

Abstract
Objective
Inflammatory responses are the driving force of atherosclerosis development. IκB kinase β (IKKβ), a central coordinator in inflammation through regulation of nuclear factor-κB, has been implicated in the pathogenesis of atherosclerosis. Macrophages play an essential role in the initiation and progression of atherosclerosis, yet the role of macrophage IKKβ in atherosclerosis remains elusive and controversial. This study aims to investigate the impact of IKKβ expression on macrophage functions and to assess the effect of myeloid-specific IKKβ deletion on atherosclerosis development.
Methods and Results
To explore the issue of macrophage IKKβ involvement of atherogenesis, we generated myeloid-specific IKKβ-deficient low-density lipoprotein receptor–deficient mice (IKKβΔMyeLDLR−/−). Deficiency of IKKβ in myeloid cells did not affect plasma lipid levels but significantly decreased diet-induced atherosclerotic lesion areas in the aortic root, brachiocephalic artery, and aortic arch of low-density lipoprotein receptor–deficient mice. Ablation of myeloid IKKβ attenuated macrophage inflammatory responses and decreased atherosclerotic lesional inflammation. Furthermore, deficiency of IKKβ decreased adhesion, migration, and lipid uptake in macrophages.
Conclusion
The present study demonstrates a pivotal role for myeloid IKKβ expression in atherosclerosis by modulating macrophage functions involved in atherogenesis. These results suggest that inhibiting nuclear factor-κB activation in macrophages may represent a feasible approach to combat atherosclerosis.
Keywords: atherosclerosis, inflammation, IκB kinase β, macrophage, nuclear factor-κB
Atherosclerosis is a chronic inflammatory disease.1,2 Many inflammatory pathways that contribute to the initiation and progression of atherosclerosis are regulated by the transcription factor nuclear factor (NF)-κB, a master regulator of innate and adaptive immune responses.3-5 The NF-κB family consists of 5 members: p65 (RelA), RelB, c-Rel, p100/p52, and p105/p50. NF-κB normally remains in the cytoplasm bound to the inhibitory protein IκB. Activating signals, such as proinflammatory cytokines, reactive oxygen species, and viral products, lead to activation of IκB kinase (IKK), which then phosphorylates IκB and promotes its degradation, allowing NF-κB to translocate to the nucleus where NF-κB can bind to the promoter regions of its target genes.3,5 IKK consists of 2 kinase subunits, IKKα and IKKβ, and a regulatory subunit NF-κB essential modulator or IKKγ.3,6 IKKβ is the predominant catalytic subunit of the IKK complex, which is required for canonical activation of NF-κB by inflammatory mediators.6
IKKβ-mediated NF-κB activation has been implicated in the pathogenesis of atherosclerosis. Activated NF-κB has been identified in human atherosclerotic plaques and was enhanced in unstable coronary plaques.7,8 NF-κB activation in human atherosclerosis was IKKβ-dependent and resulted in selective upregulation of major proinflammatory and prothrombotic mediators.8 Mapping atherosclerosis modifier loci on the apolipoprotein E–deficient (ApoE−/−) background had localized the negative regulator of NF-κB, A20 to a locus conferring sensitivity to atherosclerosis in ApoE−/− mice.9 Indeed, atherosclerosis was increased in A20 haploinsufficient and decreased in A20-overexpressing ApoE−/− mice.10 In addition, inhibiting NF-κB activity in endothelial cells (ECs) by the deletion of NF-κB essential modulator or expression of dominant-negative IκBα decreased vascular inflammation and atherosclerosis in ApoE−/− mice.11
Macrophages are the major inflammatory cells involved in the progression of atherosclerosis, yet the role of macrophage IKKβ in atherosclerosis remains elusive and controversial. NF-κB–mediated inflammatory functions by macrophages have generally been considered to be proatherogenic.2,4 interestingly, Kanters et al12 previously reported that low-density lipoprotein–deficient (LDLR−/−) mice transplanted with IKKβ-deficient macrophages had increased atherosclerosis. However, macrophage-specific inhibition of NF-κB activation by overexpressing transdominant, nondegradable forms of IκBα decreased foam cell formation,13 and myeloid-specific IκBα deletion promoted atherogenesis in LDLR−/− mice.14 Furthermore, LDLR−/− mice with hematopoietic cells deficient in the NF-κB subunit p50 also had decreased atherosclerotic lesion size.15 The inconsistent conclusions drawn from these studies suggest that further research is needed to define the role of macrophage IKKβ/NF-κB signaling in atherosclerosis.
To explore the issue of macrophage IKKβ involvement of atherogenesis, we generated myeloid-specific IKKβ-deficient LDLR−/− mice (IKKβΔMyeLDLR−/−). Here, we report that myeloid IKKβ deficiency protected LDLR−/− mice from diet-induced atherosclerosis, most likely because of modulated macrophage functions involved in atherogenesis.
Methods
Animals
Myeloid-specific IKKβ knockout (IKKβΔMye) mice were generated by crossing mice carrying loxP-flanked IKKβ alleles (IKKβF/F) with LysM-Cre transgenic mice,16 as previously described.17,18 IKKβF/F mice were kindly provided by Dr Michael Karin at the University of California, San Diego. The mice used in this study were backcrossed at least 7 additional generations onto the C57BL/6 background (>98% C57BL/6) using the marker-assisted Microsatellite Genotyping technique. To increase susceptibility to atherosclerotic lesion development, the IKKβΔMye mice were crossed to LDLR−/− mice (Jackson Laboratories) to generate IKKβΔMyeLDLR−/− and IKKβF/FLDLR−/− mice. All mice used in this study had IKKβF/FLDLR−/− double-mutant background, and IKKβΔMyeLDLR−/− mice carried heterozygous knock-in for LysM-Cre. For atherosclerosis study, 4-week-old experimental male IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− littermates were weaned and fed with a high-fat Western diet (21.2% fat, 0.2% cholesterol; TD 88137, Harlan Teklad) for 12 weeks until euthanization at 16 weeks of age. Body composition was measured by EchoMRI (Echo Medical System). All procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Blood Analysis
Plasma total cholesterol and triglyceride concentrations were determined enzymatically by a colorimetric method.19 Plasma from multiple mice (n=6) was pooled, and plasma lipoprotein cholesterol distributions were determined by fast-performance liquid chromatography.20
Atherosclerotic Lesion Analysis
Optimal cutting temperature compound–embedded hearts or brachiocephalic arteries were sectioned and stained with Oil-red-O, and atherosclerotic lesions were quantified as previously described.19 Aortas were harvested and fixed with 10% formalin, and en face analysis was performed as previously described.21
Macrophage Isolation and Function Assays
Macrophages were isolated as previously described.20,22 Bone marrow–derived macrophages (BMMs) were isolated from the femurs of mice and cultured in DMEM medium supplemented with 10 ng/mL recombinant mouse macrophage colony–stimulating factor (Invitrogen) for 7 days before the experiment. Elicited peritoneal macrophages were harvested from each genotype by peritoneal lavage with PBS 3 days after intraperitoneal injection of 1 mL of 3% thioglycollate. For adhesion assay, calcein acetoxymethyl–labeled peritoneal macrophages were incubated with primary porcine ECs, and attached cells were fixed and counted.23 Migration assays were performed using transwells with 8.0-μm pore polycarbonate membrane inserts (Corning).22 Macrophages were seeded on the transwell filters, and the lower chambers were filled with either control media or media containing 500 ng/mL lipopolysaccharide (LPS). After 5 hours, cells were removed from the upper surface of the insert by scraping using Q-Tips. The membranes were fixed with 1% glutaraldehyde (Sigma), stained with hematoxylin (Leica) and mounted on the slides using glycerol gelatin. Hematoxylin-stained cells were counted under the microscope. For lipid uptake assay, macrophages were incubated with DMEM containing 100 μg/mL of oxidized LDL (Biomedical Technologies) for 24 hours, followed by washing with PBS and staining with Oil-red-O/hematoxylin.20
RNA Isolation and Quantitative Real-Time PCR Analysis
Total RNA was isolated from mouse tissues or cells using TRIzol Reagent (Life Technologies), and quantitative real-time polymerase chain reaction (PCR) was performed using gene-specific primers and the SYBR green PCR kit (Life Technologies) as previously described.19,20 The primer sets used in this study are listed in Table I in the online-only Data Supplement.
Statistical Analysis
All data are expressed as mean±SD. Statistically significant differences between 2 groups were analyzed by 2-tailed Student t test for data normally distributed and by the Mann-Whitney test for data not normally distributed using Prism software. A P value <0.05 was considered significant.
Results
Generation of LDLR−/− Mice With Myeloid-Specific IKKβ Deficiency
To investigate the role of macrophage IKKβ in atherosclerosis, we generated IKKβΔMyeLDLR−/− mice by crossing IKKβΔMye (LysM-Cre/IKKβF/F) mice17 with LDLR−/− mice. All mice used in this study had IKKβF/FLDLR−/− double-mutant background, and IKKβΔMyeLDLR−/− mice also carried heterozygous knock-in for LysM-Cre. PCR analysis of genomic DNA indicates that the recombination was specific to the BMMs (Figure I in the online-only Data Supplement), consistent with previous reports that LysM-Cre–mediated gene excision almost exclusively occurs in macrophages and neutrophils.16,24 The mRNA levels of IKKβ were significantly decreased in peritoneal macrophages and BMMs but not in other major tissues of IKKβΔMyeLDLR−/− mice compared with IKKβF/FLDLR−/− mice (Figure 1A). Consistent with the quantitative real-time PCR results, protein levels IKKβ were also substantially reduced in both peritoneal macrophages and BMMs of IKKβΔMyeLDLR−/− mice (Figure 1B). The protein levels of IKKα were not affected in macrophages of IKKβΔMyeLDLR−/− mice, indicating the specific deletion of IKKβ (Figure 1B). To determine whether deficiency of IKKβ inhibits NF-κB activity in macrophages, BMMs isolated from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were treated with NF-κB stimulator, tumor necrosis factor-α (TNFα). TNFα-induced NF-κB subunit p65 translocation from cytoplasm to nucleus was inhibited in BMMs of IKKβΔMyeLDLR−/− mice (Figure 1C). Electrophoretic mobility shift assay confirmed that TNFα-induced DNA binding activity of NF-κB in macrophages was attenuated by IKKβ deficiency (Figure 1D). It is worth noting that NF-κB activity was not completely blocked in macrophages of IKKβΔMyeLDLR−/− mice, which may be because of the incomplete gene deletion induced by LysM-Cre/LoxP (Figure 1A and 1B). These results indicate that IKKβ expression levels were significantly decreased in macrophages of IKKβΔMyeLDLR−/− mice, and activation of NF-κB was inhibited in IKKβ-deficient macrophages.
Figure 1.

Generation of low-density lipoprotein–deficient (LDLR−/−) mice with myeloid-specific IκB kinase β (IKKβ) deficiency. A, IKKβ mRNA expression levels in major tissues and macrophages of IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were analyzed by quantitative real-time polymerase chain reaction (**P<0.01, ***P<0.001, n=5). B, Western blot analysis of IKKβ and IKKα protein levels in bone marrow–derived macrophages (BMMs) and peritoneal macrophages of IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice. C, BMMs isolated from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were stimulated with tumor necrosis factor-α (TNFα) (20 ng/mL) or vehicle for 30 minutes. Cells were then fixed and stained with anti-p65 primary antibody, followed by fluorescein-labeled secondary antibody (green). The nuclei were visualized with diamidino-2-phenylindole (blue). A representative figure from 3 independent experiments with similar results is shown (original magnification, ×400). D, Macrophages were stimulated with TNFα (20 ng/mL) or vehicle for 30 minutes. Nuclear proteins were extracted, and nuclear factor (NF)-κB binding activity was determined by electrophoretic mobility shift assay. WAT indicates white adipose tissue; BAT, brown adipose tissue.
Deficiency of IKKβ in Macrophages Does Not Affect Plasma Lipid Levels But Decreases Atherosclerosis in LDLR−/− Mice
To assess the impact of deficiency of macrophage IKKβ on atherosclerosis, 4-week-old male IKKβΔMyeLDLR−/− and IKKβF/FLDLR−/− littermates were fed a high-fat Western diet for 12 weeks. At 16 weeks of age, no significant differences in body weight, lean mass, fat mass, and fasting glucose levels were observed in IKKβΔMyeLDLR−/− and IKKβF/FLDLR−/− mice (Figure II in the online-only Data Supplement). Both IKKβΔMyeLDLR−/− and IKKβF/FLDLR−/− mice had diet-induced hyperlipidemia, but myeloid IKKβ deficiency did not affect plasma total cholesterol and triglyceride levels (Figure 2A and 2B). Furthermore, fast-performance liquid chromatography analysis showed that IKKβΔMyeLDLR−/− and IKKβF/FLDLR−/− mice had similar plasma cholesterol distribution pattern (Figure 2C).
Figure 2.
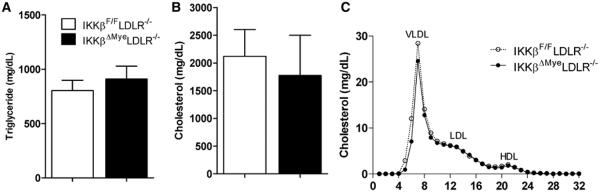
Myeloid-specific IκB kinase β (IKKβ) deficiency does not affect plasma lipid levels in low-density lipoprotein–deficient (LDLR−/−) mice. Four-week-old male IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− littermates were fed a Western diet for 12 weeks. The plasma levels of triglyceride (A) and cholesterol (B) were measured by standard method (n=9–10 per group), and plasma cholesterol distribution (C) was analyzed by fast-performance liquid chromatography. HDL indicates high-density lipoprotein; VLDL, very-low-density lipoprotein.
Quantification of cross-sectional lesion areas at the aortic root revealed that IKKβΔMyeLDLR−/− mice had 25% decreased lesion area (310 900 ± 28 310 μm2) compared with IKKβF/FLDLR−/− littermates (414 500 ± 30 940 μm2; Figure 3A). In addition to aorta, deficiency of myeloid IKKβ also inhibited atherosclerosis development in the brachiocephalic artery, an artery prone to developing advanced lesions. Brachiocephalic artery cross-sectional lesion areas were decreased 51% in IKKβΔMyeLDLR−/− mice (38 930 ± 6571 μm2) compared with IKKβF/FLDLR−/− mice (79 950 ± 15 320 μm2; Figure 3B). Furthermore, en face analysis of the aortic arch also showed 32% decreased lesions in IKKβΔMyeLDLR−/− mice (9.96 ± 0.62% in IKKβF/FLDLR−/− mice and 6.73 ± 1.01% in IKKβΔMyeLDLR−/− mice; Figure 3C). Taken together, deficiency of myeloid IKKβ significantly decreased diet-induced atherosclerosis in LDLR−/− mice.
Figure 3.

Deficiency of myeloid IκB kinase β (IKKβ) decreases atherosclerosis in low-density lipoprotein–deficient (LDLR−/−) mice. Quantitative analysis of the lesion area in the aortic root (A), brachiocephalic artery (B), and aortic arch (C) of IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice (n=11-15 per group, *P<0.05 and **P<0.01). Representative Oil-red-O–stained sections or en face aortic arches from each genotype are displayed next to the quantification data.
Deficiency of IKKβ Attenuates Macrophage Inflammatory Responses and Reduces Lesional Inflammation
In atherosclerosis, macrophages participate in inflammatory responses that contribute to expansion of the subendothelial layer and lesion formation and progression.2 To determine the role of IKKβ in the regulation of macrophage inflammatory responses, BMMs were isolated from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice and treated with endotoxin LPS. Gene expression analyses showed that the ability of LPS to induce expression of mRNAs encoding TNFα, monocyte chemotactic protein (MCP)-1, interleukin (IL)-1α, IL-1β, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 was attenuated in macrophages of IKKβΔMyeLDLR−/− mice (Figure 4A). These results suggest that ablation of IKKβ reduces NF-κB–regulated proinflammatory gene expression in macrophages. Indeed, gene profiling of peritoneal macrophages isolated from Western diet–fed IKKβΔMyeLDLR−/− mice showed downregulation of several proinflammatory molecules, including TNFα, IL-1α, IL-1β, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 (Figure 4B). Interestingly, the expression levels of IL-10, an anti-inflammatory cytokine, were also decreased in peritoneal macrophages of IKKβΔMyeLDLR−/− mice. We also noted that the expression levels of another NF-κB target gene, CCR7,25 were significantly decreased by IKKβ deficiency. Consistent with in vitro gene expression analyses, immunofluorescence staining showed that the expression levels of several key inflammatory cytokines, including MCP-1, TNFα, and IL-1β, were decreased in the atherosclerotic lesions of IKKβΔMyeLDLR−/− mice (Figure 4C). Thus, deficiency of IKKβ attenuated macrophage inflammatory responses and reduced lesional inflammation.
Figure 4.

Deficiency of IκB kinase β (IKKβ) attenuates macrophage inflammatory responses and reduces lesional inflammation. A, Bone marrow–derived macrophages isolated from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were treated with lipopolysaccharide (LPS) (500 ng/mL) or vehicle control for 4 hours. Expression of proinflammatory cytokines was analyzed by quantitative real-time polymerase chain reaction (QPCR; *P<0.05, **P<0.01, n=3). B, Gene profile by QPCR of peritoneal macrophages from Western diet (WD)–fed IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice (*P<0.05, **P<0.01, n=4–6 per group). C, Sections of atherosclerotic lesions in the aortic root of WD-fed IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were stained with antibodies against mouse monocyte chemotactic protein (MCP)-1, tumor necrosis factor-α (TNFα), or interleukin (IL)-1β followed by fluorescein-labeled secondary antibody (red). The nuclei were stained with diamidino-2-phenylindole (DAPI; blue). A representative figure from 3 mice per group and the similar result is shown (original magnification, ×100). ICAM indicates intercellular adhesion molecule; LDLR−/−, low-density lipoprotein receptor–deficient mice.
Ablation of IKKβ Modulates Macrophage Functions Related to Atherosclerosis Development
One of the earliest events in atherogenesis is the entry of monocytes, the precursors of macrophages, into the arterial wall. Several NF-κB–regulated chemokines and adhesion molecules were downregulated in macrophages of IKKβΔMyeLDLR−/− mice (Figure 4). We next investigated the effects of IKKβ deficiency on macrophage adhesion and migration properties. Incubation of freshly isolated macrophages with primary ECs showed that ablation of IKKβ significantly decreased adhesion of macrophage to ECs (Figure 5A). We also examined the effects of IKKβ deficiency on macrophage migration by transwell assay. As shown in Figure 5B, IKKβ deficiency significantly decreased the migration of peritoneal macrophages. Furthermore, LPS stimulated migration of macrophages isolated from IKKβF/FLDLR−/− mice, and this induction was abolished in those from IKKβΔMyeLDLR−/− mice. Therefore, ablation of IKKβ decreases the macrophage adhesion and migration abilities.
Figure 5.

Ablation of IκB kinase β (IKKβ) modulates macrophage function related to atherosclerosis development. A, Peritoneal macrophages isolated from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were labeled with calcein acetoxymethyl (AM) and cultured with primary porcine endothelial cell monolayer for 30 minutes. Adhered cells were then counted under a fluorescence microscope using a ×10 objective, and quantification of 3 independent experiments is presented in the bottom panel (*P<0.05). B, Peritoneal macrophages were seeded on the transwell filters and stimulated with control media or media containing 500ng/mL LPS (lower chambers) for 6 hours. Cells that migrated to the underside of transwell were stained with hematoxylin and counted under the microscope (*P<0.05, n=3). C, Fresh isolated peritoneal macrophages from IKKβF/FLDLR−/− and IKKβΔMyeLDLR−/− mice were incubated with oxidized LDL (oxLDL; 100 μg/mL) for 24 hours and stained with Oil-red-O and hematoxylin. D, Foam cell quantification from peritoneal macrophages in studies described in C (*P<0.05, *P<0.01, ***P<0.001, n=3). LDLR−/− indicates low-density lipoprotein receptor–deficient mice.
Accumulation of lipid-loaded macrophages is a hallmark of atherosclerosis.26 Interestingly, oxidized LDL uptake and foam cell formation were substantially reduced in macrophages of IKKβΔMyeLDLR−/− mice compared with that of IKKβF/FLDLR−/− mice (Figure 5C and 5D), which may be attributed to the decreased type A scavenger receptor mRNA levels in IKKβ-deficient macrophages (Figure III in the online-only Data Supplement). Taken together, ablation of IKKβ decreased adhesion, migration, and lipid uptake in macrophages, which may coordinately contribute to decreased atherosclerosis in IKKβΔMyeLDLR−/− mice.
Discussion
Although the role of IKKβ/NF-κB pathway in inflammation and immune response has been extensively studied in the past 2 decades, the contribution of cell-type–specific NF-κB activation to atherosclerosis development is still poorly understood. In this study, we generated myeloid-specific IKKβ-deficient LDLR−/− mice to investigate the impact of macrophage IKKβ expression on atherosclerosis development. Deficiency of IKKβ in myeloid cells did not affect diet-induced hyperlipidemia but significantly decreased atherosclerosis in LDLR−/− mice (Figure 3). IKKβ deficiency decreased macrophage inflammatory responses and reduced LPS-induced proinflammatory gene expression (Figure 4). Many NF-κB–regulated proinflammatory molecules were downregulated in IKKβΔMyeLDLR−/− macrophages. Furthermore, the protein levels of several proinflammatory cytokines, including MCP-1, TNFα, and IL-1β, were decreased in the atherosclerotic lesions of IKKβΔMyeLDLR−/− mice. Those molecules play important roles in atherosclerosis initiation and progression. For example, monocytes are attracted by MCP-1 to lesion-prone areas, which is considered a critical step in atherosclerosis initiation.1,26 Mice deficient in MCP-1 have significantly reduced atherosclerotic lesions.27 We also found that IKKβ-deficient macrophages adhered less robustly to ECs and showed impaired migratory responses compared with control macrophages (Figure 5). Therefore, the attenuated macrophage inflammatory responses and impaired adhesion and migration properties may coordinately contribute to the decreased atherosclerosis in IKKβΔMyeLDLR−/− mice.
In addition to decreased inflammatory responses, lipid uptake and foam cell formation were also substantially reduced in the macrophages of IKKβΔMyeLDLR−/− mice compared with that of IKKβF/FLDLR−/− mice. The decreased lipid uptake in IKKβ-deficient macrophages may be attributed to the reduced expression levels of type A scavenger receptor that plays an important role in macrophage lipid uptake and foam cell formation.28,29 Our results are consistent with a previous report that inhibition of NF-κB activity in macrophages reduces foam cell formation.13 Deficiency of NF-κB subunit p50 was also reported to decrease type A scavenger receptor expression and reduce lipid uptake in activated macrophages.15 Whereas the in vivo effects of type A scavenger receptor deficiency on atherosclerosis development remain unclear and controversial,28-30 the decreased atherosclerosis in IKKβΔMyeLDLR−/− was very likely caused by the combined effects of decreased inflammation, adhesion, migration, and lipid uptake in macrophages.
IKKβ-dependent NF-κB activation has been implicated in vascular pathologies, and many studies have implicated that inhibition of NF-κB activation in macrophages have antiatherogenic effects.2,13,14 However, Kanters et al12 previously reported that bone marrow transplantation of macrophages lacking IKKβ increased atherosclerotic lesion sizes in LDLR−/− mice. The discrepancy between our atherosclerosis results and those reported by Kanters et al12 may be attributed to differences in experimental design. Although the same LysM-Cre transgenic mice were used by both groups, Kanter et al12 used a different IKKβF/F mouse model to generate IKKβΔMye mice. In their study, 10-week-old irradiated LDLR−/− mice were transplanted with control or IKKβ-deficient macrophages from IKKβF/F and IKKβΔMye mice, respectively. After 4 weeks of recovery and 10 weeks on a high-fat diet (16% fat, 0.15% cholesterol), mice were euthanized, and the authors found that those 24-week-old LDLR−/− mice transplanted with IKKβ-deficient macrophages had increased atherosclerosis in the aortic root,12 the only location they measured atherosclerotic lesion size. The authors found that IKKβ deficiency inhibited LPS-induced production of IL-10, which has anti-inflammatory and antiatherogenic effects in mice. However, IKKβ-deficient macrophages also exhibited a strong reduction in TNFα production.12 Therefore, the definitive mechanism for increased lesion area in LDLR−/− mice transplanted with IKKβ-deficient macrophages is unclear.
In this study, we used a different approach to explore the issue of macrophage IKKβ involvement of atherogenesis. Instead of bone marrow transplantation, we brought the well-characterized IKKβΔMye mice17,18 onto a LDLR−/− background. Four-week-old IKKβΔMyeLDLR−/− mice and their control littermates were fed a standard Western diet (21.2% fat, 0.2% cholesterol) for 12 weeks. We found that IKKβΔMyeLDLR−/− mice developed smaller lesions in 3 different locations, including aortic root, brachiocephalic artery, and aortic arch, compared with their control littermates. Consistent with previous reports,17,18 IKKβ deficiency decreased macrophage inflammatory responses. Despite the decreased IL-10 expression levels, the expression levels of many NF-κB–regulated proinflammatory molecules were downregulated in IKKβΔMyeLDLR−/− macrophages. Furthermore, IKKβΔMyeLDLR−/− macrophages had impaired adhesion, migration, and lipid uptake properties, which may also contribute to decreased atherosclerosis. Whereas bone marrow transplantation has been used extensively to study macrophage function in atherosclerosis, studies have found that bone marrow cells retain the potential to differentiate into a variety of nonhematopoietic cell lineages, including epithelial cells, hepatocytes, osteoblasts, chondrocytes, adipocytes, and perivascular cells.31-34 It is plausible that those progenitor cells in bone marrow of animals on different background might affect the atherosclerosis development in LDLR−/− mice. Therefore, the methodological differences may account for the discrepant atherosclerosis outcomes between our and Kanters et al’s studies. In addition, the different diets (16% fat versus 21.2% fat), duration of high-fat feeding (10 weeks versus 12 weeks), and ages of animals (24 weeks old versus 16 weeks old) used in these studies may also contribute to the difference in atherosclerotic lesion development.
The role of IKKβ in the regulation of proinflammatory genes has been well documented. Interestingly, IKKβ-mediated NF-κB activation can also transcriptionally regulate anti-inflammatory cytokines such as IL-10 (Figure 4B) and negatively regulate IL-1β secretion in macrophages.35 For example, Greten et al35 used a high-dose LPS (30 mg/kg) to treat mice and found that mice with targeted IKKβ deletion in myeloid cells were more susceptible to endotoxin-induced shock. Consistent with our results (Figure 4A), LPS-mediated IL-1β mRNA expression levels were inhibited in IKKβ-deficient macrophages.35 However, NF-κB also controls genes such as PAI-2 and Bcl-XL, whose products inhibit caspase-1–mediated pro–IL-1β process in macrophages. Thus, NF-κB plays a dual role in the regulation of IL-1β secretion (positively regulate IL-1β mRNA transcription and negatively regulate pro–IL-1β process), and prolonged inhibition of IKKβ may enhance IL-1β secretion on endotoxin challenge. It is currently not clear whether IKKβ can negatively regulate IL-1β secretion in chronic inflammatory diseases such as atherosclerosis and arthritis. Our study showed that deficiency of IKKβ reduced protein levels of IL-1β and other key inflammatory mediators such as MCP-1 in atherosclerotic lesions of IKKβΔMyeLDLR−/− mice (Figure 4C). Future studies are required to further understand the role of IKKβ in the regulation of IL-1β secretion under chronic inflammatory conditions.
Macrophage migration plays an essential role in atherosclerotic lesion initiation and progression. The down-regulation of NF-κB–regulated chemokines and adhesion molecules such as CCR7, MCP-1, and intercellular adhesion molecule-1 may contribute to decreased adhesion and migration of IKKβ-deficient macrophages (Figure 5). Recent studies also demonstrated the possibility to decrease atherosclerosis by increasing macrophage emigration from atherosclerotic lesions.36,37 CCR7 has been identified to be an important chemokine for promoting the egress of macrophage from the arterial wall during atherosclerosis regression.36,37 It is possible that the decreased CCR7 expression in IKKβ-deficient macrophages may affect atherosclerosis regression. However, deficiency of CCR7 have been shown to decrease atherosclerotic plaque development in ApoE−/− mice38 and lead to suppressed monocyte recruitment and increased levels of circulating leukocytes in ApoE−/− mice during atherosclerosis regression.39 Very recently, Gils et al40 reported that other factors such as netrin-1 also regulate macrophage emigration, and targeted deletion of netrin-1 in macrophages promoted the emigration of macrophages from plaques and decreased atherosclerosis. Thus, the mechanisms that regulate atherosclerosis regression are not well understood, and it would be interesting to investigate the potential role of IKKβ in regulating macrophage emigration and atherosclerosis regression in the future.
In summary, we demonstrate that myeloid-specific IKKβ deficiency decreases atherosclerosis in LDLR−/− mice. IKKβ plays an important role in modulating macrophage functions related to atherosclerosis. Ablation of IKKβ attenuated macrophage inflammatory responses and decreased macrophage adhesion, migration, and lipid uptake. These findings suggest that inhibition of NF-κB activity in myeloid may represent a feasible therapeutic strategy to combat atherosclerosis.
Supplementary Material
Acknowledgments
We thank Dr Jan Breslow at Rockefeller University for invaluable advices, discussions, and technical support; Dr Michael Karin at University California, San Diego for IKKβ flox mice; and Dr Alan Daugherty and Deborah Howatt for fast-performance liquid chromatography analysis.
Sources of Funding
This work was supported, in part, by National Institutes of Health (NIH) grants P20RR021954, P20GM103527, and P30HL101300. R.N.H. was supported by NIH training grant 5T32HL072743. C.Z. was supported by American Heart Association grant 09SDG2150176.
Footnotes
This manuscript was sent to Philip S. Tsao, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures
None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. CellMetab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou C, Tabb MM, Nelson EL, Grün F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, Blumberg B. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–36. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 7.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci USA. 2004;101:5634–5639. doi: 10.1073/pnas.0401060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Idel S, Dansky HM, Breslow JL. A20, a regulator of NFkappaB, maps to an atherosclerosis locus and differs between parental sensitive C57BL/6J and resistant FVB/N strains. Proc Natl Acad Sci USA. 2003;100:14235–14240. doi: 10.1073/pnas.1835672100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci USA. 2007;104:18601–18606. doi: 10.1073/pnas.0709011104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJ, Havekes LM, Pannekoek H. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis. 2007;192:283–290. doi: 10.1016/j.atherosclerosis.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Goossens P, Vergouwe MN, Gijbels MJ, Curfs DM, van Woezik JH, Hoeksema MA, Xanthoulea S, Leenen PJ, Rupec RA, Hofker MH, de Winther MP. Myeloid IκBa deficiency promotes atherogenesis by enhancing leukocyte recruitment to the plaques. PLoS ONE. 2011;6:e22327. doi: 10.1371/journal.pone.0022327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanters E, Gijbels MJ, van der Made I, Vergouwe MN, Heeringa P, Kraal G, Hofker MH, de Winther MP. Hematopoietic NF-kappaB1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood. 2004;103:934–940. doi: 10.1182/blood-2003-05-1450. [DOI] [PubMed] [Google Scholar]
- 16.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 17.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 19.Zhou C, Pridgen B, King N, Xu J, Breslow JL. Hyperglycemic Ins2AkitaLdlr−/− mice show severely elevated lipid levels and increased atherosclerosis: a model of type 1 diabetic macrovascular disease. J Lipid Res. 2011;52:1483–1493. doi: 10.1194/jlr.M014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sui Y, Xu J, Rios-Pilier J, Zhou C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J Lipid Res. 2011;52:1652–1659. doi: 10.1194/jlr.M017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. doi: 10.1385/1-59259-340-2:293. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLoS ONE. 2011;6:e26656. doi: 10.1371/journal.pone.0026656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han SG, Eum SY, Toborek M, Smart E, Hennig B. Polychlorinated biphenyl-induced VCAM-1 expression is attenuated in aortic endothelial cells isolated from caveolin-1 deficient mice. Toxicol Appl Pharmacol. 2010;246:74–82. doi: 10.1016/j.taap.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, Mohammad L, Henstridge DC, Febbraio MA, Hewitt SC, Korach KS, Bensinger SJ, Hevener AL. Myeloid-specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci USA. 2011;108:16457–16462. doi: 10.1073/pnas.1104533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mburu YK, Egloff AM, Walker WH, Wang L, Seethala RR, van Waes C, Ferris RL. Chemokine receptor 7 (CCR7) gene expression is regulated by NF-κB and activator protein 1 (AP1) in metastatic squamous cell carcinoma of head and neck (SCCHN) J Biol Chem. 2012;287:3581–3590. doi: 10.1074/jbc.M111.294876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 29.Babaev VR, Gleaves LA, Carter KJ, Suzuki H, Kodama T, Fazio S, Linton MF. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler Thromb Vasc Biol. 2000;20:2593–2599. doi: 10.1161/01.atv.20.12.2593. [DOI] [PubMed] [Google Scholar]
- 30.Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, Alvarez-Leite JI, de Winther MP, Tabas I, Freeman MW. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alison MR, Poulsom R, Jeffery R, Dhillon AP, Quaglia A, Jacob J, Novelli M, Prentice G, Williamson J, Wright NA. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406:257. doi: 10.1038/35018642. [DOI] [PubMed] [Google Scholar]
- 32.Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 34.Lagasse E, Connors H, Al-Dhalimy M, Reitsma M, Dohse M, Osborne L, Wang X, Finegold M, Weissman IL, Grompe M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–1234. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 35.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feig JE, Pineda-Torra I, Sanson M, et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415–4424. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luchtefeld M, Grothusen C, Gagalick A, Jagavelu K, Schuett H, Tietge UJ, Pabst O, Grote K, Drexler H, Förster R, Schieffer B. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation. 2010;122:1621–1628. doi: 10.1161/CIRCULATIONAHA.110.956730. [DOI] [PubMed] [Google Scholar]
- 39.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Gils JM, Derby MC, Fernandes LR, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.