

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease which is currently untreatable. Inflammation plays a major role in the pathogenesis of motor neuron death in ALS. Pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and Fas ligand (FasL) are amongst the most important mediators of neuro-inflammation. We have previously demonstrated that elevation of these proinflammatory cytokines occurs in both ALS transgenic mice and in human ALS postmortem spinal cord tissues. Lenalidomide is a potent immunomodulatory agent, with the ability to down-regulate proinflammatory cytokines and up-regulate anti-inflammatory cytokines. We previously reported the neuroprotective effects of lenalidomide, when treatment was started 2 months prior to onset of disease in the G93A SOD1 transgenic mouse model of ALS. Since in ALS patients, treatment can only begin after the appearance of symptoms, we sought to determine the efficacy of lenalidomide administration starting at symptom onset in the G93A SOD1 mice. We found that lenalidomide treatment extended the survival interval from the age of onset by 18.3 days (~45%). Additionally, lenalidomide treatment improved rotarod performance, reduced weight loss, and attenuated neuronal cell death in the lumbar spinal cord. Qualitative histological analysis showed that lenalidomide treatment modestly reduced the expression of the proinflammatory cytokines Fas Ligand, IL-1β, TNF-α and CD40 ligand. RNA protection Assay (RPA) on a pre-selected panel of cytokines showed that proinflammatory cytokines were reduced and anti-inflammatory cytokines were up-regulated. These data encourage further clinical evaluation of lenalidomide as therapeutic strategy to block or slow disease progression in human ALS patients.
Keywords: G93A, SOD1, Lenalidomide, TNF-α, FasL, Inflammation
Introduction
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease with a prevalence of 1–2 per 100,000. The exact mechanismof ALS in causing selective degeneration of motor neurons is not fully understood, however, several hypotheses are currently under investigation. These include neuroinflammation, oxidative damage, mitochondrial dysfunction, glutamate toxicity and SOD1 aggregation (Bruijn et al., 2004; Kiaei et al., 2005b). Ninety percent of ALS cases are sporadic, 10% are familial with a mostly autosomal-dominant mode of inheritance. A major breakthrough in ALS research was the discovery that 20% of familial cases are caused by gain-of-function-mutations in the gene coding for the antioxidant enzyme superoxide dismutase 1 (SOD1) (Rosen et al., 1993). This led to the development of ALS mouse models which closely mimic the disease phenotype and neuropathological characteristics (Gurney et al., 1994).
Microglial activation and secretion of proinflammatory cytokines appears to play a major role in chronic neurodegeneration in both familial and sporadic ALS (Moreira et al., 1993; Hall et al., 1998; Hensley et al., 2002; Wilms et al., 2003; Kiaei et al., 2005b, 2006; Moreau et al., 2005). Studies in ALS transgenic mice in which mutant SOD1 gene could be deleted from specific cell types, indicate that mutant SOD1 in microglia is particularly important for disease progression (Boillee et al., 2006). A recent study on the role of microglia found that expansion of the microglial cell population is mainly attributable to proliferation of myeloid precursor cells and found that elimination of this group of microglia had no effect on motor neuron degeneration (Gowing et al., 2008). Identification of key events that link microglial activation and the secretion of proinflammatory cytokines may provide new insights into the role of microglial activation and proinflammatory cytokines in the patho- genesis of ALS. We have previously showed that immunomodulatory agents such as thalidomide and lenalidomide are neuroprotective in the G93A SOD1 mouse model of ALS, when treatment initiated at 30 days of age. Lenalidomide treatment resulted in an attenuation of spinal cord neuronal cell death, enhanced motor performance and increased survival by 17% (Kiaei et al., 2006). As the majority of ALS patients are sporadic, and treatment can only begin at the time of diagnosis, a treatment strategy that slows the progression of ALS starting at the onset of symptoms is highly desirable. In this study, we therefore tested the efficacy of lenalidomide in slowing the progression of ALS when treatment begins at the onset of symptom in the G93A SOD1 mice.
Results
Two cohorts of the G93A mice were monitored weekly until they reached the age of 60 days, and then were subjected to rotarod and weight measurements twice per week. The age of disease onset was determined as described in the Materials and methods. We found that treatment with lenalidomide starting at symptom onset reduced neuronal cell death, increased motor performance, down-regulated proinflammatory cytokines and delayed the age of death in the G93A SOD1 mice as compared with vehicle-treated littermate controls.
Motor performance
Lenalidomide treatment from the onset of ALS in G93A mice, significantly improved motor performance as compared to control G93A mice, ANOVA repeated measures, P<0.0001 (Fig. 1A). Weight loss was significantly reduced in the G93A mice treated with lenalidomide as compared to controls, P<0.0001 (Fig. 1B).
Fig. 1.

The effect of lenalidomide treatment from onset of ALS on motor performance and weights in the G93A SOD1 mice. (A) Lenalidomide-treated mice stayed on the rotarod significantly longer as compared to littermate G93A mice fed on control food. (B) Lenalidomide-treated mice had significantly less weight loss as compared to littermate G93A mice fed on control food. The onset of ALS and the start of lenalidomide treatment, indicated by black arrows. Values are mean+SD. ▲ Lenalidomide-treated G93A mice (n=18). ■ Littermate G93A control mice (n=18) were fed on control food (Chaw without lenalidomide preparation made by Dyets, Inc.).
Neuronal cell count by Nissl staining
Lenalidomide treatment significantly attenuated neuronal loss in the lumbar spinal cord of the G93A mice. The number of surviving neurons at 110 days of age was significantly higher in the G93A mice treated with lenalidomide from the age of onset (~day 85 after birth) as compared with the G93A mice fed control food (P<0.01) (Fig. 2).
Fig. 2.

The effect of lenalidomide treatment on Nissl-stained neuronal cell counts in G93A SOD1 transgenic mice at 110 days of age. Lenalidomide treatment started from the age of onset of ALS, and it significantly attenuated the neuronal cell loss as compared to G93A SOD1 mice controls (***P<0.001) by ANOVA and Student–Newman–Keuls tests. Values are mean±SEM. *P<0.05.
The neuronal cell counts at 110 days of age were 60% lower in untreated G93A SOD1 mice than wild-type controls. The G93A SOD1 mice treated with lenalidomide had 33% more neurons in the spinal cords as compared to vehicle-treated G93A SOD1 littermates (Fig. 2). Lenalidomide treatment was not able to protect 100% of neurons in the spinal cord as there is a significant reduction in Nissl counts in spinal cord sections of G93A mice treated with lenalidomide vs. wildtype mice.
Immunohistochemical analysis for pro-inflammatory cytokines
We investigated the effect of lenalidomide on the expression of pro-inflammatory cytokines, TNF-α, FasL, IL-1β and CD40 (a marker of glial activation) by qualitative immunohistochemistry. TNF-α and FasL immunoreactivity increased in the G93A mice treated with control food, is consistent with our previous findings (Kiaei et al., 2006). Qualitative analysis of spinal cord sections of lenalidomidetreated G93A mice spinal cord sections showed a modest reduction in immunoreactivity for TNF-α and FasL. IL-1β and CD40 immunoreactivities were also modestly reduced (Fig. 3). We observed that the expression of IL-1β appeared to be mainly in glial cells (Fig. 3). The tissue samples for this analysis obtained from 110-day-old mice, in which 50% of motor neurons are lost in the lumbar spinal cord of untreated G93A mice, which is best time point to determine changes due to treatment. Since treated G93A mice die at delayed age, it is expected that inflammatory markers, if measured at later time points, would gradually increase and may reach similar levels to those in untreated G93A mice.
Fig. 3.
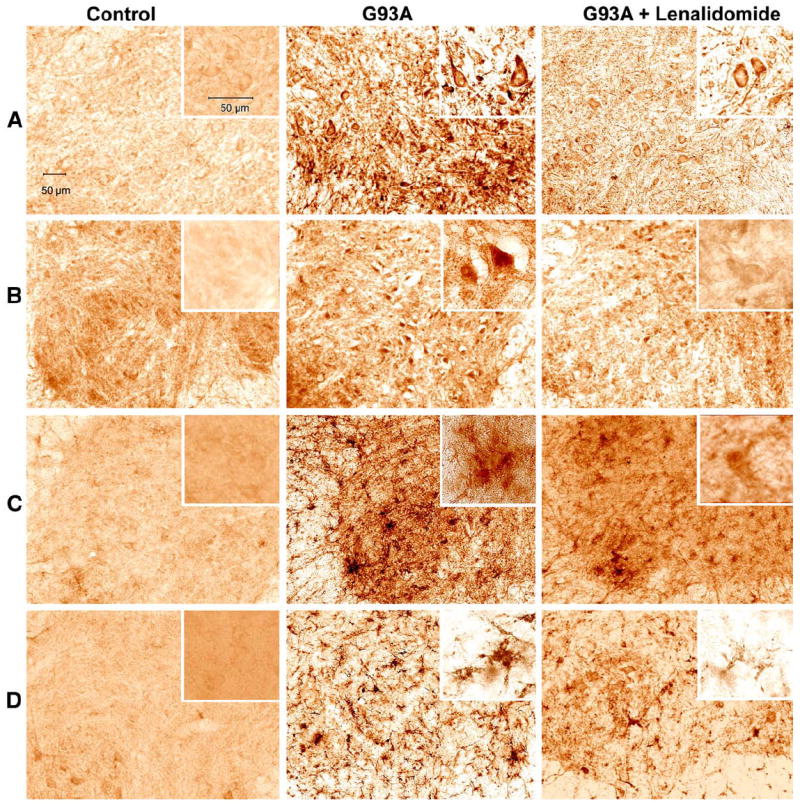
TNF-α, FasL, IL-1β and CD40 immunoreactivities in the G93A SOD1 controls and lenalidomide-treated mice. TNF-α, FasL, IL-1β and CD40 immunoreactivities were examined in the spinal cords of transgenic G93A SOD1 and lenalidomide-treated mice at 110 days old. All the panels are immunostaining in the spinal cord anterior horn sections. Lenalidomide treatment modestly reduced staining of TNF-α (A), FasL (B), IL-1β (C) and CD40 (D). Each panel has a high magnification shown in inset. Scale bar of 50 μm shown for large panels, and insets.
Pro-inflammatory cytokine levels by multiprobe RPAs
Total RNA isolated from the spinal cords of treated and control G93A mice sacrificed at 110 days of age were subjected to multiprobe RNA protection assay (RPA). RPA analysis showed that lenalidomide treatment from the age of onset of disease down-regulated proinflammatory cytokines like TNF-α from 358% to 261%, Fas associated protein (FAP) from 236% to 131%, Fas associated factor (FAF) from 264% to 105% (Table 1A). The treatment up-regulated anti-inflammatory cytokines; IL-1RA from 452% to 632%, and TGF-β3 from 122% to 139% (Table 1B). These percentages are tabulated from densitometry arbitrary values obtained from RNA band density in the RPA gel. The values obtained from non-transgenic control RNA bands were used as 100%, and the percentages of RNA bands in G93A mice and G93A plus lenalidomide were calculated as percentage of controls.
Table 1.
(A and B) RNA protection analysis by RNA protection assay (RPA) of spinal cord samples from lenalidomide-treated and control G93A SOD1 mice showing lenalidomide modulates cytokines in the spinal cord of G93A SOD1 mice.
| Cytokines | Non-Tg controls (%) n =6 | G93A Control (%) n =6 | G93A + lenalidomide (%) n=6 | Percentage of change in G93A control vs. G93A +Lenalidomide |
|---|---|---|---|---|
| (A) List of reduced cytokines | ||||
| IL-1α | 100.00 | 409 | 351 | −14% P< 0.04 |
| IL-1β | 100.00 | 379 | 330 | −13% n.s. |
| IL-18 | 100.00 | 169 | 163 | −3% n.s. |
| MIF | 100.00 | 119 | 113 | −5% n.s. |
| TNF-α | 100.00 | 358 | 261 | −25% P< 0.0001 |
| IFNβ | 100.00 | 130 | 114 | −12% n.s. |
| TGFβ1 | 100.00 | 310 | 273 | −12% P< 0.03 |
| TGFβ2 | 100.00 | 163 | 149 | −8% n.s. |
| FADD | 100.00 | 142 | 139 | −2% n.s. |
| FAP | 100.00 | 236 | 131 | −44% P< 0.0001 |
| FAF | 100.00 | 264 | 105 | −60% P< 0.0001 |
| (B) List of increased cytokines | ||||
| IL-1RA | 100.00 | 453 | 632 | 39% P< 0.0001 |
| TGFβ3 | 100.00 | 122 | 140 | 15% n.s. |
| FAS | 100.00 | 236 | 275 | 16% n.s. |
| TRAIL | 100.00 | 135 | 161 | 19% n.s. |
| TNFRp55 | 100.00 | 264 | 295 | 12% n.s. |
| TRADD | 100.00 | 100 | 108 | 8% n.s. |
| RIP | 100.00 | 148 | 165 | 11% n.s. |
Numbers are expressed as percent of change in treated vs. untreated G93A mice. Not significant (n.s.).
The effect of lenalidomide on SOD1 expression
Total protein was extracted from spinal cord of treated and control G93A mice sacrificed at 110 days and subjected to Western blot analysis. The total SOD1 protein expression level in both group were probed using anti-SOD1 antibody and bands were quantified and normalized against GAPDH protein. We found that lenalidomide treatment had no effect on SOD1 expression in the spinal cord (data not shown).
Progression of disease and survival
Lenalidomide treatment from the age of onset of ALS phenotypes significantly extended the survival of G93A mice. The time interval from symptom onset to death was significantly longer for G93A mice treated with lenalidomide (59.1±5.8 days vs. 40.8±6.7 days, the difference of 18.3 days or ~45%) as compared to controls, P<0.001 (Fig. 4). The overall survival of lenalidomide-treated G93A mice increased from a mean survival of 127.7±5.1 days (n=18) in controls to 142.7±5.4 days (n=18) P<0.001 (a difference of 15 days or 12%) as compared to vehicle-treated G93A mice (Fig. 4).
Fig. 4.
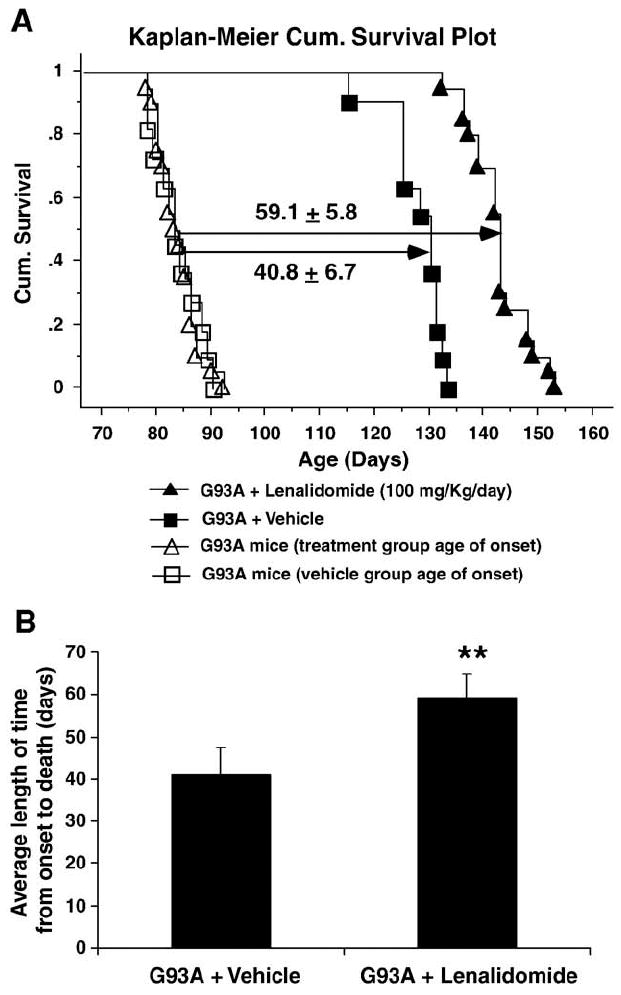
The effect of lenalidomide treatment from onset of ALS on survival in G93A SOD1 mice. (A) The cumulative (Cum.) probability of survival for G93A mice treated from disease onset with lenalidomide (100 mg/kg/day) shows significantly higher (**P<0.01) Mental–Cox, logrank. The duration from age of onset to death is significantly extended by 44.3% (P<0.001). Values are mean±SD. (B) The average length of survival time shown in histogram. The G93A SOD1 mice treated with lenalidomide (n=18) survived significantly longer from the age of onset as compared to untreated G93A littermate controls (n=18). **P<0.01.
Discussion
Several reports have demonstrated that pro-inflammatory cytokines have a toxic role in the pathogenesis of ALS (Almer et al., 2001; Hensley et al., 2003; Kiaei et al., 2006; Wu et al., 2006). Inflammatory cascades contribute to motor neuron death in the spinal cord in the G93A SOD1 transgenic mice, as well as in human ALS patients. In this study, we extended our previous findings that thalidomide and lenalidomide have neuroprotective effects in G93A mice (Kiaei et al., 2006). Now we report on the therapeutic efficacy of lenalidomide when administered from the time of symptom onset in the G93A SOD1 mice. We demonstrate that lenalidomide delays the age of death, mainly by slowing disease progression. We measured the survival interval from symptom onset to time of death, for the G93A SOD1 mice treated with lenalidomide as compared to vehicle treatment, and noted a significantly longer survival interval from onset to death (increased by 44%) in the drug-treated animals. This indicates that lenalidomide has, at least in this mouse model, the potential to double the survival time from onset of symptoms of ALS to death. In our previous study we showed that thalidomide and lenalidomide extended survival by 16% and 18%, respectively. Retrospective analysis of the onset and survival data showed that lenalidomide treatment increased survival duration from onset to death by 24 days (~60%) and thalidomide increased the survival duration by 52%. These results were achieved when treatment began about 60 days prior to symptom onset of ALS in which we observed a delay in the age of death and a slowing of the progression of ALS, perhaps via reduction in proinflammatory cytokines and an increase in anti-inflammatory cytokines (Kiaei et al., 2006). Therefore, lealidomide’s ability to slow the progression of ALS is a significant feature that may be valuable for exploration in human ALS clinical trials. Both of our studies have consistently demonstrated that lenalidomide treatment modulated proinflammatory cytokines in the spinal cord. We found that TNF-α, Fas L and IL-1β immunoreactivity were all modestly reduced in this study, although the data are more convincing when treatment was started 60 days prior to symptom onset. The modest changes in pro-inflammatory and antiinflammatory cytokines could be a reflection of the short treatment time as mice were treated for only 20 days prior to sacrifice compared to our previous study where the treatment length (from treatment start to sac date) was 80 days.
The use of mutant SOD1 transgenic mice (particularly G93A SOD1 mice) as a valid model of ALS for the testing of compounds has recently come under question. To answer this question, comparable translational and therapeutic data are required. To date, the G93A SOD1 mouse model remains as the most widely used and is perhaps the best model available to test therapeutic strategies for ALS. However, the use of other mouse models in addition to the mutant SOD1 model, may be beneficial in finding new targets for the treatment of ALS.
We have checked for microglial activation, which was attenuated in the lenalidomide-treated mice. This finding suggests that both neuronal and glial cells in the spinal cord may have benefited from lenalidomide. The mechanism of action of lenalidomide is not currently known, but since we previously demonstrated that lenalidomide modulates the expression of cytokines, and examined a panel of pre-selected cytokines by RPA analysis. We found that several pro-inflammatory cytokines were modestly decreased, and anti-inflammatory cytokines were moderately up-regulated. A panel of 18 cytokines was analyzed and we found that potent antiinflammatory cytokines, such as IL-1RA, were dramatically upregulated by lenalidomide. IL-1R agonist acts to block the actions of IL-1α and IL-1β. TNF-α was also reduced significantly. Other pro-inflammatory cytokines including Fas associated protein (FAP) and Fas associated factor (FAF) were also dramatically reduced. The mechanisms of FAP and FAF may involve direct binding to FAS. Other cytokine changes were not significant in the present study.
We noticed a marginal decrease in TGF-β1 and TGF-β2 which was not expected. However, this may be a consequence of overall modulation by lenalidomide. Additionally, Fas RNA was also increased which was not expected.
Lenalidomide, is an immunomodulatory compound with higher potency for TNF-α than its parent compound thalidomide, may potentially be suitable for the treatment of various immunopathological disorders (Corral et al., 1999; Bartlett et al., 2004). Based on this and our previous study, the effect of lenalidomide appears to be more than simply inhibiting TNF-α and we suggest that lenalidomide may act on multiple targets. The multiple effects seen in the lenalidomide-treated G93A mice suggest that lenalidomide either targets multiple proinflammatory cascades or acts on a major transcriptional pathway upstream which results in a reduction in pro-inflammatory cytokines, anti-inflammatory cytokine stimulation and collectively, blocks neuronal cell death caused by mutant SOD1. TNF-α is widely known to be involved in the cell death pathways and we showed that TNF-α and other proinflammatory cytokines are elevated in the spinal cord of ALS mice prior to onset of ALS symptoms in the G93A mice (Kiaei et al., 2006). Despite these findings, prior studies that examined a TNF-α knock out mice were crossed with G93A or G37R mice, did not find an effect on the life span (Gowing et al., 2006). Several studies demonstrated the involvement of microglia in many acute and chronic neurological diseases. (Weydt and Moller, 2005). Astrocytes are reported to display a specific interactions with motor neurons, and produce potent toxic factors that result in their death (Nagai et al., 2007). Deleting mutant SOD1 from microglia using Lox G37RSOD1 mice had little effect on the initial phase of ALS, but it sharply slowed disease progression (Boillee et al., 2006). However a recent study examined the role of microglial in ALS and ablated 50% of proliferating microglia (CD11b expression glial cells) although this produced no effect on ALS (Gowing et al., 2008).
Since lenalidomide is a new drug with FDA approval for treatment of multiple myeloma, chronic use and long-term safety of this drug must be established before considering it for the long-term treatment in humans. Furthermore, the dosage used in this study was designed for mice, and a significant difference exists between the tolerability and pharmacokinetics of drugs between mice and humans. The effective and optimum dose of lenalidomide for human use must therefore be determined in a clinical trial in ALS patients. Lenalidomide like many drugs has a variety of side effects. For a list of these effect and prescribing information refer to, http://www.revlimid.com/ or http://www.drugs.com/sfx/lenalidomide-side-effects.html.
Our data provide evidence that lenalidomide is a drug treatment that might be effective in slowing ALS progression in humans.
Materials and methods
Mice
G93A SOD1 transgenic familial ALS mice (high copy number) (Gurney et al., 1994) were obtained from The Jackson Laboratory (Bar Harbor, ME). All protocols were conducted within National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee. We maintained the transgenic G93A hemizygotes by mating transgenic males with B6SJLF1/J hybrid females. Transgenic offspring were genotyped by PCR assay of DNA obtained from tail tissue. G93A transgenic mice were assigned randomly to the control (vehicle, mouse chow only) (n=30), 100 mg/kg/day lenalidomide groups given in the diet (n=30). This dose derived by assuming each mouse would consume 5 g of food per day. This dosing regimen was used in our previous report (Kiaei et al., 2006). Treatments started from onset of ALS phenotype (80–90 days of age). Lenalidomide feed admix (0.05% or 0.5 g/kg) and control food was blended using Purina Lab Chaw #5001 as base food at Dyets (Bethlehem, PA). Based on 5 g of food consumed per mouse, we calculated the possible lenalidomide dose approximately would be 100 mg/kg/day. Six G93A mice on control food as control and six G93A mice from the lenalidomide treated groups were killed at 110 days of age for histological evaluation. Six mice per group were used for neuronal cell counts and immunohistochemistry. Six G93A mice were used from each group at 110 days of age for biochemical analysis. Because of species-specific potency, doses used in this study cannot be extrapolated for human and should be used as guidelines only. The effective and safe dose of lenalidomide for humans remains to be determined in clinical trials.
Survival
The initial sign of disease in G93A transgenic mice is a resting tremor that progresses to gait impairment, asymmetrical manifested around 80–90 days post-natal. Disease onset is when these signs appear and will extend to symmetrical paralysis of the hindlimbs, followed by complete paralysis at the end stage. Mice were killed when they were unable to roll over within 20 s after being pushed on their side, and this time point was recorded as the time of death.
Motor function testing (rotarod)
Motor function of these mice was assessed by rotarod twice per week starting at 60 days of age. Mice were trained for 2–3 days to become acquainted with the rotarod apparatus (Columbus Instruments, Columbus, OH). The testing began by placing the mice on a rod rotating at 12 rpm, and the time that mice stayed on the rod (until falling off or staying the maximum 5 min) was recorded as a measurement of the competence of their motor function. To determine the onset of symptoms, a 15 rpm for 10 min protocol was used. Three trials were performed, and the best result of the three trials.
Histological evaluation and stereological analysis
Mice were perfused transcardially with 0.1 M cold PBS for 1 min, followed by cold 4% paraformaldehyde in PBS for 10 min. The spinal cords were dissected carefully, and the lumbar segment was identified using the ribs and the vertebrae as a guide. Tissues were postfixed in 4% paraformaldehyde for 6 h. Blocks were cryoprotected in 30% sucrose for 24 h.
For stereological analysis, serial coronal sections (50 μm thick) were cut through the lumbar (L1–L4) spinal cord enlargements from wild-type controls and vehicle-, lenalidomide-treated G93A mice and stained for Nissl substance using cresyl violet as described previously (n=6) (Kiaei et al., 2005a). The optical fractionator method in the Stereo Investigator (v3.45) software program (Microbrightfield, Burlington, VT) was employed to obtain unbiased stereological counts of Nissl-stained neurons in the ventral horn of 10 serial non-adjacent sections (250 μm apart). Cell counts were made bilaterally within an area demarcated by a horizontal line drawn through the central canal and encompassing the ventral gray horn of the gray matter to include layers seven through nine. The size of the x-y sampling grid was 200 μm × 200 μm. The counting frame thickness was 14 μm with 3-μm guard zones. Neuronal counts are expressed as the means ± standard errors of the mean.
A separate set of sections was collected as free-floating sections and processed for immunohistochemistry. The sections were immunostained with antibodies to TNF- (Peprotech, NJ), FasL (Santa Cruz Biotechnology, Santa Cruz, CA), CD40 (Serotec, Oxford, UK), and glial fibrillary acid protein (GFAP; Dako, Carpinteria, CA), using a modified avidin–biotin–peroxidase technique. The immunoreaction was visualized using 3,3′-diaminobenzidine tetrahydrochloride dihydrate with nickel intensification (Vector Laboratories, Burlingame, CA) as the chromogen. The sections were mounted onto gelatin-coated slides, dehydrated, cleared in xylene, and coverslipped. The specificity of immunostaining was confirmed by omission of the primary antibody.
Multiprobe ribonuclease protection assays
Total RNA was extracted from rapidly frozen spinal cord (n=6) using a method described previously (Hall et al., 1998). Commercial ribonuclease protection assay (RPA) probe sets (PharMingen, San Diego, CA) were used to detect specific RNAs. Radiolabeled probes were synthesized from DNA templates containing a T7 RNA polymerase promoter. Radiolabeled probes were mixed and hybridized with 5–10 μg of total RNA and treated with RNase A and T1 and resolved on 5% polyacrylamide/8 M urea gels. Dried gels were developed using a PhosphorImager (Storm 840 PhosphorImager; Molecular Dynamics, Sunnyvale, CA), and bands were quantified using instrument-resident software. For internal control and normalization the sum of L32 and GAPDH were used and then bands from samples were normalized to the value of L32 + GAPDH.
Western blot analysis
Equal amounts of total protein homogenized in RIPPA buffer of each spinal cord sample were separated on 4–20% Tris-glycine polyacrylamide gel. Proteins were electroblotted to PVDF membrane, probed with sheep anti-SOD1 (Calbiochem) as the primary antibody and protein expression detected by ECL. Anti-Beta-Actin (Abcam) was used as house keeping protein.
Statistical analysis
Kaplan–Meier survival analysis and Logrank (Mantel–Cox) were used for survival comparisons. ANOVA was followed by the Newman–Keuls test to determine statistical differences in lumbar spinal cord neuron counts between groups. Repeated-measures ANOVA were used for rotarod and weight comparisons. Using GraphPad InStat software, unpaired t-test with Welch correction was used to determine the difference in cytokine RNA in control and G93A RNA samples with and without lenalidomide.
Acknowledgments
This work was supported by Research Sponsored Grant from Celgene Corporation to MK, and NIH grant (NS044154), Muscular Dystrophy Association, and Oklahoma Center for the Advancement of Science and Technology to KH.
Lenalidomide was kindly provided by Celgene.
References
- Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, Chen C, Przedborski S. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol. 2001;49:176–185. [PubMed] [Google Scholar]
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4:314–322. doi: 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing G, Philips T, Van Wijmeersch B, Audet JN, Dewil M, Van Den Bosch L, Billiau AD, Robberecht W, Julien JP. Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci. 2008;28:10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, West M, Williamson K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2002;82:365–374. doi: 10.1046/j.1471-4159.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN, Stewart C, West M, West S, Williamson KS. Message and protein-level elevation of tumor necrosis factor alpha (TNF alpha) and TNF alpha-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol Dis. 2003;14:74–80. doi: 10.1016/s0969-9961(03)00087-1. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005a;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Petri S, Choi DK, Chen J, Calingasan NY, Beal MF. Integrative role of cPLA with COX-2 and the effect of non-steriodal anti-inflammatory drugs in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2005b;93:403–411. doi: 10.1111/j.1471-4159.2005.03024.x. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology. 2005;65:1958–1960. doi: 10.1212/01.wnl.0000188907.97339.76. [DOI] [PubMed] [Google Scholar]
- Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993;177:1675–1680. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Weydt P, Moller T. The role of microglial cells in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2005;16:1081–1090 xi. doi: 10.1016/j.pmr.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Wilms H, Sievers J, Dengler R, Bufler J, Deuschl G, Lucius R. Intrathecal synthesis of monocyte chemoattractant protein-1 (MCP-1) in amyotrophic lateral sclerosis: further evidence for microglial activation in neurodegeneration. J Neuroimmunol. 2003;144:139–142. doi: 10.1016/j.jneuroim.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]