

Abstract
Due to their viscoelastic nature, white matter axons are susceptible to damage by high strain rates produced during traumatic brain injury (TBI). Indeed, diffuse axonal injury (DAI) is one of the most common features of TBI, characterized by the hallmark pathological profiles of axonal bulbs at disconnected terminal ends of axons and periodic swellings along axons, known as “varicosities.” Although transport interruption underlies axonal bulb formation, it is unclear how varicosities arise, with multiple sites accumulating transported materials along one axon. Recently, axonal microtubules have been found to physically break during dynamic stretch-injury of cortical axons in vitro. Here, the same in vitro model was used in parallel with histopathological analyses of human brains acquired acutely following TBI to examine the potential role of mechanical microtubule damage in varicosity formation post-trauma. Transmission electron microscopy (TEM) following in vitro stretch-injury revealed periodic breaks of individual microtubules along axons that regionally corresponded with undulations in axon morphology. However, typically less than a third of microtubules were broken in any region of an axon. Within hours, these sites of microtubule breaks evolved into periodic swellings. This suggests axonal transport may be halted along one broken microtubule, yet can proceed through the same region via other intact microtubules. Similar axonal undulations and varicosities were observed following TBI in humans, suggesting primary microtubule failure may also be a feature of DAI. These data indicate a novel mechanism of mechanical microtubule damage leading to partial transport interruption and varicosity formation in traumatic axonal injury.
Keywords: Diffuse axonal injury, DAI, traumatic brain injury, TBI, axons, microtubules, axon varicosities, axonal transport, amyloid precursor prtotein, axonal stretch
Introduction
Recently there has been surging interest in traumatic brain injury (TBI), in part due its high prevalence in military personnel serving in ongoing global conflicts and in participants of contact sports. In actuality, TBI has long been a leading cause of morbidity and mortality throughout society, with over 1.7 million cases in the US each year (Faul, et al., 2010). The unique biomechanical nature of TBI is thought to be responsible for the development of diffuse axonal injury (DAI) spread throughout the white matter. As one of the most common and important pathological features of TBI, DAI is produced in all levels of injury, including mild TBI also referred to as “concussion” (Adams, et al., 1982, Blumbergs, et al., 1995, Geddes, et al., 1997, Geddes, et al., 2000).
As viscoelastic structures, axons can accommodate the substantial stretch that occurs during normal daily movements (Dennerll, et al., 1989, Hammarlund, et al., 2007). In contrast, under dynamic mechanical loading, axons are thought to become brittle, predisposing them to physical damage. This viscoelastic response may be particularly critical in TBI, where axons in white matter are exposed to high strain rates as the brain is rapidly deformed (Gennarelli, et al., 1982, Meaney, et al., 1995, Smith and Meaney, 2000). Notably, this inherent mechanical vulnerability of axons may explain their relatively selective injury in TBI (Adams, et al., 1982, Geddes, et al., 1997, Geddes, et al., 2000).
A primary effect of dynamic deformation of axons during injury is the interruption of axonal transport, resulting in accumulation of transported materials in axonal swellings within just hours of trauma (Smith, et al., 1999). Commonly, swellings appear in a periodic arrangement along connected axons, like beads on a string, to form a pathological phenotype classically referred to as “axonal varicosities” (Rand and Courville, 1946). A more widely recognized, but not necessarily more common, axonal pathology found shortly after TBI is a single swelling at a disconnection point on an axon, described as a terminal “axonal bulb” (previously referred to as a “retraction ball”) (Rand and Courville, 1946, Smith and Meaney, 2000, Smith, et al., 2003, Strich, 1956). Although complete transport interruption at a single axon terminal appears to account for bulb formation in injured axons, it has remained unclear how transport interruption can occur at multiple regions along axons resulting in the periodic swellings of varicosities.
To examine the subcellular substrates that cause transport interruption after traumatic axonal injury, we recently used a model of dynamic stretch injury of axons spanning two populations of cortical neurons separated by lithographically-fabricated micro-channels (Iwata, et al., 2004, Smith, et al., 1999, Tang, et al., 2003, Tang-Schomer, et al., 2010, Wolf, et al., 2001, Yuen, et al., 2009). Employing tensile loading parameters akin to conditions of in vivo traumatic axonal injury, we found that axonal microtubules selectively undergo mechanical failure during stretch injury. Manifested as physical breaks in the microtubule filaments, this immediate damage coincided regionally with subsequent accumulation of transported materials in discrete swellings (Tang-Schomer, et al., 2010). However, the potential role of microtubule breakage in the formation of periodic axonal swellings was not examined.
In the present study, we used the same in vitro model of dynamic stretch injury of cortical axons to determine a potential association between regional microtubule integrity and the formation of axonal varicosities. To provide clinical context of evolving changes following traumatic axonal injury in vitro, we performed histopathological analyses of post-mortem brain tissue acquired acutely following severe TBI in humans.
Materials and Methods
Isolated Axonal Cultures Using Micro-Patterned Channels
In order to examine selective dynamic mechanical trauma to axons, we developed a unique system whereby micro-patterned barriers promote the separation of neuronal soma from their axons when growing in culture. Specifically, two populations of neurons were plated on a deformable silicone membrane held within a culture well (0.005 inch thickness, Specialty Manufacturing, St. Paul, MN, USA) separated by lithographically-fabricated micro-channels (Fig. 1). This micropatterned barrier provides the physical constraints necessary to direct axon-only longitudinal outgrowth between two cell populations. Microchannels (0.4 mm width × 2 mm length × 0.2 mm height) were fabricated onto the surface of a molded elastomeric stamp by casting polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, MI, USA) from patterned lithographic masters, as described previously (Tang-Schomer, et al., 2010).
Figure 1. In vitro Model: Dynamic Stretch Injury of Axons.
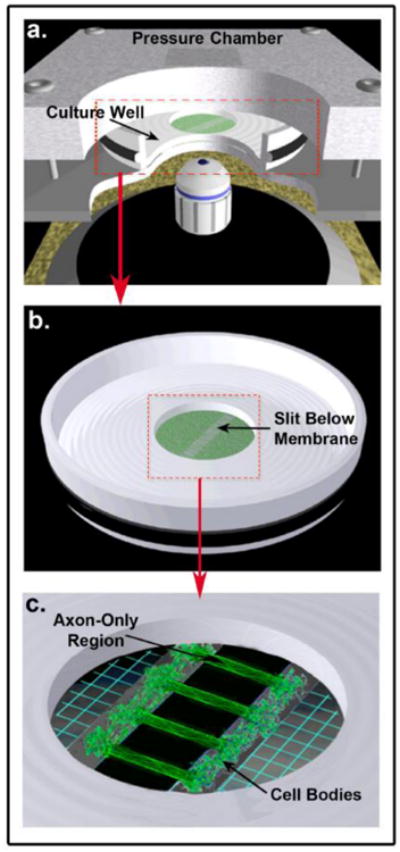
Pulsed air pressure introduces uniaxial stretch on patterned longitudinally arranged axon tracts, simulating stretch injury of CNS axons during head trauma. (a) The injury device consists of a culture well sealed within a pressure chamber into which a controlled air pulse is delivered. The culture well contains a central deformable membrane onto which primary cortical neuronal cells are plated (b). Two separate populations of cells are separated by a lithography fabricated micro-patterned barrier. Axonal processes extend through the 2mm microchannels to integrate with the opposing population of neurons, thus creating a unique axon-only region that can be placed over the slit in the device (c). Upon controlled delivery of air into the sealed chamber, the subsequent pressure change within the chamber allows the regionally specific deflection of the axon-only region.
Prior to the plating of cells, micropatterned stamps were positioned centrally on the deformable membrane (pre-coated with 1mg/mL poly-L-lysine) and filled with sterile water in order to lyse (via osmotic shock) any neuronal somata initially entering the channels upon plating. Primary cortical neurons from embryonic day 18 (E18) Sprague -Dawley rats (Charles River, Wilmington, MA, USA) were plated at a density of 375,000 -500,000 cells/cm2 on the membrane. Cells were plated and cultured in NeuroBasal medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B-27 neural supplement (Invitrogen, Carlsbad, CA, USA), 400 μM L-glutamine (GlutaMAX, Invitrogen, Carlsbad, CA, USA), and 5% fetal bovine serum (HyClone, Logan, UT, USA). After cells adhered to the deformable membrane, the channels were filled with medium allowing neurites extending from somata adjacent to the micro-patterned barrier to grow into the channels. Specifically, axonal processes started to enter the microchannels by 3-4 days in culture. By 7-10 days in vitro, axons had traversed the channels to synapse with neuronal somata on the opposite side of the 2 mm long channels. A clear ‘axon only’ region was thus defined between the independent neuronal populations, as shown previously (Tang-Schomer, et al., 2010).
In vitro Dynamic Stretch Injury of Axons
Injury was performed using the established controlled axonal injury model as described previously in detail (Iwata, et al., 2004, Smith, et al., 1999, Tang-Schomer, et al., 2010, Wolf, et al., 2001, Yuen, et al., 2009). Experiments were performed at 10-12 days in vitro in controlled saline solution (CSS; 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 15 mM glucose, and 25 mM HEPES, pH 7.4). Prior to injury, the micropatterned stamp was removed to leave the intact axons traversing two distinct populations of neuronal somata on the deformable silicone membrane. Culture wells were then placed in a sealed, pressure controlled, device at an orientation aligning the region of cultured axons directly above a machined 2 × 15 mm slit in a metal plate (Fig. 1). A controlled air pulse was then injected into the sealed pressure chamber containing the culture. This pressure change within the chamber results in the rapid downward deflection of the axon-only region of membrane through the slit positioned below. As such, only the cultured axons undergo rapid tensile elongation. Measurement of nominal uniaxial strain experienced within the axon-only region is calculated by determining the centerline membrane deflection relative to the width of the underlying slit as previously described (Iwata, et al., 2004, Smith, et al., 1999, Tang-Schomer, et al., 2010, Wolf, et al., 2001). The membrane deflection was determined by z-distance change of the center of the focal plane at a specific chamber pressure from that of the zero pressure (the default ambient pressure). Once the pressure-strain relationship was determined, pressure values were applied in a controlled fashion to axonal cultures for desired strain levels of 75%. Previous characterization of the model indicates that undulations can be observed post-injury with at least 5% strain (Yuen, et al., 2009). Moreover, strain levels of 75% have previously been demonstrated to result in immediate undulations, followed by the development of axonal swellings by 3 hours, and complete degeneration of virtually all axons by 24 hours post-injury (Tang-Schomer, et al., 2010).
Immunocytochemical Examinations
Immunocytochemistry was performed on cultures following fixation using 4% paraformalydehyde at ≤ 2 minutes and 3 hours post-injury. After permeabilization with 0.1% Triton X-100, cells were blocked with 4% goat serum and incubated in a primary antibody overnight at 4 °C. Primary antibodies included mouse monoclonal anti-β III-tubulin (1:1000; Sigma, USA), rabbit polyclonal anti-human tau (Tau, 1:250; Dako, Denmark), mouse monoclonal anti-amyloid precursor protein (22C11, 1:100; Chemicon, Temocular, CA, USA), and rabbit polyclonal anti-neurofilament 200 kDa (NF200, 1:250; Covance Research Products, Princeton, NJ, USA). Subsequently, the appropriate Alexa Fluor secondary antibody (Invitrogen, Carlsbad, CA, USA) was applied for 1 hr (1:200-500 dilution) at room temperature. Images were acquired using a 60× oil objective lens on a Nikon TE300 inverted microscope with a CCD camera (Cooke Corporation, Romulus, MI, USA) equipped with Camware v2.15 software.
Uninjured control axons of the same age were treated identically to those of the injured cultures with omission of the injury. Five independent cultures were examined for each primary antibody. Within each independent culture, there were at least 6 microchannels, each containing a minimum of 100 axons traversing the gap.
Examination using Electron Microscopy and Ultrastructural Analysis
Uninjured control axons and axons immediately (≤2 min) and at 3 hr after stretch injury were fixed and processed for analysis using transmission electron microscopic (TEM). For each experimental condition, 2 independent cultures were processed. Specifically, cells were fixed with a mixture of 2% paraformaldehyde and 2.5% glutaraldehyde overnight. After washing with 0.1 M sodium cacodylate buffer (3×10 mins), cells were post-fixed with osmium (2%) plus potassium ferricyanide (1.5%), followed with three buffer washes and two diH2O washes. To enhance the intensity of cell membranes, cells were stained with 2% uranyl acetate for 20 min, followed again with two diH2O washes. Serial dehydration was performed via 3 min incubations in 50%-100% EtOH. Post-fixed cultures were embedded in resin in situ, separated from the silicone membrane, and re-embedded.
To select axons for EM examination, a ∼1 mm × 1 mm square in the middle of the axon-only region (containing 24-26 micro-channels) was cut, encompassing all the axon processes (100-200) within one micro-channel. Longitudinal sections of 1 mm width × 1 mm length × 60 nm thickness were obtained. For each section, 100-200 high-resolution TEM images (with a dimension of ∼ 2 × 2 μm at magnification of 70-150,000×) with overlapping borders were serially acquired.
TEM images with overlapping borders were stitched together in order to reconstruct high-resolution panoramic images of axon processes up to a length of 25 μm using an average 6.7 ± 4.2 frames at 150,000×. 65 reconstructed axonal processes (19 uninjured control cultures, 16 at ≤2mins post-injury and 30 at 3 hours post-injury) were examined.
Immunohistochemical Examination of Axonal Pathology Following Acute Severe TBI in Post-Mortem Human Brain Tissue
Tissue from the brains of 4 individuals who died acutely following severe TBI were selected from the TBI tissue archive of the Department of Neuropathology, Glasgow UK. Approval for the use of tissue was granted by the South Glasgow and Clyde Research Ethics Committee. Cases were selected with a history of a single severe TBI who had previously been identified to have DAI at routine diagnostic postmortem. All cases were found deceased at the scene of the injury or died shortly after admission to hospital as a result of severe TBI. Specifically, case 1 was an 18 year-old male who died from TBI 10 hours after sustaining an assault including blunt-force trauma to the head. Case 2 was a 41 year-old male who died 16 hours after TBI caused by a fall down a flight of stairs. Case 3 was an 18 year-old female who died 22 hours after a motor vehicle collision (MVC). Case 4 was a 25 year-old male who died 12 hours, also following a MVC. None of the cases had any previous history of neurological disease or previous TBI. Case 5 was an 18 year-old female, uninjured control who died as a result of hematological malignancy and had no prior history of TBI.
Human Brain Tissue Preparation and Immunohistochemistry
Whole brains were immersion fixed in 10% formol saline for a minimum of 3 weeks then sampled using a standardized block selection protocol. After being processed to paraffin via standard techniques, 8μm sections were examined in the coronal plane from blocks containing the corpus callosum, cingulate gyrus and adjacent white matter.
Immunohistochemistry specific for the amyloid precursor protein (APP) was performed as the gold standard marker for the clinical examination of axonal injury (Gentleman, et al., 1993, Gorrie, et al., 2002, Lambri, et al., 2001, Reichard, et al., 2003, Sherriff, et al., 1994). Specifically, following deparaffinization and rehydration to dH2O, sections were immersed in 3% aqueous H2O2 (10 minutes) to quench endogenous peroxidase activity. Antigen retrieval was performed via microwave pressure cooker at high power for 10 minutes. After cooling, tissue was blocked using 1 drop of normal horse serum (Vector Labs, Burlingame, CA, USA) per 5ml of Optimax buffer (BioGenex, San Ramon, CA, USA) for 30 minutes. Incubation with the primary antibody specific for APP (22C11; Millipore, Billerica, MA, USA) was performed for 20 hours at 4°C. After rinsing (PBS/Tween), a biotinylated secondary antibody was applied for 30 minutes (Vector labs, Burlingame, CA, USA) followed again by rinsing (PBS/Tween). Next, the Avidin Biotin Horse-Radish Peroxidise (HRP) complex (10 μl Avidin DH solution and 10μl Biotinylated enzyme per 5ml Optimax Buffer) was applied for 30 minutes (Vector labs, Burlingame, CA, USA) and visualization achieved using 3,3-diaminobenzidine (DAB) (Vector Labs, Burlingame, CA, USA). Sections were analyzed using a Leica DMRB light microscope (Leica Microsystems, Wetzlar, Germany).
Results
Dynamic stretch injury of axons results in immediate (≤2 min) microtubule displacement
Immediately following injury (≤2 minutes) cultures frequently contained axons with altered morphology manifest as a series of undulations, demonstrated using both immunocytochemistry and TEM (Fig. 2). Undulations of single axons were easily identified as bending distortions, with a width of 5–8μm and amplitude of 3–6μm, and as such indicative of an increase in both regional and overall axonal length. Specifically, rapid tensile stretch of axons (75% strain) resulted in up to at least a 65% localized length increase in the region of a typical undulation. Uninjured axons, as expected, displayed a normal arrangement of microtubules traversing the axonal length like parallel cables, with no undulations or swellings (Fig. 2b-c).
Figure 2. Microtubule Displacement and Undulation Formation Immediately After Dynamic Stretch Injury of Axons.
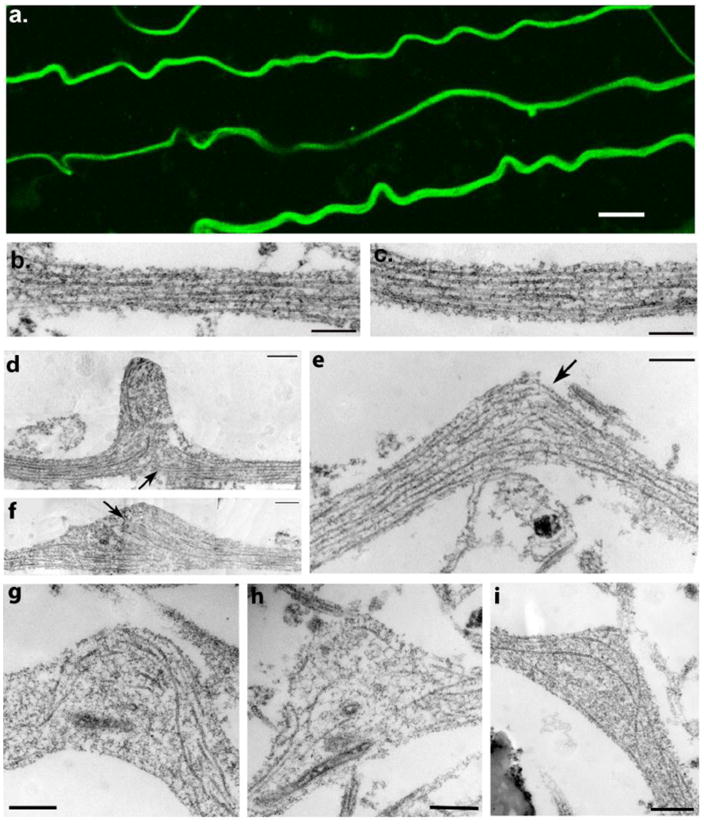
(a) Representative images displaying multiple axons with an undulating morphology and immunoreactive for βIII-tubulin immediately (≤2 minutes) following dynamic stretch injury (Scale bar approx. 5μm). (b-c) TEM images showing a normal region of axon with intact microtubules aligned in parallel cables. (d-i) TEM images within 2 minutes following dynamic stretch injury showing widening of the spaces between microtubules near the peak of axon undulations accompanied by compaction of the polymers. Microtubules are also no longer linear and aligned in parallel but rather appear distorted and twisted with discrete breaks (arrows). Breaking and twisting of microtubules was observed near the peak of axon undulations where newly generated free-ends at disconnection points failed to align. Scale bars: 500 nm.
Further examination using TEM revealed extensive disruption to the organization of microtubules within these axonal undulations (Fig. 2d-i). Microtubules could clearly be identified as filamentous structures with an approximately 24nm wide cross-sectional diameter traversing the main axis of an axon. Widening of the lateral spacing between microtubules was commonly observed, accompanied by focal constrictions of compacted polymers, indicating longitudinal microtubule displacement (Fig. 2d-i). Near the apex of axon undulations, microtubules were typically broken, and the newly generated free-ends failed to align (Fig. 2d-i). At any location, approximately less than a third of microtubules in any one region within an axon were observed to be broken as was previously characterized (Tang-Schomer, et al., 2010). Multiple points of such discrete, focal damage to microtubules along the axon length could be identified, extending at least several hundred microns.
The interpretation that microtubule breaks were indeed produced via physical rupture by tensile forces rather than normal microtubule ends was supported by serial observations. In particular, the disconnection sites were typically in close proximity to one another on a single thin section, with no indication of an intervening short segment of microtubule on any adjacent section. Moreover, the ends of broken microtubules failed to display the appearance of a typical microtubule being lost from the plane of section. Interestingly, the ends also lacked the appearance of a typical microtubule undergoing dynamics or of those produced by a severing protein. In contrast, microtubule ends were non-distinct, grainy in appearance and the microtubule itself was slightly contorted or twisted in appearance.
While undulations were prevalent, acutely injured axons rarely displayed swellings (identified by their more symmetrical shape versus the distinctly asymmetrical curvature of an axon undulation).
Varicose axonal swellings 3 hr following dynamic stretch injury of axons occur at regions of microtubule disruption and loss
By 3 hr following injury periodic swellings could be observed in axons (Fig. 3,4). Immunocytochemical examination revealed a series of swellings distributed along the axon that were immunoreactive for β III-tubulin, the microtubule-associated protein tau, the fast transport protein amyloid precursor protein (APP), and the slow transport protein neurofilament (NF200) (Fig. 3a-d). Moreover, at the same time point, axons displaying an undulating morphology were now rarely observed.
Figure 3. Immunocytochemical Staining of Axons Following Dynamic Stretch Injury.
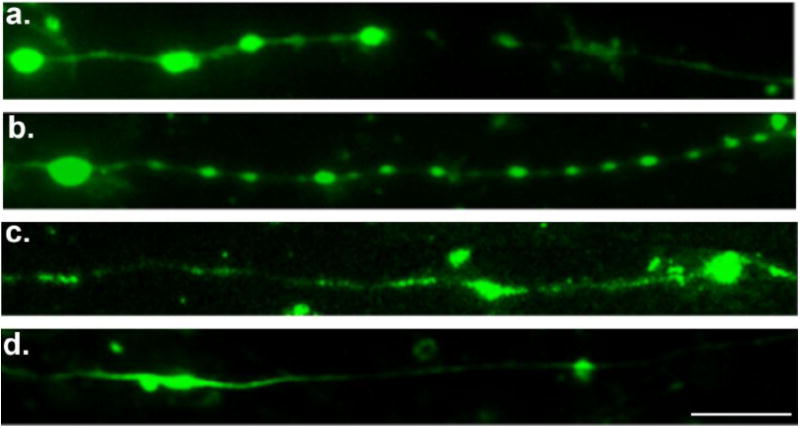
Immunofluorescent images of injured axons (3hr post-injury) displaying a series of swellings along the axonal length like beads on a string. Swellings display accumulations of (a) tubulin, (b) the microtubule-associated protein tau, (c) amyloid precursor protein (APP), and (d) neurofilament (NF200). Scale bar, 10 μm.
Figure 4. Microtubule Breakage and Loss in Varicose Axonal Swellings 3 Hours After Dynamic Stretch Injury of Axons.
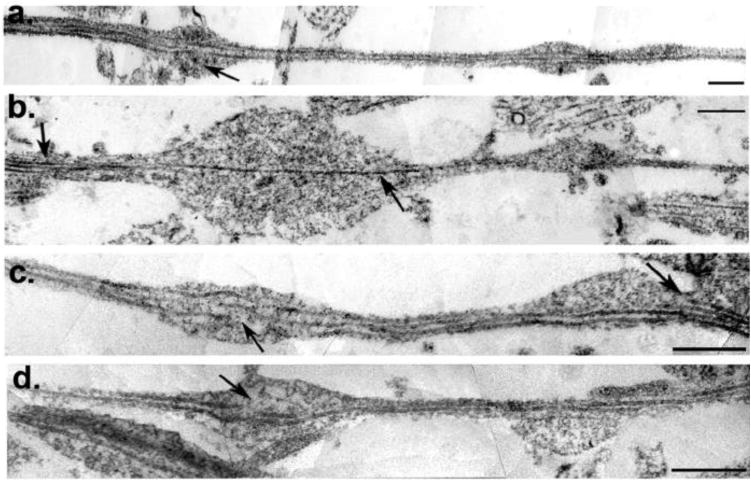
(a-d) TEM images of axons at 3hr post-injury were stitched together to reconstruct a panoramic image of an axon segment. Individual microtubules display selective breakage within axon swellings (a,c,d arrows). While breakage or loss of microtubules can be seen in association with swelling, other microtubules can be observed traversing the swollen region intact. Specifically, (b) shows a solitary intact microtubule traversing a large swelling (arrow) whereas multiple microtubules can be observed in the adjacent non-swollen region from which it emerged (arrow). This sole remaining microtubule feeds into a subsequent but separate swelling. Scale bars: 500 nm.
Reconstructed panoramic TEM images of axonal processes confirmed immunocytochemical observations of multiple serial swellings with a “beads on a string” appearance (Fig. 4a-d). Notably, TEM also revealed focal breakage of microtubules observed almost exclusively within the swollen regions of axons when compared to adjacent, non-swollen region of axons with a normal linear morphology. Furthermore, microtubules were commonly less frequent and in many cases completely absent within swellings when compared to both the non-swollen region of injured axons and indeed normal uninjured axons (Fig 2b-c, 4).
Axonal Pathology Following Acute Severe TBI in Humans
TBI cases displayed widespread and extensive axonal pathology within the corpus callosum and adjacent white matter, while no axonal pathology was observed in the uninjured control case (Fig. 5a). Following acute, severe TBI in humans, immunohistochemistry revealed APP accumulating within damaged axons in all 4 cases throughout the regions examined. Axonal morphology closely resembled the morphological appearance of axons following dynamic stretch injury in vitro including undulations and varicosities (Fig. 5,6). Damaged axons displayed the classic morphological appearance of axonal bulbs at disconnected axon terminals and varicose swellings along the length of axons, often within the same microscopic field (Fig. 6a,d). Moreover, axons with an undulating morphology could occasionally be observed (Fig. 5b, 6a). Accumulating APP also revealed some axons to be intact, yet displaying a series of discrete but connected swellings, whilst others were undergoing overt degeneration, with beading of axons into distinct fragments (Fig. 6a-f). Occasionally singular swellings along an otherwise intact axon could be observed, distinct from the disconnected, terminal axonal bulbs. (Fig 6e).
Figure 5. Undulations and Varicosities Following Acute Traumatic Brain Injury in Humans at Post-Mortem.
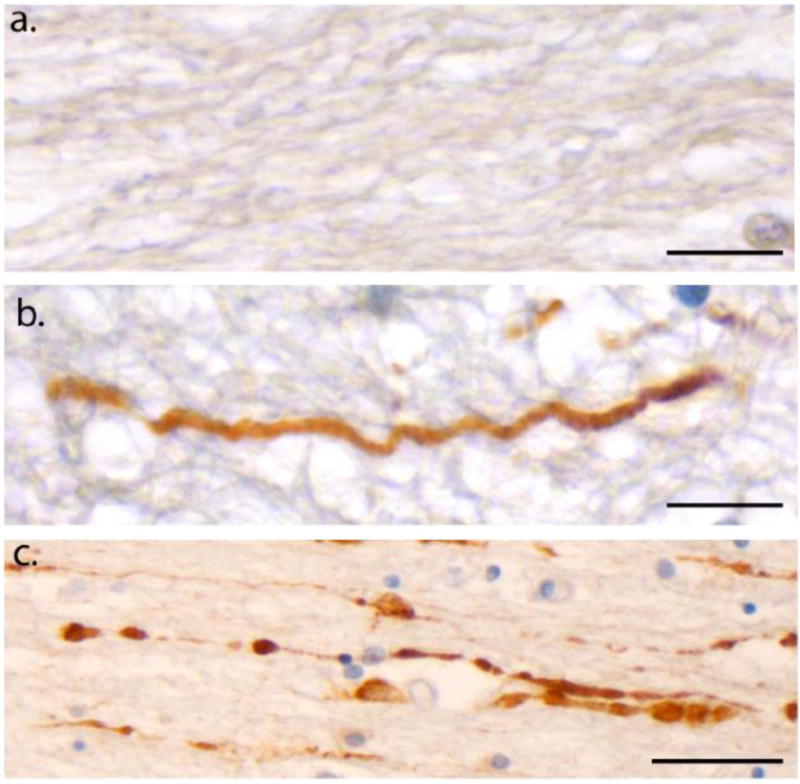
(a) An absence of APP immunoreactivity in normal white matter within the corpus callosum of an 18F with no history of TBI who died as a result of haematological malignancy (case #5). Scale bar: 25μm (b) APP Immunoreactivity within the corpus callosum of an 18M who died 10 hours following acute severe traumatic brain injury caused by an assault (blunt force trauma to the head) (case #1). Axons display an undulating morphology akin to what is observed in vitro following dynamic stretch injury. Scale bar: 25μm (c) APP Immunoreactivity within the corpus callosum, also in case #1, displaying a classic varicose morphology with multiple individual swellings along the length of an individual axon can be observed. Scale bar: 30μm.
Figure 6. Varicose Axons within the Corpus Callosum of 4 Human Cases of Acute Severe TBI Examined at Post-Mortem via APP Immunohistochemistry.
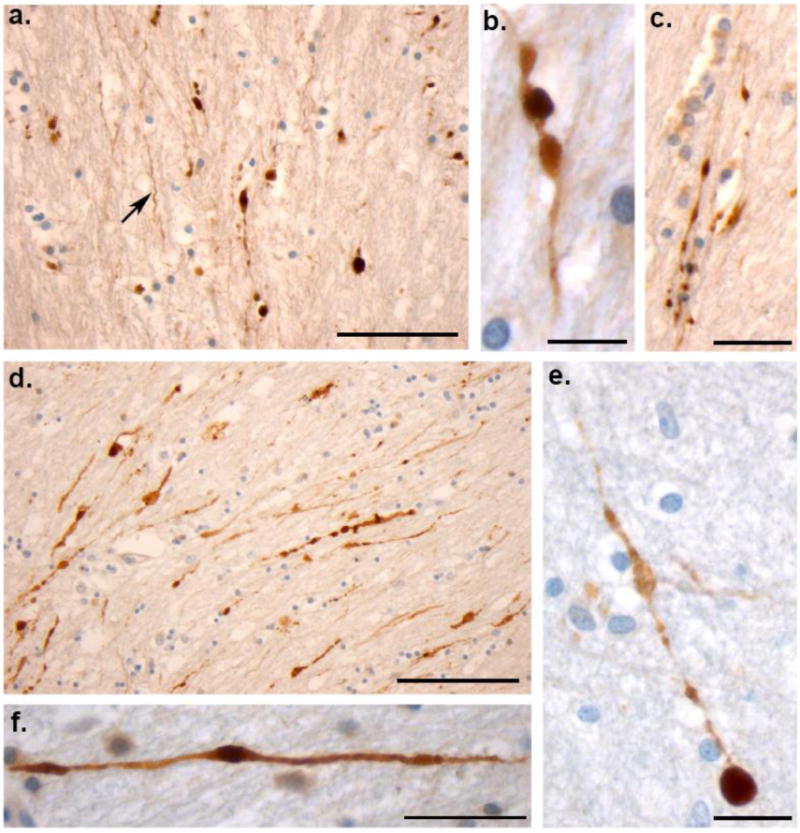
(a) Extensive axonal pathology with classic varicosities and axonal bulb formation within the corpus callosum of an 18F who died 22 hours following a MVC (case #3). Axons with an undulating morphology can be observed within the same field (arrow). Scale bar: 100μm (b and c) High magnification axonal varicosities in the corpus callosum of a 41 year-old male who died 16 hours following a fall (case #2). Note the serial swellings indicating multiple points of partial transport interruption. Scale bars (b):100μm, (c)30μm. (d) Extensive axonal pathology with classic varicosities and axonal bulb formation within the corpus callosum of an 18M who died 10 hours following blunt force trauma to the head (case #1). Scale bar: 100μm (e) High magnification of a single axon accumulating APP in a 25 year old male following a MVC (case #4). Note the multiple points of swelling along the visible axon length culminating in a large axonal swelling (axonal bulb) at the disconnected axon terminal. Scale bar: 90μm (f) Individual axon displaying a varicose morphology with isolated serial swellings, aslso from case #3. Scale bar: 50μm
Discussion
These results indicate a novel mechanism of partial interruption of axonal transport due to traumatic axonal injury, which appears to account for the formation of axonal varicosities. Periodic breaks of individual microtubules were observed along axons following stretch-injury in vitro, demonstrated by electron microscopy. However, typically only a few microtubules were broken in any specific region along the axon. These breaks corresponded regionally with undulations in the axons' morphology, which within hours evolved into periodic swellings. In human brains, similar undulations along axons and axonal varicosities were observed at postmortem acutely following severe TBI. These data suggest a process whereby transport within an axon may be halted along broken microtubules, yet can proceed through the same region via other intact microtubules. Accordingly, partial interruption of transport at multiple sites along an injured axon may give rise to the serial varicose swellings commonly found after TBI.
Diffuse axonal injury in TBI occurs due to dynamic mechanical loading of axons in white matter tracts during head rotational acceleration (Smith and Meaney, 2000) resulting in the hallmark pathologies of axonal varicosities and terminal axonal bulbs (Adams, et al., 1982, Geddes, et al., 1997, Geddes, et al., 2000). Since the diameter of these swellings can exceed 30-fold the normal caliber of the axons, this huge increase in volume implicates interruption of axonal transport as the underlying source of the swelling rather than representing a simple redistribution of local axonal constituents. Moreover, dense accumulations of transported proteins are found within the swellings, further supporting localized interruption of axonal transport.
The historical use of the term “interruption of axonal transport” for TBI has typically implied complete derailment of transported cargoes at a specific site at the level of the whole axon, akin to the interruption of transport that occurs upon axonal transection. Although this process appears responsible for axonal bulb formation, it cannot explain the formation of axonal varicosities. Indeed, axonal varicosities are often observed to have swellings separated by lengths of axon with a relatively normal diameter, suggesting they emerge via a distinct mechanism.
In the present study, TEM examination of the axonal ultrastructure shortly after dynamic stretch injury of axons in vitro revealed evidence for direct mechanical breakage of microtubules at multiple sites along axons. This microtubule breakage was typically observed in regions where the axon had taken on an undulating morphology, as described previously (Tang-Schomer, et al., 2010). Notably, however, undulations occurred in association with breakage of individual microtubules. Specifically, within any one axonal undulation, typically less than a third of the microtubules were found to be immediately broken upon injury, while the remaining microtubules appeared intact. As such, the traumatic breakage of individual microtubules appears to block relaxation to the pre-stretch length of axons within that region, potentially by impeding sliding of adjacent microtubules. Subsequent depolymerization of the broken microtubules may release this mechanical block, allowing undulating axons to relax back to a linear geometry (Fig 7).
Figure 7. Proposed Mechanism of Varicosity Formation after Traumatic Axonal Injury.
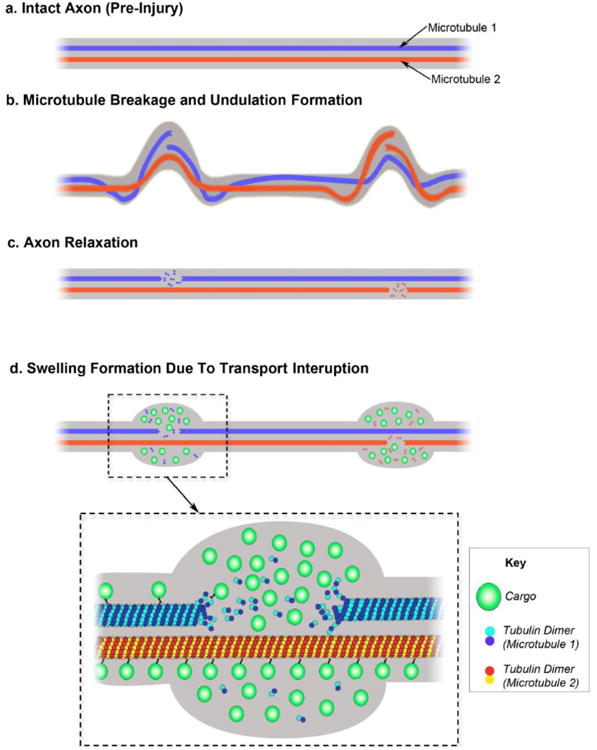
(a) Illustration displaying two illustrated microtubules (MT1 and MT2) within an intact axon (pre-injury). (b) Following injury, mechanical breaking occurs at different sites in both microtubule 1 and microtubule 2. Misalignment of broken microtubules causes deformation of the axon observed as two discrete undulations. (c) Shortly afterward, catastrophic depolymerization from the broken ends of the microtubules allows the undulations to collapse and the axon recovers its linear morphology. (d) Microtubule breakage leads to impairment of axonal transport and subsequent accumulation of transported cargos near the microtubule breaking point. By contrast, axon transport on the intact microtubules remains normal. This ‘partial transport impairment’ may account for the formation of serial swellings that give axons a varicose appearance following traumatic axonal injury.
By examining the subsequent temporal evolution of axonal pathology with combined TEM and immunocytochemical analyses, regions with broken microtubules were observed to swell and undergo further structural disorganization. By three hours post-injury, accumulation of transported proteins had formed in large, periodic swellings at sites of individual microtubule damage. Importantly, these observations of mechanical microtubule breakage and morphological alterations after traumatic axonal injury in vitro may be extrapolated to human TBI. While undulations and varicosities of axons have long been recognized as acute pathological profiles in human TBI, no underlying cause has previously been identified. Here, the virtually identical morphological changes of undulations and varicosities of axons observed in the corpus callosum of humans shortly after severe TBI, compared to in vitro traumatic axonal injury, suggests a shared mechanistic basis of primary microtubule rupture. However, acutely following TBI in humans, the progression of morphological changes of injured axons may proceed at varying rates between axons since undulations, varicosities and bulb formations could often be observed together in the same tissue section.
The selective subcellular disruption of axonal microtubules appears to be due to an inherent viscoelastic response that occurs uniquely as a consequence of the dynamic mechanical loading experienced during traumatic injuries (Gennarelli, et al., 1982, Meaney, et al., 1995, Smith and Meaney, 2000). With slow stretching, axons can easily endure 100% strain and return to their original length unharmed (Galbraith, et al., 1993, Tang-Schomer, et al., 2010). However, dynamic stretch was found to induce microtubule breakage leading to varicose swellings. As the stiffest structures in axons (Gittes, et al., 1993), microtubules may be selectively vulnerable to breakage as a consequence of high strain. Moreover, while all microtubules in axons underwent an identical tensile strain at the time of injury, mechanical failure occurred at varying locations for each individual microtubule at different points along the axon. This suggests that there may be mechanically weak portions along individual microtubules that do not typically line up between different microtubules within one axon.
Notably, the axons within the model described are unmyelinated. Recent work indicates that myelinated fibers are less susceptible to mechanical strain when compared to unmyelinated fibers, using both in vivo and in vitro TBI models. Specifically, smaller unmyelinated axons were more likely to suffer dysfunction of conduction pathways in vivo (Reeves, et al., 2007, Reeves, et al., 2005), while non-myelinated axons were more prone to secondary disconnection following an in vitro model of axonal stretch-injury (Staal and Vickers, 2011). With approximately 50% of axons within the human brain being unmyelinated, the stretch-injury model represents a predominant axonal subtype that is highly relevant in TBI. Examination of the potentially protective role of myelin in microtubule integrity post injury will be an important future consideration.
Taken together, these data suggest that axonal trauma triggers a unique scenario that results in partial transport interruption. A key event appears to be the staggered location of mechanical failure between individual microtubules along axons. With this arrangement, some transported cargo may be derailed at a breakage point of one individual microtubule, while other cargo may continue through this same axonal region via other intact microtubules. Consequently, successive breaks of the remaining microtubules distally, may give rise to the periodic swellings that make up axonal varicosities.
Interestingly, even the breakage of relatively small number of microtubules in any one section was associated with a notable diminution in axonal transport. Within axons, microtubules normally exist in a variety of different lengths (Yu and Baas, 1994) and the severing of microtubules by enzymes such as katanin is a normal ongoing process (Yu, et al., 2005). Thus, it is unclear why just a few additional breaks are detrimental and why there is not relatively rapid repair through normal microtubule dynamics. Given the often twisted and contorted appearance of damaged microtubules, we would contend that microtubules are not exclusively damaged at the point of breakage, but rather a significant portion of the surrounding region is impacted by the breakage event. Moreover, such structural damage may occur in the absence of a complete physical break. As such, this would likely impact the interaction of molecular motors and the movement of cargo along the length of the polymers. Moreover, such extensive damage may not be so readily repaired by endogenous mechanisms, accounting for the longterm impact on axonal transport. Indeed, if damaged microtubules persist, physical severing by proteins such as katanin, may give rise to new microtubules with a flawed lattice, perpetuating and even escalating the problem.
Conclusion
Identification of microtubule damage in traumatic axonal injury may have important implications for treatment and repair in TBI. Indeed, partial transport interruption due to microtubule damage may not represent an irreversible process. Injured axons may be able to contend with small accumulations of transported material as long as there is limited progression of cytoskeletal disorganization and proteolysis. We have previously shown that stabilization of microtubules at the time of injury using the tubulin-binding drug, taxol, significantly deters axon degeneration (Tang-Schomer, et al., 2010). Accordingly, stabilization of the axonal cytoskeleton after mechanical trauma has been suggested as a new therapeutic target in TBI. Even so, if mechanical damage of microtubules is more complex, involving structural flaws to the lattice itself, the most potent therapies may lie in either ridding the axon of damaged microtubules, or attempting to repair them.
Highlights.
Diffuse axonal injury results in transport interruption observed as varicosities along axons > Using a model of axonal stretch injury the mechanism of axonal varicosity formation was examined > Individual microtubule breakage results in partial transport interruption and varicose swelling > Similar varicosities found in humans indicate primary microtubule failure may be a feature of DAI
Acknowledgments
We thank Kevin Browne of the Dept. Neurosurgery and Center for Brain Injury and Repair, University of Pennsylvania for his technical assistance; Ray Meade from the Biomedical Imaging Core at University of Pennsylvania for EM assistance, and Janice E. Stewart, Dept. Neuropathology, Southern General Hospital, Glasgow, UK for her assistance with immunohistochemical procedures for human studies. This work is supported by the National Institute of Health grants: NS056202, NS038104, NS048949 (all DHS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J Neurotrauma. 1995;12:565–572. doi: 10.1089/neu.1995.12.565. [DOI] [PubMed] [Google Scholar]
- Dennerll TJ, Lamoureux P, Buxbaum RE, Heidemann SR. The cytomechanics of axonal elongation and retraction. J Cell Biol. 1989;109:3073–3083. doi: 10.1083/jcb.109.6.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta (GA): 2010. [Google Scholar]
- Galbraith JA, Thibault LE, Matteson DR. Mechanical and electrical responses of the squid giant axon to simple elongation. J Biomech Eng. 1993;115:13–22. doi: 10.1115/1.2895464. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Beer TW, Ellison DW. The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol. 1997;23:339–347. [PubMed] [Google Scholar]
- Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol. 2000;26:105–116. doi: 10.1046/j.1365-2990.2000.026002105.x. [DOI] [PubMed] [Google Scholar]
- Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Gittes F, Mickey B, Nettleton J, Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J Cell Biol. 1993;120:923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite PM. Axonal injury in children after motor vehicle crashes: extent, distribution, and size of axonal swellings using beta-APP immunohistochemistry. J Neurotrauma. 2002;19:1171–1182. doi: 10.1089/08977150260337976. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, Bastiani MJ. Axons break in animals lacking beta-spectrin. J Cell Biol. 2007;176:269–275. doi: 10.1083/jcb.200611117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambri M, Djurovic V, Kibble M, Cairns N, Al-Sarraj S. Specificity and sensitivity of betaAPP in head injury. Clin Neuropathol. 2001;20:263–271. [PubMed] [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Rand CW, Courville CB. Histologic changes in the brain in cases of fatal injury to the head; alterations in nerve cells. Arch Neurol Psychiatry. 1946;55:79–110. doi: 10.1001/archneurpsyc.1946.02300130003001. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Lee NN, Povlishock JT. Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res. 2007;1154:225–236. doi: 10.1016/j.brainres.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Reichard RR, White CL, 3rd, Hladik CL, Dolinak D. Beta-amyloid precursor protein staining of nonaccidental central nervous system injury in pediatric autopsies. J Neurotrauma. 2003;20:347–355. doi: 10.1089/089771503765172309. [DOI] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol (Berl) 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF. Axonal damage in traumatic brain injury. The neuroscientist. 2000;6:483–495. [Google Scholar]
- Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. 2003;18:307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal JA, Vickers JC. Selective vulnerability of non-myelinated axons to stretch injury in an in vitro co-culture system. J Neurotrauma. 2011;28:841–847. doi: 10.1089/neu.2010.1658. [DOI] [PubMed] [Google Scholar]
- Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163–185. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MD, Golden AP, Tien J. Molding of three-dimensional microstructures of gels. J Am Chem Soc. 2003;125:12988–12989. doi: 10.1021/ja037677h. [DOI] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 2010;24:1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Baas PW. Changes in microtubule number and length during axon differentiation. J Neurosci. 1994;14:2818–2829. doi: 10.1523/JNEUROSCI.14-05-02818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Solowska JM, Qiang L, Karabay A, Baird D, Baas PW. Regulation of microtubule severing by katanin subunits during neuronal development. J Neurosci. 2005;25:5573–5583. doi: 10.1523/JNEUROSCI.0834-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen T, Browne K, Iwata A, Smith D. Sodium Channelopathy Induced by Mild Axonal Trauma Worsens Outcome After a Repeat Injury. J Neuroscience Research. 2009 doi: 10.1002/jnr.22161. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]