

Abstract
Traumatic brain injury (TBI) has devastating acute effects and in many cases seems to initiate long-term neurodegeneration. Indeed, an epidemiological association between TBI and the development of Alzheimer's disease (AD) later in life has been demonstrated, and it has been shown that amyloid-β (Aβ) plaques — one of the hallmarks of AD — may be found in patients within hours following TBI. Here, we explore the mechanistic underpinnings of the link between TBI and AD, focusing on the hypothesis that rapid Aβ plaque formation may result from the accumulation of amyloid precursor protein in damaged axons and a disturbed balance between Aβ genesis and catabolism following TBI.
Traumatic brain injury (TBI) is a common and often devastating health problem1,2. Despite its prevalence, TBI has only recently become widely recognized as a major health issue — in part due to the intense media attention on the high incidence of TBI in ongoing military conflicts. In addition, there has been a growing awareness of the epidemiological association between a history of TBI and the development of Alzheimer's disease (AD) later in life3–12. This link is supported by the identification of acute and chronic AD-like pathologies in the brains of TBI patients and in animal models of TBI.
There are several possible mechanisms linking an episode of TBI to later development of neurodegenerative disease, such as neuronal loss13–15, persistent inflammation16,17 and cytoskeletal pathology18,19. However, the pathophysiological link that has received the most attention is the production, accumulation and clearance of amyloid-β (Aβ) peptides following TBI. Here, we will examine the current understanding of how a single TBI can trigger both rapid and insidiously progressive AD-like pathological changes. In particular, we will examine the association between TBI and Aβ turnover.
TBI and AD: epidemiological link
Compelling data from several studies demonstrate that a history of TBI is one of the strongest epigenetic risk factors for AD3–12,20. However, there is not a complete consensus, as some epidemiological studies have failed to find such an association21–28. A major point of contention has been the retrospective nature of some reports that may have led to recall bias — a systematic error due to inaccuracies in subjects' ability to recall their history of TBI. This is of particular concern when gathering information from patients with cognitive impairments or from secondary informants. Nevertheless, larger, more controlled studies, including level 1 evidence (which requires prospective examination and randomization)11, has led to a general acceptance that TBI is a risk factor for developing AD29.
It has also been suggested that a history of TBI accelerates the onset of AD10,30–32, and that the more severe the injury, the greater the risk of developing AD9,11. Indeed, because TBI is a complex and heterogeneous disorder, the type and extent of the acute pathology probably has an important role in determining the risk of developing AD. In addition, the baseline susceptibility of the patient may be predetermined by multiple factors such as age, sex and the interplay of several known or unknown genetic factors. For example, there is evidence that genetic predisposition, as a result of an apolipoprotein E (APOE) polymorphism, may influence the likelihood of developing AD after TBI (BOX 1).
Box 1. The effects of apolipoprotein E genotype in traumatic brain injury.
As with Alzheimer's disease (AD)137,138, the lipid transport protein apolipoprotein E (APOE) has been implicated in influencing amyloid pathology and outcome following traumatic brain injury (TBI). A series of studies have found that individuals carrying the APOE ε4 allele were more likely to have a poor outcome following injury139–147. However, there have also been reports that failed to show any association between APOE ε4 carriers and outcome148–150. Indeed, a recent prospective study examining 984 cases only found an association with possession of an APOE ε4 allele and outcome in younger adults and children, with the association being strongest in patients aged less than 15 years150. Thus, despite a general acceptance that possession of an APOE ε4 allele worsens outcome after TBI, there is renewed debate in this regard.
Epidemiological data have provided additional information by implicating APOE4 genotype as a risk factor for the later development of AD following TBI7,9,11,25,151–153. However, considerable debate remains over whether APOE and TBI operate in a synergistic manner to increase the risk of AD development or, alternatively, act as independent but additive risk factors.
Carriers of the APOE ε4 allele were found to be at increased risk of amyloid-β (Aβ) deposition following TBI154. Aβ deposition was also significantly increased following head trauma in PDAPP (platelet-derived growth factor promoter expressing amyloid precursor protein) mice carrying the human APOE ε4 allele versus those carrying APOE ε3 or no APOE155. The mechanism by which APOE is able to exert an effect on Aβ deposition remains elusive. In addition, the interplay of APOE polymorphism with the microsatellite polymorphism in neprilysin, also shown to contribute to Aβ deposition112, will be of interest. Indeed, when combined, these polymorphisms could potentially provide useful predictive information.
TBI and AD: pathological links
Human TBI and Aβ plaques
The first clue indicating a pathological link between TBI with AD was the observation that Aβ plaques, a hallmark of AD33, are found in up to 30% of patients who die acutely following TBI34,35. Notably, these plaques were found in all age groups, even in children. By comparison, in control cases (individuals that died from non-neurological causes), plaques were found almost exclusively in elderly individuals35. Plaques have even been observed in peri-contusional tissue surgically excised from survivors of TBI36,37.
The plaques found in TBI patients are strikingly similar to those observed in the early stages of AD (FIG. 1). However, plaques in AD develop slowly and are predominantly found in the elderly, whereas TBI-associated plaques can appear rapidly (within just a few hours) after injury35,36. In addition, plaques were identified following a range of traumatically induced pathologies that resulted from various causes of injury35.
Figure 1. Immunohistochemical findings exploring mechanisms of amyloid-β plaque formation following traumatic brain injury.
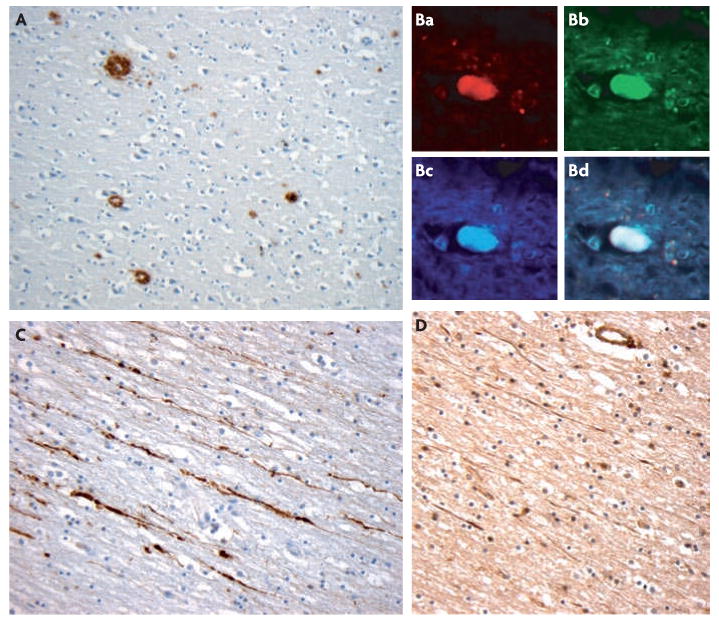
A | representative amyloid-β (Aβ) plaques (brown) found acute following a single incidence of traumatic brain injury (TBI) caused by a fall in an 18 year old male. The survival time from injury was just 10 hours. Plaques were identified using an antibody specific for Aβ. B | representative immunohistochemistry showing amyloid precursor protein (APP) (Ba), β-site-APP-cleaving enzyme (BACE) (Bb) and presenilin-1(PS-1) (Bc) co-accumulating (Bd) in the disconnected terminal of an axon, known as an axon bulb. C | Demonstration of axonal pathology using APP immunohistochemistry. APP (brown) accumulates within the tortuous varicosities along the length of damaged axons. D | Increased neprilysin immunoreactivity (brown) is also observed in damaged axons following TBI. Panel B is reproduced, with permission, from REF. 42 © (2009) International society of Neuropathology.
While TBI-associated plaques largely appear in the grey matter, they have also been identified in white matter38. The predominant type of Aβ peptide in the plaques formed after TBI, and in the soluble Aβ found in the brains of these patients, is Aβ42, the AD-associated form of Aβ that is prone to aggregation37,39. Although the plaques observed following trauma are typically diffuse40, like those observed in early AD, it is not known whether these plaques mature over time into the denser, neuritic plaques typical of advanced AD.
Although the existence of Aβ plaques following trauma in humans is generally well established34–36,38,41–43, it is only in recent years that the mechanisms driving plaque development have begun to be elucidated.
Human TBI and unaggregated Aβ peptides
In contrast to the well-characterized formation of Aβ plaques after TBI, comparatively little is known about how total brain concentrations of Aβ, including both soluble and oligomeric forms of the peptide, vary following injury.
An initial study reported an increase in the presence of Aβ in ventricular cerebrospinal fluid (CSF) following severe head trauma44, although it is important to note that control cases in this study were elderly, which makes interpretation of these findings difficult. Levels of Aβ were elevated for the first week after TBI and then declined towards control levels in the subsequent 2 weeks44. In addition, the same Aβ peptide that is predominant in plaques following TBI, Aβ42, was found in the CsF of these patients39,44,45. By contrast, a later study reported a persistent decrease in Aβ concentration in ventricular CsF from days 1–5 following severe TBI46. This contradictory finding was supported by a further study47, although in this case progression over time was not investigated and the collection time points ranged from an acute measurement to more than 9 months after injury. A further caveat of this work was that the ventricular CsF from TBI cases was compared with lumbar CsF in controls47. Clearly there is a lack of consensus on this issue. What influences the movement of Aβ from the extracellular space to cerebrospinal circulation is also unknown, particularly following TBI in which blood-brain barrier permeability and vascular integrity may be dramatically altered. Therefore, it is not apparent whether Aβ in CSF, either ventricular or lumbar, reflects Aβ concentrations in the interstitial space within the brain parenchyma.
Recent studies have used invasive intracranial microdialysis to obtain direct measurements of brain parenchymal Aβ concentrations in humans following severe TBI48,49. Although no baseline (pre-injury) data were available, one such study found that microdialysate Aβ concentrations steadily increased over time following TBI, and were correlated with improved global neurological status48. One interpretation of this finding is that depressed neuronal function following TBI decreases Aβ genesis, which subsequently returns to baseline as recovery ensues. Another study that used similar techniques failed to demonstrate any overt change in post-TBI Aβ concentrations between 27 h and 99 h after TBI49. However, they did find that patients with diffuse axonal injury (DAI) had elevated Aβ levels when compared with those with focal injuries, suggesting that the type of injury may be an important influence on Aβ dynamics.
Axonal pathology: a source of Aβ?
Axonal injury after TBI
Although it is likely that multiple sources contribute to Aβ plaque formation after TBI, axonal swellings represented an obvious pathology for examination in initial investigations. Notably, DAI is one of the most common pathologies of TBI, and independently contributes to significant morbidity and mortality50–52. A key feature of DAI is interruption of axonal transport due to cytoskeletal disruption. This causes an accumulation of proteins in the axon, including amyloid precursor protein (APP), which can be cleaved to form Aβ53–57. These accumulations occur in tortuous varicosities along the length of the axon or at the disconnected axon terminals, known as axonal bulbs. Although originally described as diffuse, the actual distribution of axonal pathology is multifocal, with varicosities and bulbs occurring throughout the deep and subcortical white matter. Swollen axons are particularly common in midline structures such as the corpus callosum58. Eventual structural disorganization of the axon can lead to secondary axonal disconnection, which ultimately culminates in a progressive, degenerative axonal pathology 59–61.
Owing to the rapid and abundant accumulation of APP in damaged axons after TBI, APP immunostaining is used for the pathological assessment of DAI in humans53–58 (FIG. 1). Accordingly, it was suspected that this large reservoir of APP in injured axons might be aberrantly cleaved to rapidly form Aβ.
Acute Aβ formation in rodent TBI models
Following the observation of acute Aβ plaque formation in humans, and speculation that the source of Aβ may be damaged axons, attention was turned to animal models of TBI to explore this process (TABLE 1). Studies using non-transgenic rat models of TBI investigated these pathologies in the acute phase following injury62,63. However, although these studies consistently found extensive intra-axonal accumulation of APP, they failed to identify Aβ, via staining, in either plaques or axons62,63.
Table 1. Animal models of traumatic brain injury and amyloid pathology.
| Species (strain) | Injury | Rummary of findings | Ref. |
|---|---|---|---|
| Mouse (APP–YAC) | Controlled cortical impact |
|
64 |
| Mouse (APPNLh/NLh) | Controlled cortical impact |
|
68 |
| Mouse (APPNLh/NLh) | Controlled cortical impact |
|
69 |
| Mouse (BACE–/–) | Controlled cortical impact |
|
70 |
| Mouse (PDAPP) | Controlled cortical impact at 4 months old |
|
65 |
| Mouse (PDAPP) | Controlled cortical impact at 4 months old |
|
66 |
| Mouse (PDAPP) | Controlled cortical impact at 2 years old |
|
67 |
| Mice (PDAAP, expressing Apoe3 or Apoe4, or Apoe–/–) | Controlled cortical impact |
|
155 |
| Rat (Sprague Dawley) | Weight drop (open skull) |
|
62 |
| Rat (Sprague Dawley) | Lateral fluid percussion |
|
63 |
| Rat (Sprague Dawley) | Weight drop (closed skull) |
|
73 |
| Rat (Sprague Dawley) | Lateral fluid percussion |
|
74 |
| Swine | Rotational acceleration (model of DAI) |
|
18 |
| Swine | Rotational acceleration (model of DAI) |
|
75 |
Aβ, amyloid-β; APP, amyloid precursor protein; BACE, β-site APP-cleaving enzyme; DAI, diffuse axonal injury; DAPT, N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2-phenyl] glycin e-1,1-dimethylethyl ester; PDAPP, platelet-derived growth factor promoter expressing amyloid precursor protein; YAC, yeast artificial chromosome.
The lack of evidence of Aβ deposition in non-transgenic rats was attributed, in part, to differences in the Aβ peptides found in different species. Accordingly, various transgenic TBI models were utilized in an attempt to replicate plaque pathology. Strains of mice were selected for their predisposition to the eventual development of Aβ plaques with ageing. In a study using mice that overexpress normal human APP, there was an increase in tissue concentrations of Aβ40 acutely after injury. However, there was no increase in plaque formation, overall pathology or altered functional outcome64. Further studies used PDAPP mice, which overexpress a mutant form of APP and develop Aβ plaque pathology as they age. TBI before plaque formation induced a surge in the tissue concentration of Aβ that was associated with an increase in hippocampal neuronal death and memory impairment, indicating the potential toxicity of Aβ65. However, even in these mice, TBI did not induce acute plaque formation. Furthermore, there was a paradoxical reduction of plaques in the PDAPP mice 8 months after TBI66. A similar reduction in plaques was seen when aged mice were subjected to injury, suggesting that regression of plaques is possible67. It was thought that the loss of hippocampal neurons after injury actually decreased the overall capacity to generate Aβ. Alternatively, trauma may have induced a surge in Aβ concentrations, which in turn upregulated the mechanisms by which Aβ is cleared.
Acute elevations of hippocampal Aβ levels in the absence of plaques were also found following injury in APPNLh/NLh mice, which have both the swedish familial AD mutation and the human Aβ sequence knocked in to their endogenous APP gene68,69. Of note, unlike the PDAPP mice, APP expression in this model is dependant on the endogenous promoter and thus continuous overexpression of APP does not occur. Using this model, it was demonstrated that, by inhibiting caspase-3 activity, injury-induced elevations in Aβ levels could be reduced in association with improved histological outcomes68. In addition to providing a potential mechanism of Aβ formation (see below), this study further supported the idea that post-TBI increases in Aβ concentrations without plaque formation may be detrimental. Similarly, mice deficient in the rate-limiting enzyme for Aβ genesis — β-amyloid converting enzyme (BACE) — were found to have significantly improved histological, radiological and behavioural outcomes following injury70.
Together, these data provide important information regarding the potentially harmful consequences of elevated Aβ levels following TBI. However, they also consistently demonstrate that rodents fail to recapitulate the plaque pathology observed acutely following human TBI.
Acute Aβ formation in a swine TBI model
Why rodent TBI models failed to recapitulate the Aβ plaque pathology found acutely after human TBI was a puzzle. Initially it was thought that differences in the Aβ sequence in rodents might prevent its aggregation into plaques. However, even mice modified to generate the human Aβ sequence failed to develop plaques. It was therefore suggested that Aβ production and deposition after TBI may depend on brain anatomy as well as the type of injury. Notably, rodents have relatively small lissencephalic brains in which white matter is sparse, meaning that only limited axonal pathology can be produced in rodent TBI models. Furthermore, most rodent models utilize impact forces to induce TBI, whereas a common cause of human DAI is rotational acceleration, such as occurs in automobile crashes.
Therefore, to more closely examine the role of DAI on Aβ generation and deposition, a swine model of head rotational acceleration was used. This animal species was selected because of its relatively large gyrencephalic brain with extensive white matter. Notably, the swine Aβ sequence is identical to that of humans. This model produces DAI that is very similar in appearance to that found clinically71,72. In addition to the anticipated accumulation of APP in swollen axons, co-accumulation of Aβ was also found18. Furthermore, this model also induced the formation of parenchymal Aβ diffuse plaques in both grey and white matter. Although the number of plaques was small compared to those found in human TBI, this model finally enabled Aβ deposition to be induced in an experimental model. Furthermore, the co-accumulation of Aβ and APP in swollen axons hinted that the potential mechanism of Aβ production after TBI is linked to traumatic axonal injury.
Persistence of Aβ formation after TBI
Following the identification and axonal accumulation of Aβ plaques in the swine TBI model, rodent TBI models were re-examined both acutely and with an expanded time frame. Initially, Aβ accumulation was identified in damaged axons shortly after injury in a rat model of TBI, albeit still in the absence of Aβ plaques73. Although there was concern about the potential cross-reactivity of the primary Aβ antibody with APP in this study, another study using a different rat model of TBI used multiple specific antibodies to confirm the axonal accumulation of Aβ74. However, in this case only limited axonal Aβ was found acutely, whereas greater axonal accumulations were found 1 month after injury and persisted for at least 1 year. Increased Aβ presence could also be seen within the neuronal somata of these animals74. Notably, these chronic increases in Aβ levels were not associated with increased expression of APP and no plaque formation was found at any post-injury time point74. These findings clearly indicated the potentially chronic nature of the trauma response with ongoing development of axonal pathology and accompanying Aβ accumulation.
Interestingly, when the swine model of head rotational acceleration injury was examined up to 6 months after injury, evidence of ongoing axonal pathology was also revealed75. Again, this was characterized by APP accumulation, often with co-accumulation of Aβ. Although it was generally thought that axonal pathology is only observed in the acute phase of injury and is cleared within a few months of trauma, these results demonstrated that axonal degeneration may chronically persist42,74,75. Moreover, the continued presence of damaged axons offers a potential mechanism by which Aβ is chronically generated. However, despite this long-term production of Aβ, the quantity of Aβ plaques in the tissue observed at 6 months following trauma in swine had not increased compared to that observed following acute injury75.
Axon pathology and Aβ in humans
Examination of human brains also confirmed the accumulation of Aβ in swollen axons shortly after TBI38. More recently long-term progressive axonal degeneration and intra-axonal Aβ accumulation was also identified following human TBI, and persisted for years following the initial trauma42. These findings demonstrate that TBI can trigger long-term neurodegenerative processes in humans. This process may account for the progressive selective atrophy of white matter found after TBI in humans76. However, the mechanisms governing this protracted disconnection and degeneration of axons are unknown. It is possible that injured axons that do not degenerate in the acute phase are nonetheless prone to later degeneration with a secondary mild insult.
Aβ genesis and plaque formation in TBI
Immunohistochemical analyses revealed that the enzymes necessary for Aβ cleavage also accumulate in injured axons after TBI. Both presenilin-1 (PS-1) and BACE were found in swollen axons in the swine model of DAI75 and in humans42,43 (FIG. 1). It seems that trauma creates a unique situation whereby all the necessary substrates for Aβ formation are forced to coexist in the same place at the same time. It has been postulated that the eventual lysis and breakdown of these damaged axons may allow the expulsion of Aβ into the parenchyma where it aggregates to cause plaque formation18,75 (FIG. 2). Interestingly, at a much slower pace, this general process of axonal transport failure has been implicated as a mechanism of plaque formation in AD77.
Figure 2. Potential mechanisms of post-traumatic amyloid-β formation and clearance.

a | The mechanical forces that axons are subjected to during a traumatic event can damage axons by directly altering their structure or by initiating detrimental secondary cascades. Failure of axonal transport in these injured axons results in accumulation of multiple proteins that form swellings at their disconnected terminals known as axon bulbs. b | such protein accumulation has been demonstrated to include the enzymes necessary for the cleavage of amyloid precursor protein (APP) to amyloid-β (Aβ), including presenilin-1 (PS-1) and β-site APP-cleaving enzyme (BACE). c–d | Although the precise intracellular mechanism of Aβ genesis remains unclear, lipid rafts have been suggested to be important in allowing APP processing and thus Aβ accumulation within the axonal compartment. e–f | Injured axons that go on to degenerate and lyse will expel the accumulated Aβ into the brain parenchyma where it is at risk of aggregating into plaques. g | The enzyme that clears Aβ, neprilsyin (NeP), also accumulates in damaged axons and probably mitigates the effects of enhanced Aβ production. The balance of genesis versus catabolism will ultimately determine Aβ build-up. NeP may potentially act to clear Aβ within the axonal compartment or in the extracellular space.
Although there is strong evidence to support the idea that axons are a source of post-traumatic Aβ, it remains unknown why plaque formation is more prevalent in the grey matter following TBI. It is clear from the multiple studies discussed above that Aβ is not only present in white matter, but may undergo a post-TBI surge at these sites. In addition, increased soluble Aβ has been found in CSF following injury. Nevertheless, there seems to be a predilection for grey matter deposition, which also seems to be the case in AD. There may be specific extracellular attributes of the grey matter that selectively promotes aggregation of Aβ into plaques. Alternatively, it is possible that Aβ genesis also occurs via a different mechanism in the grey matter. Observations of APP accumulation in synaptic terminals led to the suggestion that this may be another potential site of Aβ genesis, leading to deposition in the grey matter53. In addition, there may be multiple mechanisms driving increased Aβ production following trauma at any location. It has been postulated that elevated APP production in the neuronal soma following TBI may saturate the normal α-secretase processing pathway, resulting in increased β-secretase processing and Aβ genesis53,78. Furthermore, studies of hypoxiaischaemia suggest that Aβ genesis can be increased via oxidative-stress-mediated upregulation of BACE79,80. This BACE upregulation was later shown to be mediated by γ-secretase activity, which was also enhanced due to oxidative stress81. As oxidative stress is a well-established consequence of TBI82, its role in promoting Aβ genesis after injury warrants exploration.
Aβ dynamics in the various compartments, and their relative contributions to plaque formation and pathogenicity, are largely unknown. It is possible that TBI can result in distinct Aβ dynamics as a result of different mechanisms within the various intracellular and extracellular compartments. Elucidating these potential complexities in neuronal Aβ dynamics will be an important consideration for future studies.
Intra-axonal Aβ formation
Cleavage of APP to form Aβ within the axonal membrane compartment is not consistent with the classical description of Aβ genesis in AD. As APP is a transmembrane protein, it has long been assumed that amyloidogenic processing results in the extracellular deposition of Aβ. However, increasing evidence suggests that intracellular accumulation of Aβ is possible and potentially pathogenic83.
Recent studies have described Aβ production by BACE and PS-1 in the axonal membrane compartment of peripheral nerves in mice84,85. The authors suggested that APP, β-secretase and PS-1 are transported within the axonal compartment through a direct interaction between kinesin-1 and APP84,85. The observation of Aβ within the axonal compartment led to the suggestion that amyloidogenic cleavage of APP can occur during transportation84. However, these findings have been directly challenged by another study that failed to demonstrate co-transportation of either PS-1 or BACE1 with APP in the same murine peripheral nerve86. Furthermore, they were unable to detect Aβ peptides within this nerve. The authors suggest that, at least within the peripheral nervous system, intraxonal transport of the machinery of Aβ genesis is unlikely. Whether this is also true for the CNS is unknown.
Intra-axonal processing of membranebound APP may involve lipid rafts — cholesterol-rich microdomains of the plasma membrane that are thought to compartmentalize cellular processes87, 88. Indeed, there is evidence indicating that lipid rafts may be important in amyloidogenic processing of APP in neurons89. Axons are also abundant with lipid-rich invaginations of plasma membrane known as caveolae, which may provide another intra-axonal site of APP processing. It is possible that mechanical damage to axons due to trauma may cause changes in linear lipid rafts or caveolae that promote abnormal APP processing. A recent study using APPNLh/NLh mice showed that administration of the cholesterol-lowering drug simvastatin diminished increases in Aβ levels following injury69. Associated histological and behavioural outcomes were also improved. Although the mechanisms by which simvastatin modulates Aβ dynamics may be complex, further exploration of its possible role in post-traumatic Aβ processing will be of interest.
Caspases, TBI and Aβ processing
Cysteine-dependent aspartate-specific proteases (caspases) have been identified as important in Aβ genesis90. This finding is of particular interest with regards to TBI, in which increased caspase activation has been described in humans and in animal models75,91–94.
Following controlled cortical impact in APP mice, pharmacological inhibition of injury-induced caspase-3 activation using a pan-caspase inhibitor (Boc-Asp(OMe)-CH2F) reduced both caspase-3-mediated APP processing and acute elevations in Aβ concentrations68. In addition, there was decreased neuron degeneration and tissue loss. A further study demonstrated caspase-3-mediated APP proteolysis in traumatically injured axons following head impact in a rat73.
Although the precise mechanisms by which caspases act to increase Aβ levels are not clear, recent work indicates that caspase-3 may increase APP processing by increasing the availability of BACE via an adaptor molecule (GGA3) that interrupts BACE trafficking and prevents its degradation95. Elevated BACE levels and activity would promote amyloidogenic processing of APP, potentially leading to increased Aβ genesis. Interestingly, elevated BACE levels and activity have been found following injury in a rat model of TBI96.
This work highlights the need for better understanding of the mechanisms of Aβ genesis in the post-trauma situation. Little is known about how, or if, β-secretase and γ-secretase activity are altered by trauma. Furthermore, how this influences the role of the non-amyloidogenic α-secretase pathway is largely unknown.
Aβ catabolism in TBI
The observed variations in plaque pathology in humans and in animal models of TBI provided valuable clues regarding potential mechanisms of plaque genesis. They suggested that some species, and perhaps certain individuals, are able to clear Aβ more efficiently than others, potentially because of differences in the extent or activity of Aβ degrading mechanisms. This revelation turned attention to potential mechanisms of Aβ clearance after TBI.
Neprilysin, a membrane zinc metallo-endopeptidase, has emerged as a significant Aβ degrading enzyme in vivo97,98, although there are others including insulin degrading enzyme99. Neprilysin is transcribed in a tissue-specific manner100,101, operates as a transmembrane glycoprotein102,103 and is capable of degrading monomeric and potentially oligomeric forms of Aβ104. Furthermore, neprilysin knockout mice have been shown to accumulate Aβ40 and Aβ42 in a gene dose-dependant manner105. Neprilysin has been increasingly implicated in the pathogenesis of AD106. Indeed, patients with sporadic AD were found to have up to a 50% reduction in neprilysin mRNA in areas associated with plaque pathology107.
Neprilysin following human TBI
It has recently been observed that immuno-reactivity to neprilysin increases for many months following TBI in humans42. Extensive neprilysin immunoreactivity was found in the neuronal soma and axonal bulbs of long-term survivors of TBI (FIG. 1). Interestingly, these cases comprised the same group that have a virtual absence of Aβ plaques. These findings suggest that these individuals may have a long-term upregulation of neprilysin that continually clears both intracellular and/or extra-cellular Aβ, thereby promoting plaque regression. Moreover, the findings suggest that neprilysin may have an important role in mitigating chronic Aβ production induced by trauma. Although the site at which neprilysin acts to achieve this has not been identified, the presence of neprilysin within axons suggests that intra-axonal clearance is one possible mechanism. However, it remains to be determined whether neprilysin is able to function extracellulary to clear plaques directly or whether plaque turnover is regulated by some other means.
Notably, microglia containing Aβ have been found in association with plaques after TBI42, suggesting that phagocytic clearance of plaques may occur. It is possible that the extent of the microglial response following injury may influence plaque burden. Like neprilysin, this could potentially contribute to variations in plaque pathology between individuals and indeed animal species. In support of this idea, recent work suggests that increased microglial activation is the mechanism by which passive Aβ immunization therapies act to clear plaques in AD models108. Interestingly, microglia also appear to utilize neprilysin to process Aβ109.
Neprilysin polymorphisms and Aβ plaques
Neprilysin expression is probably regulated by multiple mechanisms, including the production of Aβ itself, which has been suggested to trigger a positive-feedback loop110,111. Genetic variation may also influence the extent of expression or activity of neprilysin, which potentially accounts for the presence of Aβ plaques in only 30% of TBI patients. Indeed a recent study identified a relationship between a neprilysin polymorphism and Aβ plaque pathology acutely following TBI112, although whether this association is functionally significant or a result of genetic linkage is unknown. Further studies may be of value in determining how this polymorphism affects both short-term and long-term clinical outcome. As such, a genetic screening test of this neprilysin polymorphism could potentially help to identify individuals at risk of plaque formation, which may be an important consideration for those involved in military action or contact sports (BOX 2).
Box 2. Repetitive traumatic brain injury.
In 1928, the term ‘punch drunk syndrome’ was introduced to describe a disorder of progressive dementia that develops after repetitive traumatic brain injury (TBI) from boxing156. Now termed ‘dementia pugilistica’157, this syndrome can present many years after retiring from boxing158, affecting as many as 17% of retired boxers159. The incidence of dementia pugilistica increases with duration of time spent boxing159, number of knockouts experienced160 and apolipoprotein E (APOE) genotype161. Although this work has been centred on boxers, increasing evidence suggests that repetitive TBI experienced from playing professional American football can result in increased rates of late-life cognitive impairment162. Furthermore, recent pathological studies of the brains of former players in the National Football League in the United States of America revealed extensive Alzheimer's disease (AD)-like neurofibrillary tangles (NFTs) and, in some cases, amyloid-β (Aβ) plaques163–165.
Although the predominant pathology of dementia pugilistica is the formation of AD-like NFTs, Aβ pathologies later emerged as a potentially important finding. As with single TBI and AD, diffuse Aβ plaques are found in the brain158,166. Deposits of Aβ have also been observed in both leptomeningeal and cortical blood vessels166. More recently, accelerated Aβ deposition was observed following a model of mild repetitive TBI in Tg2576 mice (known to develop Aβ plaques with ageing)167,168. This increased deposition was found in association with evidence of increased lipid peroxidation and could be reversed by pretreatment with oral vitamin E168.
NFTs and neuropil threads are an important feature of both AD and dementia pugilistica19,158,165,169–173. These intracellular structures contain abnormal (hyperphosphorylated) forms of the microtubule-associated protein tau. One study found NFTs and neuropil threads in the neocortex of five cases of repetitive, mild head trauma in young patients19. In addition, the molecular profile and ubiquitylation of tau in dementia pugilistica was similar to that observed in the filamentous tau inclusions seen in AD173,174.
In contrast to well-developed tau pathology in dementia pugilistica, NFTs were not found to be acutely increased after a single TBI175. Although tau has also been observed accumulating in axons following trauma, this was only found in a small subset of patients36,43. In a study in which pigs received single experimental brain injuries, accumulations of tau were observed in a limited number of neuronal perikarya18. Rats also demonstrated phosphorylated tau accumulation in neurons at 6 months after injury176.
App and Aβ in TBI pathophysiology
It is unclear whether the accumulation of large quantities of APP in damaged axons after TBI serves a mechanistic role or is simply an epiphenomenon. It has been suggested that APP and its non-amyloidogenic processing have beneficial effects with respect to neuroprotection, neurite outgrowth and synaptogenesis113–120. In addition, the administration of soluble APPα (the product of non-amyloidogenic processing via the α-secretase pathway) improved functional outcome and reduced neuronal cell loss and axonal injury following TBI in rats120.
The role of Aβ plaques in TBI outcome has also not yet been established. Even in AD, considerable debate remains over the pathogenicity of Aβ plaques (as opposed to its unaggregated soluble forms)121–123. Indeed, a recent investigation in which apparent Aβ plaque clearance followed Aβ immunization in patients with AD failed to demonstrate altered clinical outcome124. Furthermore, recent evidence suggests that unaggregated oligomeric forms of Aβ may contribute to toxicity125–130. As such, rapid aggregation of Aβ into plaques may be a protective event. An evaluation of the role of oligomeric Aβ following TBI will therefore be an important future consideration. So far, only one study has implicated unaggregated Aβ toxicity in neuron death after TBI. As described above, TBI in PDAPP mice resulted in extensive hippocampal neuron death and associated neurocognitive impairment in association with a local surge in unaggregated Aβ levels in the hippocampus65. This may suggest that the toxic properties of Aβ only emerge when levels exceed a certain threshold131.
Unaggregated Aβ could have other deleterious effects in TBI. Solubilized Aβ injected into the brains of rodents has profound effects on cognition and long-term potentiation — an electrophysiological correlate of some forms of learning and memory132–134. Thus, after TBI an acute surge in Aβ concentrations, as well as its continued production for years, may influence functional recovery.
Conclusions and future directions
The link between trauma and the later development of neurodegenerative diseases such as AD is likely to be extremely complex, and work in this field remains in its infancy. One recurring issue in this field of research is the involvement of axons and the pathological accumulation of multiple proteins within axons damaged by trauma. This parallels the increasing emphasis on axonal involvement in the pathogenesis of AD, particularly axonal transport defects.
TBI is a common, devastating disorder and is the leading cause of death in children and young adults135. The later development of AD or neurodegenerative disease comes not only at a human cost, but also constitutes a considerable socioeconomic burden. Hence, this is an avenue of research of potential importance to almost everyone, and may have particular significance to those at high risk of TBI, such as those involved in contact sports players or in the military.
A mechanistic understanding of what drives the risk of developing AD following TBI will be imperative for the development of post-trauma interventions aimed at halting the onslaught of such debilitating neurodegeneration. Furthermore, the advancement of drug discoveries in the field of AD, such as BACE and γ-secretase inhibitors70,136 or neprilysin replacement strategies, may have potentially important roles in both the acute and chronic management of TBI.
Databases
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
UniProtKB: http://ca.expasy.org/sprot
APP | BACE1 | caspase-3 | neprilysin | PS-1
Further Information
Penn center for Brain injury and repair: http://www.med.upenn.edu/cbir
Smith Laboratory Homepage: http://www.uphs.upenn.edu/neurosurgery/smithlab
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Acknowledgments
This work was supported by US National Institutes of Health grants NS038104 and NS056202.
Footnotes
Competing interests statement: The authors declare no competing financial interests
Contributor Information
Victoria E. Johnson, Penn Center for Brain Injury and Repair, Department of Neurosurgery, University of Pennsylvania School of Medicine, 3320 Smith Walk, Hayden Hall 105, Philadelphia, Pennsylvania 19104, USA, and at the University of Glasgow, University Avenue, Glasgow G12 8QQ,UK
William Stewart, Department of Neuropathology, Southern General Hospital, 1345 Govan Road, Glasgow G51 4TF, UK, and at the University of Glasgow.
Douglas H. Smith, Penn Center for Brain Injury and Repair, Department of Neurosurgery, University of Pennsylvania School of Medicine, 3320 Smith Walk, Hayden Hall 105, Philadelphia, Pennsylvania 19104, USA
References
- 1.Langlois JA, Rutland-Brown W, Thomas KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Centers for Disease Control and Prevention Nation Center for Injury Prevention and Control; Atlanta, Georgia: 2006. [Google Scholar]
- 2.Thurman D, Alverson C, Dunn K, Guerrero J, Sniezek J. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Molgaard CA, et al. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 1990;9:233–242. doi: 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- 4.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer's disease. Neurology. 1985;35:264–267. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- 5.Mortimer JA, et al. Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case–control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl. 2):28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 6.Graves AB, et al. The association between head trauma and Alzheimer's disease. Am J Epidemiol. 1990;131:491–501. doi: 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- 7.O'Meara ES, et al. Head injury and risk of Alzheimer's disease by apolipoprotein E genotype. Am J Epidemiol. 1997;146:373–384. doi: 10.1093/oxfordjournals.aje.a009290. [DOI] [PubMed] [Google Scholar]
- 8.Salib E, Hillier V. Head injury and the risk of Alzheimer's disease: a case control study. Int J Geriatr Psychiatry. 1997;12:363–368. doi: 10.1002/(sici)1099-1166(199703)12:3<363::aid-gps515>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 9.Guo Z, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 10.Schofield PW, et al. Alzheimer's disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997;62:119–124. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plassman BL, et al. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 12.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotapka MJ, Graham DI, Adams JH, Gennarelli TA. Hippocampal pathology in fatal non-missile human head injury. Acta Neuropathol. 1992;83:530–534. doi: 10.1007/BF00310031. [DOI] [PubMed] [Google Scholar]
- 14.Smith DH, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 15.Maxwell WL, Mackinnon MA, Stewart JE, Graham DI. Stereology of cerebral cortex after traumatic brain injury matched to the Glasgow Outcome Score. Brain. 2010;133:139–160. doi: 10.1093/brain/awp264. [DOI] [PubMed] [Google Scholar]
- 16.Gentleman SM, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 17.Nicoll JA, et al. Association of interleukin-1 gene polymorphisms with Alzheimer's disease. Ann Neurol. 2000;47:365–368. [PMC free article] [PubMed] [Google Scholar]
- 18.Smith DH, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 20.van Duijn CM, et al. Head trauma and the risk of Alzheimer's disease. Am J Epidemiol. 1992;135:775–782. doi: 10.1093/oxfordjournals.aje.a116364. [DOI] [PubMed] [Google Scholar]
- 21.Chandra V, Philipose V, Bell PA, Lazaroff A, Schoenberg BS. Case–control study of late onset “probable Alzheimer's disease”. Neurology. 1987;37:1295–1300. doi: 10.1212/wnl.37.8.1295. [DOI] [PubMed] [Google Scholar]
- 22.Amaducci LA, et al. Risk factors for clinically diagnosed Alzheimer's disease: a case–control study of an Italian population. Neurology. 1986;36:922–931. doi: 10.1212/wnl.36.7.922. [DOI] [PubMed] [Google Scholar]
- 23.Broe GA, et al. A case–control study of Alzheimer's disease in Australia. Neurology. 1990;40:1698–1707. doi: 10.1212/wnl.40.11.1698. [DOI] [PubMed] [Google Scholar]
- 24.Ferini-Strambi L, Smirne S, Garancini P, Pinto P, Franceschi M. Clinical and epidemiological aspects of Alzheimer's disease with presenile onset: a case control study. Neuroepidemiology. 1990;9:39–49. doi: 10.1159/000110750. [DOI] [PubMed] [Google Scholar]
- 25.Katzman R, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol. 1989;25:317–324. doi: 10.1002/ana.410250402. [DOI] [PubMed] [Google Scholar]
- 26.Launer LJ, et al. Rates and risk factors for dementia and Alzheimer's disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups European Studies of Dementia. Neurology. 1999;52:78–84. doi: 10.1212/wnl.52.1.78. [DOI] [PubMed] [Google Scholar]
- 27.Williams DB, Annegers JF, Kokmen E, O'Brien PC, Kurland LT. Brain injury and neurologic sequelae: a cohort study of dementia, parkinsonism, and amyotrophic lateral sclerosis. Neurology. 1991;41:1554–1557. doi: 10.1212/wnl.41.10.1554. [DOI] [PubMed] [Google Scholar]
- 28.Mehta KM, et al. Head trauma and risk of dementia and Alzheimer's disease: the Rotterdam study. Neurology. 1999;53:1959–1962. doi: 10.1212/wnl.53.9.1959. [DOI] [PubMed] [Google Scholar]
- 29.Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- 30.Sullivan P, Petitti D, Barbaccia J. Head trauma and age of onset of dementia of the Alzheimer type. JAMA. 1987;257:2289–2290. doi: 10.1001/jama.1987.03390170045014. [DOI] [PubMed] [Google Scholar]
- 31.Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E. Severe head injury hastens age of onset of Alzheimer's disease. J Am Geriatr Soc. 1989;37:970–973. doi: 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 32.Nemetz PN, et al. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am J Epidemiol. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- 33.Victor MR, Ropper AH. Adams and Victor's Principles of Neurology. McGraw-Hill; 2001. pp. 1–1692. [Google Scholar]
- 34.Roberts GW, Gentleman SM, Lynch A, Graham DI. βA4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 35.Roberts GW, et al. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikonomovic MD, et al. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 37.DeKosky ST, et al. Association of increased cortical soluble Aβ42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64:541–544. doi: 10.1001/archneur.64.4.541. [DOI] [PubMed] [Google Scholar]
- 38.Smith DH, Chen XH, Iwata A, Graham DI. Amyloid β accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 39.Gentleman SM, et al. Aβ42 is the predominant form of amyloid β-protein in the brains of short-term survivors of head injury. Neuroreport. 1997;8:1519–1522. doi: 10.1097/00001756-199704140-00039. [DOI] [PubMed] [Google Scholar]
- 40.Horsburgh K, et al. β-amyloid (Aβ)42(43), Aβ42, Aβ40 and apoE immunostaining of plaques in fatal head injury. Neuropathol Appl Neurobiol. 2000;26:124–132. doi: 10.1046/j.1365-2990.2000.026002124.x. [DOI] [PubMed] [Google Scholar]
- 41.Huber A, Gabbert K, Kelemen J, Cervod-Navarro J. Density of amyloid plaques in brains after head trauma. J Neurotrauma. 1993;10(Suppl. 1):180. [Google Scholar]
- 42.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid β plaques despite persistent accumulation of amyloid β in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uryu K, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raby CA, et al. Traumatic brain injury increases β-amyloid peptide 1–42 in cerebrospinal fluid. J Neurochem. 1998;71:2505–2509. doi: 10.1046/j.1471-4159.1998.71062505.x. [DOI] [PubMed] [Google Scholar]
- 45.Emmerling MR, et al. Traumatic brain injury elevates the Alzheimer's amyloid peptide Aβ42 in human CSF. A possible role for nerve cell injury. Ann NY Acad Sci. 2000;903:118–122. doi: 10.1111/j.1749-6632.2000.tb06357.x. [DOI] [PubMed] [Google Scholar]
- 46.Kay AD, et al. Alterations in cerebrospinal fluid apolipoprotein E and amyloid β-protein after traumatic brain injury. J Neurotrauma. 2003;20:943–952. doi: 10.1089/089771503770195795. [DOI] [PubMed] [Google Scholar]
- 47.Franz G, et al. Amyloid β 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60:1457–1461. doi: 10.1212/01.wnl.0000063313.57292.00. [DOI] [PubMed] [Google Scholar]
- 48.Brody DL, et al. Amyloid-β dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marklund N, et al. Monitoring of brain interstitial total tau and β amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227–1237. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- 50.Geddes JF, Vowles GH, Beer TW, Ellison DW. The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol. 1997;23:339–347. [PubMed] [Google Scholar]
- 51.Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol. 2000;26:105–116. doi: 10.1046/j.1365-2990.2000.026002105.x. [DOI] [PubMed] [Google Scholar]
- 52.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 53.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 54.Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite PM. Axonal injury in children after motor vehicle crashes: extent, distribution, and size of axonal swellings using β-APP immunohistochemistry. J Neurotrauma. 2002;19:1171–1182. doi: 10.1089/08977150260337976. [DOI] [PubMed] [Google Scholar]
- 55.Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- 56.Lambri M, Djurovic V, Kibble M, Cairns N, Al-Sarraj S. Specificity and sensitivity of βAPP in head injury. Clin Neuropathol. 2001;20:263–271. [PubMed] [Google Scholar]
- 57.Reichard RR, White CL, 3rd, Hladik CL, Dolinak D. Beta-amyloid precursor protein staining of nonaccidental central nervous system injury in pediatric autopsies. J Neurotrauma. 2003;20:347–355. doi: 10.1089/089771503765172309. [DOI] [PubMed] [Google Scholar]
- 58.Adams JH, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 59.Povlishock JT, Becker DP. Fate of reactive axonal swellings induced by head injury. Lab Invest. 1985;52:540–552. [PubMed] [Google Scholar]
- 60.Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- 61.Maxwell WL, Domleo A, McColl G, Jafari SS, Graham DI. Post-acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma. 2003;20:151–168. doi: 10.1089/08977150360547071. [DOI] [PubMed] [Google Scholar]
- 62.Lewen A, Li GL, Nilsson P, Olsson Y, Hillered L. Traumatic brain injury in rat produces changes of β-amyloid precursor protein immunoreactivity. Neuroreport. 1995;6:357–360. doi: 10.1097/00001756-199501000-00032. [DOI] [PubMed] [Google Scholar]
- 63.Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and β-amyloid peptide after experimental brain injury in the rat. J Neurosci. 1996;16:1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murai H, et al. Twofold overexpression of human β-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J Comp Neurol. 1998;392:428–438. doi: 10.1002/(sici)1096-9861(19980323)392:4<428::aid-cne2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 65.Smith DH, et al. Brain trauma induces massive hippocampal neuron death linked to a surge in β-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol. 1998;153:1005–1010. doi: 10.1016/s0002-9440(10)65643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakagawa Y, et al. Traumatic brain injury in young, amyloid-β peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Aβ deposition during aging. J Comp Neurol. 1999;411:390–398. [PubMed] [Google Scholar]
- 67.Nakagawa Y, et al. Brain trauma in aged transgenic mice induces regression of established Aβ deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 68.Abrahamson EE, et al. Caspase inhibition therapy abolishes brain trauma-induced increases in Aβ peptide: implications for clinical outcome. Exp Neurol. 2006;197:437–450. doi: 10.1016/j.expneurol.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 69.Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST. Simvastatin therapy prevents brain trauma-induced increases in β-amyloid peptide levels. Ann Neurol. 2009;66:407–414. doi: 10.1002/ana.21731. [DOI] [PubMed] [Google Scholar]
- 70.Loane DJ, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature Med. 2009;15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith DH, et al. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol. 1997;56:822–834. [PubMed] [Google Scholar]
- 72.Meaney DF, et al. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- 73.Stone JR, et al. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid β peptide in traumatic axonal injury. J Neurotrauma. 2002;19:601–614. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- 74.Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-β in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 2002;61:1056–1068. doi: 10.1093/jnen/61.12.1056. [DOI] [PubMed] [Google Scholar]
- 75.Chen XH, et al. Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bigler ED. Quantitative magnetic resonance imaging in traumatic brain injury. J Head Trauma Rehabil. 2001;16:117–134. doi: 10.1097/00001199-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 77.Stokin GB, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 78.Graham DI, et al. Altered β-APP metabolism after head injury and its relationship to the aetiology of Alzheimer's disease. Acta Neurochir Suppl. 1996;66:96–102. doi: 10.1007/978-3-7091-9465-2_17. [DOI] [PubMed] [Google Scholar]
- 79.Tamagno E, et al. β-Site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 80.Guglielmotto M, et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1α. J Neurochem. 2009;108:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- 81.Tamagno E, et al. Oxidative stress activates a positive feedback between the γ- and β-secretase cleavages of the β-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Povlishock JT, Kontos HA. The role of oxygen radicals in the pathobiology of traumatic brain injury. Hum Cell. 1992;5:345–353. [PubMed] [Google Scholar]
- 83.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nature Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 84.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 85.Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 86.Lazarov O, et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simons K, Toomre D. Lipid rafts and signal transduction. Nature Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 88.Pike LJ. Rafts defined: a report on the Keystone symposium on lipid rafts and cell function. J Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 89.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gervais FG, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 91.Clark RS, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–821. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- 92.Knoblach SM, et al. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19:1155–1170. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- 93.Yakovlev AG, et al. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1399. doi: 10.1097/00006123-200106000-00051. discussion 1399–1401. [DOI] [PubMed] [Google Scholar]
- 95.Tesco G, et al. Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blasko I, et al. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease β-secretase (BACE-1) J Neural Transm. 2004;111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- 97.Shirotani K, et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem. 2001;276:21895–21901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- 98.Iwata N, et al. Identification of the major Aβ1– 42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 99.Miners JS, et al. Aβ-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li C, et al. Comparison of the structure and expression of the human and rat neprilysin (endopeptidase 24.11)-encoding genes. Gene. 1995;164:363–366. doi: 10.1016/0378-1119(95)00464-h. [DOI] [PubMed] [Google Scholar]
- 101.Li C, Hersh LB. Characterization of the promoter region of the rat neprilysin gene. Arch Biochem Biophys. 1998;358:189–195. doi: 10.1006/abbi.1998.0855. [DOI] [PubMed] [Google Scholar]
- 102.Roques BP, Noble F, Dauge V, Fournie-Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- 103.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 104.Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113–116. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- 105.Iwata N, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 106.Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-β peptide and Alzheimer's disease. Pharmacol Ther. 2005;108:129–148. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 107.Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- 108.Koenigsknecht-Talboo J, et al. Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pardossi-Piquard R, et al. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron. 2005;46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 111.Mohajeri MH, Wollmer MA, Nitsch RM. Aβ 42-induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J Biol Chem. 2002;277:35460–35465. doi: 10.1074/jbc.M202899200. [DOI] [PubMed] [Google Scholar]
- 112.Johnson V, et al. A neprilysin polymorphism and amyloid β plaques following traumatic brain injury in humans. J Neurotrauma. 2009 Mar 27; doi: 10.1089/neu.2008-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mattson MP, et al. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 114.Milward EA, et al. The amyloid protein precursor of Alzheimer's disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 115.Roch JM, Jin LW, Ninomiya H, Schubert D, Saitoh T. Biologically active domain of the secreted form of the amyloid β/A4 protein precursor. Ann NY Acad Sci. 1993;695:149–157. doi: 10.1111/j.1749-6632.1993.tb23044.x. [DOI] [PubMed] [Google Scholar]
- 116.Small DH, et al. Neurite-outgrowth regulating functions of the amyloid protein precursor of Alzheimer's disease. J Alzheimers Dis. 1999;1:275–285. doi: 10.3233/jad-1999-14-508. [DOI] [PubMed] [Google Scholar]
- 117.Small DH, et al. A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morimoto T, Ohsawa I, Takamura C, Ishiguro M, Kohsaka S. Involvement of amyloid precursor protein in functional synapse formation in cultured hippocampal neurons. J Neurosci Res. 1998;51:185–195. doi: 10.1002/(SICI)1097-4547(19980115)51:2<185::AID-JNR7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 119.Van den Heuvel C, et al. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp Neurol. 1999;159:441–450. doi: 10.1006/exnr.1999.7150. [DOI] [PubMed] [Google Scholar]
- 120.Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. doi: 10.1016/j.brainres.2006.03.107. [DOI] [PubMed] [Google Scholar]
- 121.Terry RD, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 122.Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 123.Engler H, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 124.Holmes C, et al. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 125.Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hartley DM, et al. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 128.Cleary JP, et al. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nature Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 129.Lee EB, et al. Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 130.Lesne S, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 131.Pearson HA, Peers C. Physiological roles for amyloid β peptides. J Physiol. 2006;575:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mi to gen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Walsh DM, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 134.Shankar GM, et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 136.Bateman RJ, et al. A γ-secretase inhibitor decreases amyloid-β production in the central nervous system. Ann Neurol. 2009;66:48–54. doi: 10.1002/ana.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 138.Saunders AM, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 139.Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–1071. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 140.Sorbi S, et al. ApoE as a prognostic factor for post-traumatic coma. Nature Med. 1995;1:852. doi: 10.1038/nm0995-852. [DOI] [PubMed] [Google Scholar]
- 141.Friedman G, et al. Apolipoprotein E-ε4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–248. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 142.Liberman JN, Stewart WF, Wesnes K, Troncoso J. Apolipoprotein E ε4 and short-term recovery from predominantly mild brain injury. Neurology. 2002;58:1038–1044. doi: 10.1212/wnl.58.7.1038. [DOI] [PubMed] [Google Scholar]
- 143.Sundstrom A, et al. APOE influences on neuropsychological function after mild head injury: within-person comparisons. Neurology. 2004;62:1963–1966. doi: 10.1212/01.wnl.0000129268.83927.a8. [DOI] [PubMed] [Google Scholar]
- 144.Lichtman SW, Seliger G, Tycko B, Marder K. Apolipoprotein E and functional recovery from brain injury following postacute rehabilitation. Neurology. 2000;55:1536–1539. doi: 10.1212/wnl.55.10.1536. [DOI] [PubMed] [Google Scholar]
- 145.Liaquat I, Dunn LT, Nicoll JA, Teasdale GM, Norrie JD. Effect of apolipoprotein E genotype on hematoma volume after trauma. J Neurosurg. 2002;96:90–96. doi: 10.3171/jns.2002.96.1.0090. [DOI] [PubMed] [Google Scholar]
- 146.Smith C, Graham DI, Murray LS, Stewart J, Nicoll JA. Association of APOE ε4 and cerebrovascular pathology in traumatic brain injury. J Neurol Neurosurg Psychiatry. 2006;77:363–366. doi: 10.1136/jnnp.2005.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Diaz-Arrastia R, et al. Increased risk of late posttraumatic seizures associated with inheritance of APOE ε4 allele. Arch Neurol. 2003;60:818–822. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- 148.Chamelian L, Reis M, Feinstein A. Six-month recovery from mild to moderate traumatic brain injury the role of APOE-ε4 allele. Brain. 2004;127:2621–2628. doi: 10.1093/brain/awh296. [DOI] [PubMed] [Google Scholar]
- 149.Nathoo N, Chetry R, van Dellen JR, Connolly C, Naidoo R. Apolipoprotein E polymorphism and outcome after closed traumatic brain injury: influence of ethnic and regional differences. J Neurosurg. 2003;98:302–306. doi: 10.3171/jns.2003.98.2.0302. [DOI] [PubMed] [Google Scholar]
- 150.Teasdale GM, Murray GD, Nicoll JA. The association between APOE ε4, age and outcome after head injury: a prospective cohort study. Brain. 2005;128:2556–2561. doi: 10.1093/brain/awh595. [DOI] [PubMed] [Google Scholar]
- 151.Mayeux R, et al. Synergistic effects of traumatic head injury and apolipoprotein-ε4 in patients with Alzheimer's disease. Neurology. 1995;45:555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 152.Mayeux R, et al. Genetic susceptibility and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first-degree relatives. Ann Neurol. 1993;33:494–501. doi: 10.1002/ana.410330513. [DOI] [PubMed] [Google Scholar]
- 153.Mauri M, et al. Interaction between Apolipoprotein ε4 and traumatic brain injury in patients with Alzheimer's disease and mild cognitive impairment. Funct Neurol. 2006;21:223–228. [PubMed] [Google Scholar]
- 154.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E ε4 allele is associated with deposition of amyloid β-protein following head injury. Nature Med. 1995;1:135–137. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 155.Hartman RE, et al. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer's disease. J Neurosci. 2002;22:10083–10087. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Martland H. Punch drunk. J Am Med Assoc. 1928;91:1103–1107. [Google Scholar]
- 157.Millspaugh J. Dementia pugilistica. US Naval Med Bull. 1937;35:297–303. [Google Scholar]
- 158.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–378. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roberts A. Brain Damage in Boxers. Pitman Publishing; London: 1969. [Google Scholar]
- 160.Jordan BD, et al. CT of 338 active professional boxers. Radiology. 1992;185:509–512. doi: 10.1148/radiology.185.2.1410364. [DOI] [PubMed] [Google Scholar]
- 161.Jordan BD, et al. Apolipoprotein E ε4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–140. [PubMed] [Google Scholar]
- 162.Guskiewicz KM, et al. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. 2005;57:719–726. doi: 10.1093/neurosurgery/57.4.719. [DOI] [PubMed] [Google Scholar]
- 163.Omalu BI, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. [DOI] [PubMed] [Google Scholar]
- 164.Omalu BI, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. discussion 1092–1093. [DOI] [PubMed] [Google Scholar]
- 165.McKee AC, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG. Re-examination of ex-boxers' brains using immunohistochemistry with antibodies to amyloid β-protein and tau protein. Acta Neuropathol. 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 167.Uryu K, et al. Repetitive mild brain trauma accelerates Aβ deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Conte V, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 169.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 170.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 171.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nature Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 172.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 173.Dale GE, Leigh PN, Luthert P, Anderton BH, Roberts GW. Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J Neurol Neurosurg Psychiatry. 1991;54:116–118. doi: 10.1136/jnnp.54.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 175.Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 2003;29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 176.Hoshino S, et al. Emergence of immunoreactivities for phosphorylated tau and amyloid-β protein in chronic stage of fluid percussion injury in rat brain. Neuroreport. 1998;9:1879–1883. doi: 10.1097/00001756-199806010-00039. [DOI] [PubMed] [Google Scholar]