fibronectin fragments. Arthritis Rheum. 2013;15:1561–1568. doi: 10.1002/art.37922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000;15:144–147. doi: 10.1126/science.290.5489.144. [DOI] [PubMed] [Google Scholar]
- Faruqi TR, Gomez D, Bustelo XR, Bar-Sagi D, Reich NC. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci USA. 2001;15:9014–9019. doi: 10.1073/pnas.161281298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, Hatori T, Tsuchiya A, Kiyono M, Nosaka T, Nakajima H, Williams DA, Kitamura T. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol. 2006;15:937–946. doi: 10.1083/jcb.200604073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, Inoue M, Sato T, Kukimoto-Niino M, Shirouzu M, Yokoyama S, Kitamura T. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009;15:1796–1813. doi: 10.1128/MCB.01423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;15:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Atreya R, Atreya I, Neurath MF. Novel signal transduction pathways: analysis of STAT-3 and Rac-1 signaling in inflammatory bowel disease. Ann N Y Acad Sci. 2006;15:98–113. doi: 10.1196/annals.1326.001. [DOI] [PubMed] [Google Scholar]
- Razzouk S, Lieberherr M, Cournot G. Rac-GTPase, osteoclast cytoskeleton and bone resorption. Eur J Cell Biol. 1999;15:249–255. doi: 10.1016/S0171-9335(99)80058-2. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lebowitz D, Sun C, Thang H, Grynpas MD, Glogauer M. Identifying the relative contributions of Rac1 and Rac2 to osteoclastogenesis. J Bone Miner Res. 2008;15:260–270. doi: 10.1359/jbmr.071013. [DOI] [PubMed] [Google Scholar]
- Felson DT, Lawrence RC, Hochberg MC, McAlindon T, Dieppe PA, Minor MA, Blair SN, Berman BM, Fries JF, Weinberger M, Lorig KR, Jacobs JJ, Goldberg V. Osteoarthritis: new insights. Part 2: treatment approaches. Ann Intern Med. 2000;15:726–737. doi: 10.7326/0003-4819-133-9-200011070-00015. [DOI] [PubMed] [Google Scholar]
- Dervin GF, Stiell IG, Rody K, Grabowski J. Effect of arthroscopic debridement for osteoarthritis of the knee on health-related quality of life. J Bone Joint Surg Am. 2003;15:10–19. doi: 10.2106/00004623-200301000-00003. [DOI] [PubMed] [Google Scholar]
- LaPrade RF, Swiontkowski MF. New horizons in the treatment of osteoarthritis of the knee. JAMA. 1999;15:876–878. doi: 10.1001/jama.281.10.876. [DOI] [PubMed] [Google Scholar]
- Gillogly SD, Voight M, Blackburn T. Treatment of articular cartilage defects of the knee with autologous chondrocyte implantation. J Orthop Sports Phys Ther. 1998;15:241–251. doi: 10.2519/jospt.1998.28.4.241. [DOI] [PubMed] [Google Scholar]
- Lee J, Lee E, Kim HY, Son Y. Comparison of articular cartilage with costal cartilage in initial cell yield, degree of dedifferentiation during expansion and redifferentiation capacity. Biotechnol Appl Biochem. 2007;15:149–158. doi: 10.1042/BA20060233. [DOI] [PubMed] [Google Scholar]
- Shibuya M, Suzuki Y. [Treatment of cerebral vasospasm by a protein kinase inhibitor AT 877] No To Shinkei. 1993;15:819–824. [PubMed] [Google Scholar]
- Doggrell SA. Rho-kinase inhibitors show promise in pulmonary hypertension. Expert Opin Investig Drugs. 2005;15:1157–1159. doi: 10.1517/13543784.14.9.1157. [DOI] [PubMed] [Google Scholar]